
Abstract
Neurodegenerative disorders are typically characterized by late onset progressive damage to specific (sub)populations of cells of the nervous system that are essential for mobility, coordination, strength, sensation, and cognition. Addressing this selective cellular vulnerability has become feasible with the emergence of single-cell-omics technologies, which now represent the state-of-the-art approach to profile heterogeneity of complex tissues including human post-mortem brain at unprecedented resolution. In this review, we briefly recapitulate the experimental workflow of single-cell RNA sequencing and summarize the recent knowledge acquired with it in the most common neurodegenerative diseases: Parkinson’s, Alzheimer’s, Huntington’s disease, and multiple sclerosis. We also discuss the possibility of applying single-cell approaches in the diagnostics and therapy of neurodegenerative disorders, as well as the limitations. While we are currently at the point of deeply exploring the transcriptomic changes in the affected cells, further technological developments hold a promise of manipulating the affected pathways once we understand them better.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Single-cell sequencing enables profiling of cellular heterogeneity within a tissue. |
Vulnerability of specific cell types is a hallmark of neurodegenerative diseases and can be addressed by this technology. |
This review provides a summary of the recent knowledge in Parkinson’s, Alzheimer’s, Huntington’s disease, and multiple sclerosis, acquired through single-cell sequencing. |
1 Introduction
Neurodegenerative disorders (ND) are caused by a progressive loss of cells in the central nervous system (e.g., neurons), in a process known as neurodegeneration [1]. They affect millions of people worldwide, and the likelihood of developing a ND rises dramatically with age, especially as life expectancy increases. The most common ND include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS) [2,3,4,5,6]. While in some cases the cause of ND can be explained by a well-known genetic alteration, others are idiopathic, or even caused by environmental factors [7, 8]. Given that most of these disorders display a later age at onset (AAO) and a progressive course, understanding the cellular mechanisms that precede the pathological changes has been a long-standing goal in neuroscience, with the final aim being to correct and/or prevent them [9].
Initial neuroanatomical and molecular investigations established the brain regions affected in these disorders as well as the pathogenic events: the entorhinal cortex is primarily affected in AD, with deposits of amyloid beta and tau protein [2], and a progressive loss of dopaminergic neurons in the substantia nigra occurs in PD [3], while striatum is the affected tissue in HD that is caused by CAG repeat expansion in the huntingtin gene (HTT) [4] (Fig. 1). However, each of these brain regions consists of multiple cell types, and often more than a single region is affected—also in varying degrees. Traditional approaches such as immunostaining and histology often lack the necessary throughput and the resolution to obtain an unbiased global view of all cell types and genes involved in ND [10, 11]. The recent introduction of single-cell genomics approaches offers powerful tools to quantify cellular composition changes and gene expression alterations in any tissue, organ, or entire organism (https://www.humancellatlas.org/) [12,13,14,15].

Affected brain regions in the most common common neurodegenerative disorders, along with significant publications on sc/snRNA-seq
In this review article, we summarize the recent advances on using single-cell genomics approaches to understand the most common neurodegenerative disorders in human post-mortem brain tissue, induced pluripotent stem cells (iPSCs), and animal models, with a focus on PD.
2 Single-Cell and Single-Nuclei RNA Sequencing
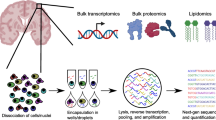
Recently, the advancements in single-cell and single-nucleus RNA sequencing have allowed both fresh and frozen tissue profiling at high resolution, enabling a simultaneous assessment of transcriptomes of thousands of cells [16]. In contrast to the traditional bulk RNA sequencing that provides only an average gene expression signal for a sequenced population of cells, single-cell sequencing distinguishes gene expression profiles of each individual cell. By preserving cellular heterogeneity, this method allows the detection of even small, disease-specific subpopulations of cells [17]. In general, the workflow starts with tissue acquisition (Fig. 2a), which then determines whether individual cells or nuclei will be isolated: cells are isolated from fresh tissues, while nuclei are isolated from frozen tissues (e.g., brains). Tissue acquisition is the crucial step in the workflow, because it determines the RNA quality—in the case of human brains, post-mortem intervals should be as short as possible, tissue dissection should be performed at low temperatures, and samples should be snap-frozen, with the aim to avoid RNA degradation [15]. Next, individual cells or nuclei are isolated from the tissue (Fig. 2b) and encapsulated into wells or barcoded droplets (Fig. 2c). Within each encapsulated entity, reverse transcription occurs, followed by cDNA amplification (Fig. 2d) and next-generation sequencing (Fig. 2e). Reads are then aligned to the reference genome/transcriptome/epigenome and quantified [17]. The quantification data are organized into the feature–barcode matrix, which upon normalization allows for data comparison between cells (Fig. 2f). The features (often several thousands of them) are then condensed, using methods such as principal component analysis, which reduces the data dimensionality. These condensed data are then clustered, which refers to the grouping of cells with similar features, e.g., according to their gene expression profile. The clustering results are usually visualized by further reducing the dimensionality, using methods such as uniform manifold approximation and projection (UMAP) or t-distributed stochastic neighbor embedding (t-SNE; Fig. 2g). Both methods visualize the results as a scatter plot, where each dot represents a cell, and those cells in close proximity would have a similar gene expression profile. Typically, each cluster represents a cell type and can be further sub-clustered into cell subpopulations. Annotation of the cell (sub)clusters is performed based on highly expressed genes in the given cluster that are well-known from the literature (e.g., oligodendrocytes are characterized by high expression of MOPB; astrocytes by AQP4; dopaminergic neurons by TH, etc.) [15, 18,19,20]. A detailed review of the best practices for single-cell analysis across modalities has recently been published by Heumos et al. [21].

Workflow of the scRNA-seq, including a tissue acquisition and b isolation of single cells/nuclei, c barcoding and cell lysis, d RNA isolation and reverse transcription, e sequencing, f data analysis, and g visualization of results
3 Parkinson’s Disease
Parkinson's disease (PD) is a progressive neurodegenerative disease characterized by a combination of motor (bradykinesia combined with rest tremor and/or rigidity) and non-motor symptoms (cognitive decline, depression, pain) [3]. The genetic cause of the disease can be found in only up to 5% of patients, with the remaining majority not having a clear genetic cause and thus classified as idiopathic PD [22]. A neuropathological hallmark of the disease is the presence of Lewy bodies and Lewy neurites, neuronal inclusions that contain the protein α-synuclein. The Lewy pathology consists of vesicular structures, dysmorphic organelles such as mitochondria, and high lipid content [23]. Dysfunction of these structures and neuroinflammation result in death of dopaminergic neurons (DaNs) that reside in the substantia nigra [3]. Although most of the PD research has focused on the DaNs, other cell types such as microglia are relevant for PD as well, consistent with their role in neuroinflammation [24]. Besides the substantia nigra, other midbrain and brain regions in general have been implicated in PD, but the exact disease etiology remains largely unknown [25].
Smajić et al. performed single-nucleus sequencing (snRNA-seq) of frozen human post-mortem midbrains (six idiopathic PD patients and five controls) to decipher the major cell types associated with idiopathic Parkinson’s disease: oligodendrocytes (ODC), oligodendrocyte precursor cells (OPC), microglia, astrocytes, ependymal cells, pericytes, endothelial cells, DaNs, and excitatory, inhibitory, and GABAergic neurons [15]. Besides these 11 cell types, the study identified an additional cluster of neuronal cells, originating almost exclusively from idiopathic PD midbrains, characterized by high expression of CADPS2 that encodes a calcium-dependent activator of secretion (Fig. 3a). These cells showed a similar expression profile to DaNs, except for low TH levels (TH is a marker gene for DaNs and encodes tyrosine hydroxylase that is involved in the conversion of tyrosine to dopamine). These CADPS2high cells are likely to represent degenerating DaNs in PD, but further investigations are needed to confirm this hypothesis. Furthermore, this study found decreased ODC levels in PD, as well as increased astrocytes and microglia. Imaging analyses of the PD-derived microglia showed their decreased branching and an amoeboid shape, indicating an activated state (Fig. 3a). In line with their role as the primary immune cells in the brain, microglia showed an upregulation of genes associated with the inflammatory trajectory in idiopathic PD (IL1B, GPNMB, HSP90AA1). By contrast, ODC showed a stress-induced upregulation of S100B.

Major findings in the most common neurodegenerative disorders from sc/snRNA-seq studies, including a Parkinson’s disease, b Alzheimer’s disease, c Huntington’s disease, and d multiple sclerosis
Another study focused on DaNs and developed a protocol to enrich for this cell type from human post-mortem substantia nigra, using fluorescence-activated nuclei sorting that enriched for NR4A2-positive cells (this gene was identified as a marker of mammalian midbrain neurons from the scRNA-seq data of mouse midbrain) [26, 27]. Kamath and colleagues identified 10 DaN subpopulations that expressed CALB1 (6 subpopulations) and SOX6 (4 subpopulations) as some of the major DaN marker genes [26]. To investigate the evolutionary conservation of DaNs, they integrated the human dataset with other species (macaque, northern treeshrew, rat, and mouse) and found a CALB1_GEM population present only in the human and macaque data, in line with higher cognitive functions in these species. They also found an increased subpopulation of astrocytes expressing VIM_LHX2 that responds to degenerative changes in PD, and an increased subset of microglia that express GPNMB, in accordance with the findings reported by Smajić et al. [15]. Importantly, a PD-vulnerable population of DaNs expressing SOX6_AGTR1 was identified by snRNA-seq and confirmed by single molecule fluorescence in situ hybridization (smFISH), and these cells were enriched for PD risk genes (SNCA, MAPT, GAK, WNT3, IGSF9B; Fig. 3a). An independent study on matched substantia nigra and cortex samples from PD donors found an association between PD risk and both DaNs and ODCs [28]. Nigral DaN modules were associated with mitochondrial processes, endocytosis, protein ubiquitination, and macroautophagy; the ODC module was related to gene regulation, kinase activity, phosphorylation, neurogenesis, and metabolic processes, while the OPC risk-associated module was enriched in gene regulation, cell differentiation, and metabolic processes. Interestingly, PD genetic risk appears to manifest not only through DaNs that are primarily affected but also through ODC and OPC, in support of the growing evidence of the importance of glia cells in neurodegenerative diseases. In line with this, the known PD gene LRRK2 showed a higher expression in OPC than other nigral cell types in this dataset [28]. These observations are further supported by a study that integrated genome-wide association study (GWAS) results with scRNA-seq data from the mouse nervous system, and found that PD is associated not only with cholinergic and monoaminergic neurons (dopamine is a monoamine neurotransmitter) but also with ODC and enteric neurons [29]. The relevance of enteric neurons in PD supports the Braak’s hypothesis, which states that idiopathic PD could start with an ingested pathogen, which then spreads from the gut to the brain via the vagus nerve [30].
Another popular approach to studying the molecular etiology of PD is by using iPSC-derived DaNs [31]. Lang and colleagues investigated iPSC-derived DaNs from individuals with the PD risk variant in the glucocerebrosidase gene, GBA-N370S [32, 33]. They found that histone deacetylase 4 (HDAC4) represses a number of genes (TSPAN7, ATP1A3, RTN1, PRKCB), leading to upregulation of the endoplasmic reticulum (ER) stress genes (PDI, FKBP9, ERO1A) [33]. Interestingly, while total HDAC4 levels were unchanged in PD, the authors identified its increased nuclear localization specifically in DaNs, in contrast to the physiological conditions where it shuffles between the nucleus and the cytosol (Fig. 3a). Pharmacological modulation of HDAC4 localization or activity restored expression levels of the mis-regulated genes, finally correcting the ER stress phenotype and perturbations in the autophagy and lysosomal pathway. These findings on the cell lines with the GBA variant could be extended to additional idiopathic PD cases, suggesting a promising new therapeutic strategy for PD [33]. Another study investigated effects of rotenone (a toxic pesticide that causes PD-like symptoms by inducing oxidative stress), tunicamycin (that causes ER stress), and a specific SNCA variant on the development of PD phenotype in iPSC-derived DaNs, and determined a population of mature DaNs vulnerable to cell death [34]. These vulnerable DaNs were characterized by expression of dopaminergic neuronal lineage genes (NR4A2, LMO3, POU2F2, LMX1A, DDC, DRD2), PD genes (SNCA, MAPT, UCHL1, ATP13A2), cytoskeleton and neuron projection genes, tubulin genes, etc. Upon the rotenone treatment, this population of cells showed decreased expression of SNCA, oxidative phosphorylation (MT-CO1, MT-CO2, NDUFA5, NDUFB6) and cholesterol biosynthesis genes (SQLE, ACAT2, ACSL3, HMGCR). Induction of the ER stress with tunicamycin leads to upregulation of ER stress response genes (as expected) and translation-associated and heat shock genes. On the other hand, genes associated with cholesterol synthesis were downregulated, similar to the rotenone treatment. The oxidative stress phenotype (caused by rotenone) but not the ER stress phenotype (caused by tunicamycin) could be rescued by felodipine, which had previously been reported to improve the phenotype in SNCA-A53T DaNs [35]. Using an isogenic SNCA-A53T cell line created by genome editing, the authors again found perturbations in cholesterol metabolism, along with changes in synaptic signaling, glycolysis, and ubiquitin–proteosomal degradation. The major findings on PD are summarized in Fig. 3a.
4 Alzheimer’s Disease
Alzheimer's disease (AD) is typically characterized by a cognitive decline and is the most common cause of dementia (although dementia can be caused by other neurodegenerative or cardiovascular pathologies) [36]. The main cognitive domains affected in AD are memory, language, visuospatial, and executive functions [2]. Besides the amnestic presentations, younger persons can also present with non-amnestic deficits—known as posterior cortical atrophy—such as challenges in reading, face recognition, or difficulties in processing complex visual scenes [37]. Genetic risk factors for AD include rare dominant variants in APP (encoding amyloid precursor protein), PSEN1, and PSEN2 (encoding presenilin 1 and 2, respectively), and more common but not completely penetrant variants in APOE, as well as variants in TREM2 and MS4 [2, 38, 39]. Pathologically, AD is characterized by the presence of β-amyloid-containing plaques in the cerebral cortex and tau-containing neurofibrillary tangles [40, 41].
One of the first and the largest studies that applied snRNA-seq on post-mortem AD brains profiled prefrontal cortex samples from 48 individuals with varying degrees of AD pathology and found that myelination-related processes were perturbed in multiple cell types [42]. The study by Mathys et al. revealed several unexpected findings including that large transcriptional changes occur early in the disease before the development of severe pathological features and that several AD-pathology-associated cell subpopulations were enriched in female cells. The authors also demonstrated that the AD risk factor APOE was upregulated in microglia, while it was downregulated in astrocytes (Fig. 3b). This finding emphasized the value of scRNA-seq methods as compared with bulk RNA sequencing, given that microglia were underrepresented in bulk data [42]. Some of these findings were replicated in an independent study, such as APOE upregulation in microglia and its downregulation in astrocytes and OPC, and increased LINGO1 levels in AD-specific subclusters [43]. Furthermore, Grubman and colleagues found that the transcription factor EB (TFEB), a regulator of lysosomal function and autophagy, acts upstream of 10 GWAS loci for AD (BIN1, CLDN11, POLN, STK32B, EDIL3, AKAP12, HECW1, WDR5, LEMD2, DLC1) in a disease-specific astrocyte subpopulation (Fig. 3b). Another study reported the relevance of endothelial cells in AD, in addition to the previously mentioned cell types [44]. Lau et al. observed an enhanced angiogenesis in endothelial cells, together with aberrant immune response in endothelial cells and microglia, reduced myelination in ODC, and impaired synaptic signaling in neurons and astrocytes (Fig. 3b) [44]. Three AD-upregulated subpopulations expressed genes associated with angiogenesis (CLDN5, ERG, FLT1, VWF) and antigen presentation (MHC-I complex). Analysis of bulk microarray data from a mouse AD model revealed a similar transcriptomic profile, suggesting that the activation of endothelial cells in neurodegeneration is conserved between humans and mice [44]. A study on 10 male individuals with APOE ε3/ε3 genotype that sequenced a large amount of nuclei (10,000 per individual) in entorhinal cortex (affected early in the disease progression) and superior frontal gyrus (affected late in the disease progression) identified RORB as a marker of selectively vulnerable excitatory neurons in the entorhinal cortex (Fig. 3b) [45]. The same study found an increased amount of microglia with AD progression (microgliosis) and reactive astrocytes that showed downregulation of genes associated with homeostasis. A simultaneous profiling of chromatin accessibility and gene expression on post-mortem prefrontal cortex samples identified cell-type-specific cis and trans regulatory elements and their target genes in AD [46]. Morabito and colleagues examined the regulatory roles of transcription factors SPI1 in microglia and NRF1 in ODC and found that SPI1 acts as a transcriptional repressor in late-stage AD, while NRF1 may contribute to neuronal dysfunction through the disruption of myelination. SREBF1 that regulates cholesterol and fatty acid metabolism was identified as a transcriptional activator throughout the ODC trajectory, while the APOE locus had cis-regulatory chromatin networks altered in AD in microglia and astrocytes [46]. Additional transcription factors that might play a role in AD-specific gene regulation include ZEB1 in neurons and MAFB in microglia (Fig. 3b) [47]. A very recent study on parietal cortex samples from AD autosomal dominant (APP and PSEN1) and risk-modifying variant (APOE, TREM2, MS4A) carriers detected the affected pathways: APOEε4 inhibitory neurons displayed signs of ferroptosis (an iron-dependent form of cell death), TREM2 ODC showed a dysregulated autophagy-lysosomal pathway, while MS4A microglia had dysregulated genes of the complement cascade [48]. The relevance of the human brain vasculature in AD has also been demonstrated in a study that found selective vulnerability of extracellular matrix-maintaining pericytes and gene expression patterns that implicated dysregulated blood flow [49]. Moreover, many of the AD GWAS genes were found to be expressed in the brain vasculature, and they were associated with endothelial protein transport, adaptive immune system, and extracellular matrix pathways [49]. scRNA-seq of peripheral blood mononuclear cells (PBMC) reported a decrease in B cells in individuals with AD, where the reduction in B cells correlated with the patients’ clinical dementia rating scores. These results were confirmed in a mouse AD model, where the B cell depletion accelerated cognitive dysfunction and worsened the phenotype [50]. Mice with tauopathy (but not with amyloid beta deposition) developed a unique innate and adaptive immune response, where numbers of T cells were increased in areas with tau pathology and could be correlated with the extent of neuronal loss. Depletion of T cells by peritoneal administration of neutralizing antibodies led to strong depletion of CD4+ and CD8+ T cells in brain parenchyma, meninges, and peripheral blood, blocking tau-mediated neurodegeneration. Furthermore, microglia shifted from activated toward homeostatic state after T cell depletion [51]. Another successful approach in treating AD-specific cellular phenotypes in mice with tauopathy was a selective removal of neuronal APOE4, which led to a reduction in tau pathology, gliosis, neurodegeneration, neuronal hyperexcitability, and myelin deficits [52]. All major findings on AD are summarized in Fig. 3b.
5 Huntington’s Disease
Huntington's disease (HD) is characterized by a hyperkinetic movement disorder known as chorea, together with dementia, behavioral, and psychiatric disturbances [4]. It is caused by a dominantly inherited CAG repeat expansion in exon 1 of the huntingtin gene (HTT). Medium spiny neurons from the striatum are the most vulnerable cell population to the presence of mutant HTT, although neuronal dysfunction and death occur in the cerebral cortex as well. snRNA sequencing on striatum and cortex of human post-mortem and mouse HD samples found that oligodendrocyte lineages—ODC and OPC—are arrested in intermediate maturation stages [53]. Lim and colleagues identified PRKCE (encoding protein kinase C epsilon) and TPK1 (encoding thiamine pyrophosphokinase 1) as central genes in these processes (Fig. 3c). High-dose thiamine and biotin treatment rescued transcriptional dysregulation in neurons and altered ODC and OPC developmental genes in a mouse model of HD [53]. Increased sensitivity of the medium spiny neurons to mutant HTT could also be explained by the downregulation of the mitochondrial oxidative phosphorylation pathway, release of mitochondrial RNA, and activation of innate immune signaling [54]. In the striatum of the mouse HD model, the innate immune sensor protein kinase R (PKR) was upregulated and phosphorylated, indicating its activation (Fig. 3c). Furthermore, mitochondrial RNA could be directly purified from a complex with PKR with immunoprecipitation, in line with previous findings [55]. A study that focused on astrocytes found that they show variable transcriptional phenotypes in HD: some of them had upregulated metallothionein and heat shock genes, while others upregulated glial fibrillary acidic protein, a reactive astrocyte marker. On the other hand, expression of several normal protoplasmic astrocyte genes and lipid synthesis genes was decreased [56]. A study that focused on the importance of the human brain vasculature found that highly expressed genes in a subcluster of astrocytes and a subcluster of microglia were associated with regulation of angiogenesis and blood vessel endothelial cell migration, suggesting that these glial cells could be relevant for vasculature regulation [57]. This study also found that several innate immune activation genes were upregulated in endothelial cells (IKBKB, IRF2/3, STAT3), astrocytes, and microglia. On the other hand, they observed a reduction of the blood–brain barrier (BBB) tight junction proteins (CLDN5 and TJP1), which is known to lead to the loss of BBB integrity. A snRNA-seq study on striatum from HD mouse model detected the genes dysregulated in the vulnerable cell types (medium spiny neurons, ODC, and astrocytes), and found that many of the cell type identity genes were downregulated in their primary cell type, while they were aberrantly upregulated in other striatal cell types [58]. Major findings on HD are summarized in Fig. 3c.
6 Multiple Sclerosis
Multiple sclerosis (MS) is an inflammatory, demyelinating neurodegenerative disease, influenced by both genetic and environmental factors. Pathologically, it is characterized by the formation of demyelinating lesions in the brain and spinal cord [5]. The main cell type affected in this disease are oligodendrocytes, as they provide metabolic support and myelinate the axons. This disease is less studied than the previously mentioned common ND, especially using the single-cell sequencing methods. A study that applied snRNA-seq on white matter areas of post-mortem human brains found decreased amounts of nuclei from OPC (expressing PDGFRA, BCAN, and SOX6) and an intermediate ODC subpopulation (that expressed OPALIN and LINC00844) in MS lesions. On the other hand, other ODC clusters (cluster 2 expressing LURAPIL-AS1 and CDH19; cluster 5 expressing KLK6, GJB1) and oligodendroglia (expressing APOE and CD74) were enriched in MS. Interestingly, several myelin protein genes were upregulated in mature ODC in MS, suggesting that a subset of these cells contributes to remyelination [59]. Another study observed a selective vulnerability and decrease in numbers of CUX2-expressing excitatory neurons in MS samples with cortical demyelination. Upregulated genes in the vulnerable excitatory neurons included oxidative stress, mitochondrial dysfunction, and cell death pathway genes as well as long non-coding RNAs (BCYRN1 and LINC00657). Upregulated genes in the MS-myelinating ODC were associated with the heat-shock response, cell stress, iron accumulation, ubiquitin-mediated protein degradation, etc., indicating a severe stress in ODC in MS. Furthermore, the study identified a cluster of phagocyting microglia that were enriched for ODC-specific markers [60]. Major findings on MS are summarised in Fig. 3d.
7 Concluding Remarks
Selective vulnerability of specific cellular (sub)populations is a hallmark of neurodegenerative diseases, but so far technologies are lacking in the necessary throughput and resolution to study these changes at scale [61]. Sc/snRNA-omics approaches offer powerful methods to study these cell-type-specific changes [62,63,64]. The recent development of high throughput single-cell sequencing workflows and computational tools made this technology applicable in numerous fields of life sciences, which together with decreasing sequencing costs holds a great promise for its wider use. The technology is at the doorsteps of human genetics and pathology diagnostics, with a possibility for its integration into routine clinical diagnostics and personalized medicine [16, 17]. However, several challenges remain for its application specifically in neurodegenerative disorders: The first of these is tissue acquisition, given the low accessibility of the brain tissue for sampling. Second, sc-RNA seq is still difficult on tissues that have been formalin-fixed and paraffin-embedded, as it is often the case with postmortem samples [65, 66]. These problems could be at least partially overcome by using patient-derived iPSC and organoids, especially as differentiation protocols are being developed and constantly improved [67]. Third, a considerable disadvantage of sc-seq is the loss of topological information during the isolation of single cells/nuclei, but the recently emerging spatial transcriptomics methods can add this layer of missing information on tissue complexity. Finally, although multiple studies have sequenced the same tissues, the lack of a universal nomenclature for individual cell (sub)types represents a challenge that needs to be addressed in the future [68].
Through joint sc-modalities (e.g., assay for transposase-accessible chromatin (ATAC) and RNA, DNA methylation and ATAC, spatial transcriptomics, sc-proteomics, sc-metabolomics, etc.) and larger sample sizes, sc-seq omics approaches will further expand our knowledge on how cell types and entire organs contribute to health and disease. Importantly, the introduction of whole-brain and even whole-organism single-cell atlases, accessible to everyone for browsing (even without prior bioinformatics knowledge), represents a major step forward [12, 69, 70]. Together, these efforts will enable a better understanding of the mechanisms underlying a disease and enable eventual treatments that target the affected populations and pathways. Once the affected cell types and their specific genes/proteins are known, their levels could be modulated by changing the gene expression (e.g., by using viral- and non-viral vectors or antisense oligonucleotides) or protein levels (e.g., by using target-specific antibodies), or replacing the damaged cells by means of regenerative medicine. Although these approaches still remain challenging for ND given the accessibility of the brain tissue and the permeability of the BBB, therapeutic options will become available as the scientific knowledge expands.
References
Gao H-M, Hong J-S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357.
Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33.
Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397(10291):2284–303.
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005.
Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, et al. Multiple sclerosis. Nat Rev Dis Primers. 2018;4:1–27.
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:1–19.
Pang SY-Y, Ho PW-L, Liu H-F, Leung C-T, Li L, Chang EES, et al. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl Neurodegener. 2019;8:23.
Gan L, Cookson MR, Petrucelli L, La Spada AR. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci. 2018;21:1300–9.
Wareham LK, Liddelow SA, Temple S, Benowitz LI, Di Polo A, Wellington C, et al. Solving neurodegeneration: common mechanisms and strategies for new treatments. Mol Neurodegener. 2022;17:23.
Vandereyken K, Sifrim A, Thienpont B, Voet T. Methods and applications for single-cell and spatial multi-omics. Nat Rev Genet. 2023;24(8):495–515.
Rao A, Barkley D, França GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596:211–20.
Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502.
Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, et al. A human cell atlas of fetal gene expression. Science. 2020;370(6518):eaba7721.
Huang X, Henck J, Qiu C, Sreenivasan VKA, Balachandran S, Behncke R, et al. Single cell, whole embryo phenotyping of pleiotropic disorders of mammalian development. bioRxiv. 2022. Available from: https://doi.org/10.1101/2022.08.03.500325v1.
Smajić S, Prada-Medina CA, Landoulsi Z, Ghelfi J, Delcambre S, Dietrich C, et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain. 2022;145:964–78.
Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single-cell RNA sequencing technologies and applications: a brief overview. Clin Transl Med. 2022;12: e694.
Sreenivasan VKA, Balachandran S, Spielmann M. The role of single-cell genomics in human genetics. J Med Genet. 2022;59:827–39.
Mitkus SN, Hyde TM, Vakkalanka R, Kolachana B, Weinberger DR, Kleinman JE, et al. Expression of oligodendrocyte-associated genes in dorsolateral prefrontal cortex of patients with schizophrenia. Schizophr Res. 2008;98:129–38.
Ikeshima-Kataoka H. Neuroimmunological implications of AQP4 in astrocytes. Int J Mol Sci. 2016;17(89):1306.
Thompson L, Barraud P, Andersson E, Kirik D, Björklund A. Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections. J Neurosci. 2005;25:6467–77.
Heumos L, Schaar AC, Lance C, Litinetskaya A, Drost F, Zappia L, et al. Best practices for single-cell analysis across modalities. Nat Rev Genet. 2023;24(8):550–72.
Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, et al. Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the shaking palsy. Mov Disord. 2017;32:1264–310.
Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22:1099–109.
Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat Disord. 2009;15(Suppl 3):S200–4.
Huynh B, Fu Y, Kirik D, Shine JM, Halliday GM. Comparison of locus coeruleus pathology with nigral and forebrain pathology in Parkinson’s disease. Mov Disord. 2021;36:2085–93.
Kamath T, Abdulraouf A, Burris SJ, Langlieb J, Gazestani V, Nadaf NM, et al. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat Neurosci. 2022;25:588–95.
Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174:1015-30.e16.
Agarwal D, Sandor C, Volpato V, Caffrey TM, Monzón-Sandoval J, Bowden R, et al. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat Commun. 2020;11:4183.
Bryois J, Skene NG, Hansen TF, Kogelman LJA, Watson HJ, Liu Z, et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat Genet. 2020;52:482–93.
Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517–36.
Xiao B, Ng HH, Takahashi R, Tan E-K. Induced pluripotent stem cells in Parkinson’s disease: scientific and clinical challenges. J Neurol Neurosurg Psychiatry. 2016;87:697–702.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–61.
Lang C, Campbell KR, Ryan BJ, Carling P, Attar M, Vowles J, et al. Single-cell sequencing of iPSC-dopamine neurons reconstructs disease progression and identifies HDAC4 as a regulator of Parkinson cell phenotypes. Cell Stem Cell. 2019;24:93-106.e6.
Fernandes HJR, Patikas N, Foskolou S, Field SF, Park J-E, Byrne ML, et al. Single-cell transcriptomics of Parkinson’s disease human in vitro models reveals dopamine neuron-specific stress responses. Cell Rep. 2020;33: 108263.
Siddiqi FH, Menzies FM, Lopez A, Stamatakou E, Karabiyik C, Ureshino R, et al. Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat Commun. 2019;10:1817.
Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–8.
Crutch SJ, Schott JM, Rabinovici GD, Murray M, Snowden JS, van der Flier WM, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13:870–84.
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–35.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27.
Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103–16.
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–7.
Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22:2087–97.
Lau S-F, Cao H, Fu AKY, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci. 2020;117:25800–9.
Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci. 2021;24:276–87.
Morabito S, Miyoshi E, Michael N, Shahin S, Martini AC, Head E, et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet. 2021;53:1143–55.
Anderson AG, Rogers BB, Loupe JM, Rodriguez-Nunez I, Roberts SC, White LM, et al. Single nucleus multiomics identifies ZEB1 and MAFB as candidate regulators of Alzheimer’s disease-specific cis-regulatory elements. Cell Genom. 2023;3: 100263.
Brase L, You S-F, D’Oliveira Albanus R, Del-Aguila JL, Dai Y, Novotny BC, et al. Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers. Nat Commun. 2023;14:2314.
Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. 2022;603:885–92.
Xiong L-L, Xue L-L, Du R-L, Niu R-Z, Chen L, Chen J, et al. Single-cell RNA sequencing reveals B cell-related molecular biomarkers for Alzheimer’s disease. Exp Mol Med. 2021;53:1888–901.
Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615:668–77.
Koutsodendris N, Blumenfeld J, Agrawal A, Traglia M, Grone B, Zilberter M, et al. Neuronal APOE4 removal protects against tau-mediated gliosis, neurodegeneration and myelin deficits. Nat Aging. 2023;3:275–96.
Lim RG, Al-Dalahmah O, Wu J, Gold MP, Reidling JC, Tang G, et al. Huntington disease oligodendrocyte maturation deficits revealed by single-nucleus RNAseq are rescued by thiamine-biotin supplementation. Nat Commun. 2022;13:7791.
Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, et al. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA Release and neuronal innate immune activation. Neuron. 2020;107:891-908.e8.
Kim Y, Park J, Kim S, Kim M, Kang M-G, Kwak C, et al. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol Cell. 2018;71:1051-63.e6.
Al-Dalahmah O, Sosunov AA, Shaik A, Ofori K, Liu Y, Vonsattel JP, et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun. 2020;8:19.
Garcia FJ, Sun N, Lee H, Godlewski B, Mathys H, Galani K, et al. Single-cell dissection of the human brain vasculature. Nature. 2022;603:893–9.
Malaiya S, Cortes-Gutierrez M, Herb BR, Coffey SR, Legg SRW, Cantle JP, et al. Single-nucleus RNA-seq reveals dysregulation of striatal cell identity due to Huntington’s disease mutations. J Neurosci. 2021;41:5534–52.
Jäkel S, Agirre E, Mendanha Falcão A, van Bruggen D, Lee KW, Knuesel I, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 2019;566:543–7.
Schirmer L, Velmeshev D, Holmqvist S, Kaufmann M, Werneburg S, Jung D, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature. 2019;573:75–82.
Fu H, Hardy J, Duff KE. Selective vulnerability in neurodegenerative diseases. Nat Neurosci. 2018;21:1350–8.
Luquez T, Gaur P, Kosater IM, Lam M, Lee DI, Mares J, et al. Cell type-specific changes identified by single-cell transcriptomics in Alzheimer’s disease. Genome Med. 2022;14:136.
Kamath T, Macosko EZ. Insights into neurodegeneration in Parkinson’s disease from single-cell and spatial genomics. Mov Disord. 2023;38:518–25.
Ahmadi A, Gispert JD, Navarro A, Vilor-Tejedor N, Sadeghi I. Single-cell transcriptional changes in neurodegenerative diseases. Neuroscience. 2021;479:192–205.
Lin X, Qiu L, Song X, Hou J, Chen W, Zhao J. A comparative analysis of RNA sequencing methods with ribosome RNA depletion for degraded and low-input total RNA from formalin-fixed and paraffin-embedded samples. BMC Genomics. 2019;20:831.
Mirzazadeh R, Andrusivova Z, Larsson L, Newton PT, Galicia LA, Abalo XM, et al. Spatially resolved transcriptomic profiling of degraded and challenging fresh frozen samples. Nat Commun. 2023;14:509.
Zhao Z, Chen X, Dowbaj AM, Sljukic A, Bratlie K, Lin L, et al. Organoids Nat Rev Methods Primers. 2022;2:1–21.
Domcke S, Shendure J. A reference cell tree will serve science better than a reference cell atlas. Cell. 2023;186:1103–14.
Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y, et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303–9.
Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018;174:1309-24.e18.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
JP is supported by research grants from the Else Kröner-Fresenius-Stiftung (2022_EKEA.55) and the University of Lübeck, Germany (J14-2021). MS is supported by the Deutsche Forschungsgemeinschaft (SP1532/3-1, SP1532/4-1, and SP1532/5-1), Max Planck Society, Deutsches Zentrum für Luft- und Raumfahrt (DLR 01GM1925).
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Availability of data and material
No original data were included in this review.
Code availability
Not applicable.
Author contributions
JP and MS designed the outline, JP wrote the first draft, MS critically edited the manuscript, and both authors approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Pozojevic, J., Spielmann, M. Single-Cell Sequencing in Neurodegenerative Disorders. Mol Diagn Ther 27, 553–561 (2023). https://doi.org/10.1007/s40291-023-00668-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-023-00668-9