
Abstract
Alzheimer’s disease (AD) is the most common form of dementia, characterized by progressive cognitive impairment and neurodegeneration. Extensive clinical and genomic studies have revealed biomarkers, risk factors, pathways, and targets of AD in the past decade. However, the exact molecular basis of AD development and progression remains elusive. The emerging single-cell sequencing technology can potentially provide cell-level insights into the disease. Here we systematically review the state-of-the-art bioinformatics approaches to analyze single-cell sequencing data and their applications to AD in 14 major directions, including 1) quality control and normalization, 2) dimension reduction and feature extraction, 3) cell clustering analysis, 4) cell type inference and annotation, 5) differential expression, 6) trajectory inference, 7) copy number variation analysis, 8) integration of single-cell multi-omics, 9) epigenomic analysis, 10) gene network inference, 11) prioritization of cell subpopulations, 12) integrative analysis of human and mouse sc-RNA-seq data, 13) spatial transcriptomics, and 14) comparison of single cell AD mouse model studies and single cell human AD studies. We also address challenges in using human postmortem and mouse tissues and outline future developments in single cell sequencing data analysis. Importantly, we have implemented our recommended workflow for each major analytic direction and applied them to a large single nucleus RNA-sequencing (snRNA-seq) dataset in AD. Key analytic results are reported while the scripts and the data are shared with the research community through GitHub. In summary, this comprehensive review provides insights into various approaches to analyze single cell sequencing data and offers specific guidelines for study design and a variety of analytic directions. The review and the accompanied software tools will serve as a valuable resource for studying cellular and molecular mechanisms of AD, other diseases, or biological systems at the single cell level.
Similar content being viewed by others

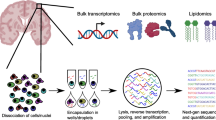
Background
Alzheimer’s disease (AD) is one of the most devastating forms of dementia common in the elderly, estimated to affect over 6.2 million individuals in the United States and 24 million worldwide [1, 2]. Clinically, AD patients present amnestic multidomain progressive dementia. A more definitive AD diagnosis requires evidence of amyloid-beta (Aβ) plaques and Tau neurofibrillary tangle (NFT) accumulation within the neurodegenerative brain [3].
AD is a highly complex and heterogeneous disease caused by various pathophysiologic mechanisms. AD can be classified by heritable cause and age of onset, i.e., rare familial AD, sporadic early-onset (EOAD), and late-onset (LOAD) [4]. While AD often progresses through a period of mild cognitive impairment (MCI), not all patients with MCI develop AD, hinting at protective or causal factors that may differentially affect subsets of patients even within traditional subtypes. Postmortem evaluations revealed that AD brains may include depositions of additional pathologies (i.e., beyond Aβ and phosphorylated tau), such as Lewy bodies, alpha-synuclein, transactive response DNA-binding protein and/or vascular-related brain lesions [5]. Further, the recently discovered five molecular subtypes of AD were associated with unique molecular signatures and distinct sets of brain cell type-specific key regulators [6].
Understanding the cell-type-specific changes and regulations at the single-cell level will enable us to decode the molecular mechanisms underlying the pathophysiologic processes contributing to dementia. Indeed, recent single-cell sequencing studies of aged and AD brains revealed a series of brain cell clusters involved in AD [7, 8]. However, these studies primarily focused on clustering and differential analyses but did not fully exploit the single-cell sequencing data to explore, for example, pseudo-temporal dynamics. To mitigate these gaps, we reviewed the state-of-the-art bioinformatics approaches to analyze single-cell transcriptome (single-cell/−nuclei RNA sequencing (sc/snRNA-seq)) and epigenome (single-cell assay for transposase-accessible chromatic sequencing (scATAC-seq)) in AD, and integrate single-cell features with abundantly available AD bulk sequencing data. Specifically, we reviewed the following 15 topics (Fig. 1): 1) quality control and normalization, 2) dimension reduction and feature extraction, 3) cell clustering analysis, 4) cell type inference and annotation, 5) differential expression for disease gene identification, 6) trajectory inference, 7) copy number variation (CNV) analysis, 8) integration of single-cell multi-omics (e.g., expression associated quantitative trait loci (eQTL) and expression associated CNVs (eCNVs)), 9) epigenomic (scATAC-seq) analysis, 10) gene network inference, 11) prioritization of cell clusters, 12) integration of single cell and bulk RNA-seq data, 13) spatial single-cell transcriptomics, and 14) comparison between single cell AD mouse model studies and single cell human AD studies. For future directions, we discuss experimental validation strategies of single-cell based findings, and translations to drug discoveries. Notably, we implemented our recommended workflow for each major analytic direction and applied them to a large snRNA-seq dataset in AD. Key analytic results were reported while the scripts and the data were shared with the research community through GitHub (see the section "Availability of data and software code" for the details). We hope that the guidelines will accelerate AD research by leveraging the power of single-cell sequencing.

Overview of the bioinformatics approaches to analyze scRNA-seq, scATAC-seq, and spatial transcriptomics data with a focus on scRNA-seq data. scRNA-seq and scATAC-seq data (A) go through appropriate quality control (QC) to remove outliers and cells with low-quality sequencing data (B), followed by normalization (B). QC-ed and normalized data are then used for dimension reduction, and feature extraction (C) clustering analysis to identify cell clusters (D). Marker genes for each cell cluster will then be identified to infer its association to known or novel cell type (E). Meanwhile, differential gene expression is performed between cell groups of interest (e.g., AD and Control) in each cell cluster to identify gene expression changes associated with the disease (F). Trajectory inference can be performed on all cells, cells in each cluster or the cells from multiple closely related cell clusters to infer cellular dynamics during developmental or disease progression (G). Copy number variations (CNVs) can also be inferred from scRNA-seq data (H). Integration of gene expression and genomic (SNPs & CNVs) data leads to the identification of expression-associated quantitative trait loci (eQTLs) (I). Epigenomic analysis by scATAC-seq can study gene expression regulatory elements in open chromatin regions (J) and will be detailed in Fig. 7. Gene coexpression and causal networks will be constructed for each cell cluster or multiple closely related cell clusters, while priors from eQTLs and epigenomic analyses can be developed for assisting causal network inference (K). Cell clusters can be prioritized based on the number of differentially expressed genes between disease and control across all cell clusters (L). scRNA-seq data can also be integrated with bulk RNA-seq data to robustly identify key molecular changes and network structures (M). Finally, cell cluster-based networks will be analyzed to prioritize key subnetworks (e.g., coexpressed gene modules) and potential network regulators for a disease (e.g., AD) under study (N). Novel cell clusters, key subnetworks and key driver genes can be validated through single-cell spatial transcriptomics analysis which offers more insights into spatially distributed molecular signals in a system or a disease under study (O). Key findings from human AD single cell sequencing data will be validated in AD mouse models and integration of mouse and human single cell data is critical for informing the correspondence between AD mouse models and human AD (P)
Overview of single-cell sequencing study design
High-throughput sequencing of bulk tissue measures the average signals of various cell types, thus falls short of dissecting the cellular heterogeneity in brain tissues. To address this issue, single-cell sequencing has been recently developed to elucidate the cell-type specificity and identify the transcriptome, epigenome, and genome changes among various cellular populations. Recently, scRNA-seq studies have been widely conducted in many research fields, such as oncology [9], developmental biology [10], immunology [11], and neurosciences [12]. Several protocols have been developed to measure mRNAs and non-coding RNAs from single cells, such as Smart-seq [13], Quartz-seq [14], CEL-seq [15], RamDa-seq [16], Drop-seq [17], sci-RNA-seq [18] and Chromium (10X Genomics).
Single-cell genome and epigenome including single-cell ChIP-seq [19] and scATAC-seq have also emerged to investigate the genomic and epigenomic status associated with the transcriptome of cells [20, 21]. Single-cell genome sequencing captures de novo germline mutations, somatic mutations, and copy number alterations to dissect the genetic heterogeneity at the cellular level [22]. scATAC-seq is useful in analyzing the patterns of open chromatin, a hallmark of active regulatory elements, in single cells. Moreover, several advanced spatial sequencing techniques (for example, 10x Visium) have included spatial dimensions of the molecular features at a near-cellular resolution [23, 24]. Although the sequencing protocols have improved platforms and reagent kits to increase the detection sensitivity, the sequencing coverage is still limited, and present challenges to robustly analyzing single-cell sequencing data.
Several considerations should be taken into account for designing single-cell sequencing studies. The first one is tissue requirement. For example, whole single-cell RNA sequencing requires fresh samples, thus requiring the study to implement a seamless process from obtaining patients’ consent, acquiring the autopsy samples, to preparing a single-cell library even sequencing within a few hours [7, 25]. On the contrary, fresh frozen samples in a tissue repository can be preserved for a substantial amount of time, providing the freedom to select samples with relevant clinical, molecular characteristics such as gender and APOE genotype, and their nuclei can be isolated for snRNA-seq data analysis. The second consideration is cellular coverage. Several cell types, especially neurons, are underrepresented in scRNA-seq dataset, due to technical issues relating to size selection during the tissue dissociation process [26, 27], while snRNA-seq covers more cell types [28, 29]. However, in selecting single-cell or single-nuclei based approaches for transcriptomic profiling, it is important to recognize that each has its strengths and limitations. Brakken et al. compared these two approaches side by side by generating matched datasets from the mouse visual cortex [30]. They found that scRNA-seq analysis has the strengths of unbiased transcriptomic profiling, a higher gene coverage rate, and a higher-power for distinguishing similar cell types. However, the tissue dissociation and cell-isolation protocols are too harsh for certain cell types, leading to significant under-representation. In contrast, snRNA-seq has the strengths of less biased cellular coverage, resistance to cell isolation-associated perturbations, and applicability to both fresh and archived frozen specimens. Single-nuclei detected transcripts are also enriched for intronic reads, whereas the majority of the single-cell detected transcripts are from exons. Interestingly, the nuclear proportion of total cellular mRNA varies significantly in a cell-type and cortical-layer-specific manner, although the biological significance of such variation is still unknown. However, Thrupp et al. [31] found that snRNA-seq data is depleted of an activated microglial subpopulation expressing the activation signature, including APOE, CST3, SPP1 and CD74, but the absence of the microglial subpopulation was likely due to the low sequencing depth. Despite these differences, it is important to note that the overall cell-type landscapes captured by these two approaches are similar [30]. Power analysis is a critical step to rationalize scRNA-seq study design to ensure robustness and reproducibility of scientific findings. In the companion GitHub repository (see the section "Availability of data and software code" for details), we provided a comprehensive review of the power analysis approaches for single cell studies and shared the script for applying a recommended approach to an AD snRNA-seq study.
Different sequencing protocols are optimized for different biological aspects. PCR plate-based sequencing protocols (e.g. Smartseq2 [32], CEL-seq [15], and MARS-seq [33]) capture cells through cell sorter or microfluidics and offer high read depth per cell with less effective cell captures [34]. Thus, these protocols provide high sensitivity to discriminate subpopulations of similar cell types with subtle differences [35]. On the contrary, droplet-based protocols (e.g. InDrop [36], Drop-seq [17], and 10x Chromium [37]) capture thousands to millions of cells with low sequencing depths per cell [34], and can offer exogenous spike-ins to handle technical noises systematically [38, 39]. Large numbers of cells in these protocols enable the detection of rare cell populations such as neuronal subtypes [40]. However, Alsema et al. 2020 report that single-cell sequencing of FACS sorted microglia by droplet-based 10x Chromium and PCR plate-based Smart-seq2 only displayed marginal differences, most likely arising from technical noises by plate-based protocols [25]. These indicate targeted studies for cell type of interest may not require large-sequencing depths to uncover distinct sub-populations. So far, the droplet-based 10x Chromium snRNA-seq, which can sequence over 10,000 nuclei per library, is the most widely used sequencing platform for human cohort studies including AD (Table 1).
Quality control and normalization
Data quality control (QC) and normalization are the essential steps to remove systematic sources of technical variations introduced during the single-cell data generation process while preserving the true biological variations. Due to the low amount of RNA in a single cell and the stochastic sampling process of sequencing, scRNA-seq data are much noisier than bulk-tissue sequencing data [46, 47]. Excessive zero or near-zero counts by the so-called “dropout” events [48], often lead to highly sparse data, shadow the biological variations in individual cells and require dedicated QC metrics to ensure that only high-quality data are selected for downstream analysis. Starting from a count matrix of unique molecular identifiers (UMIs), a typical data preprocessing workflow generally contains several steps for QC to remove low-quality cells and genes, and normalize cell-specific biases.
Quality control on the cells
Two common quality measures are the number of expressed features (i.e., features detected with non-zero counts) and the library size (i.e., the sum of counts across all features). Violin plots are used to visualize the distribution of these cell-specific measures in each donor sample [49, 50]. Cells with very few expressed features or small library size indicate low RNA-capture efficiency and are hence considered poor quality. On the other hand, cells with abnormally a large number of expressed features suggest doublets or multiplets (i.e., two or more cells mistakenly captured as a single cell) [51], hypothesizing that doublets or multiplets would have higher total RNA content (see below for a review of more elegant doublet detection methods). Thus, a lower and an upper bound for the number of expressed features can be specified for cell filtering. However, determining the bounds for the number of expressed features or library size is not trivial as both biological and technical factors need be taken into account. For example, sequencing with deeper depth leads to more reads and more expressed features, irrespective of the cell quality. Another filtering approach is to detect outliers. For instance, it has been proposed to remove cells with log-library size greater than 3 median absolute deviations (MADs) or below the median log-library size [52, 53].
The presence of doublets or multilets may severely confound the downstream analysis and interpretation. This can lead to, for example, spurious cell clusters, both false positive and false negative prediction of cell cluster markers or disease genes, biased cell-state trajectories, and misrepresented gene-gene correlation structure and gene regulatory networks [54,55,56]. Doublet detection can be facilitated through appropriate experimental design. These include species mixing (mixing of cells from different species), mixing of cells from samples with different genotypes or genetic labels, and cell “hashing” (pooling of cells from separately barcoded samples) (see [55] for a summary of the experimental assay-based methods, including their limitations, for doublet detection). However, most of the existing AD single-cell datasets have not implemented the experimental design features. This review will focus on the model-based doublet detection approaches that are applicable to all AD scRNA-seq datasets currently available.
Assuming that doublets have more RNAs than singlets, the simplest approach is to threshold overall expression content (such as the number of detected genes and total UMI counts) to classify cells with unusually high UMI or gene number as potential doublets [51, 55]. However, the assumption that cells contain similar amount of RNA is unlikely to be true due to diverse cell types or different cell cycle states. Another simple approach is to look for cells expressed with marker genes of more than one distinct cell type [51, 55, 57]. However, this requires expert knowledge of the cell types and the associated markers in the data. There are more advanced and potentially more powerful computational algorithms for doublet detection in scRNA-seq data. In a recent benchmarking study, Xi and Li evaluated nine existing doublet detection methods [54], including Scrublet [55], scran/doubletCells [58], cxds [56], bcds [56], hybrid (combination of cxds and bcds) [56], DoubletDetection [59], DoubletFinder [60], Solo [61], and DoubletDecon [62]. Seven out of the eight standalone methods (except cxds which detects co-expression of markers that are supposedly to be mutually exclusive in the same cell) first generate artificial doublets by mixing observed gene expression profiles from randomly selected droplet pairs. The major difference among these methods is the choice of embedding/dimension reduction and classifier. Via a comprehensive benchmarking on 16 real datasets with experimentally annotated doublets and 112 realistic synthetic datasets, DoubletFinder showed the best prediction accuracy while cxds had the highest computational efficiency. However, one caveat of these computational algorithms is that they were designed to identify “neotypic” doublets, which consist of cells of distinct cell types, and hence difficult to capture “embedded” doublets that encapsulate cells from the same or highly similar cell types [55].
An important cell quality measure is the percentage of reads mapped to the mitochondrial genome in each library. As increased mitochondrial fraction indicates increased apoptosis, increased cell stress, and/or loss of cytoplasmic RNA from lysed cells [52, 63], cells with a high proportion of reads allocated to mitochondrial genomes are deemed poor-quality. A recent systematic survey of scRNA-seq data suggested that a mitochondrial proportion threshold of 10% is appropriate to distinguish between healthy and low-quality cells in most human tissues, while in mouse tissues, the recommended threshold is 5% [64]. However, just like the number of expressed features, selection of a threshold for this parameter is highly dependent on tissue type and experimental setting. For example, 30% of mitochondrial mRNA, which would otherwise indicate cell stress or apoptosis in tissues with low energy need, is normal for a healthy heart muscle cell due to high energy demand [65, 66]. Mitochondrial transcripts are not expressed in nuclei. Yet, variable amounts of mitochondrial transcripts were associated with the snRNA-seq data [8, 41, 42, 67, 68]. For example, in the first snRNA-seq transcriptomic analysis of AD, the fraction of mitochondrial reads exhibited a highly skewed empirical distribution, with an elbow shape which distinctly separates cells with high and low ratios for further classification and removal by k-means clustering (k = 2) on the mitochondrial ratio [8].
Another source of noises in the droplet-based scRNA-seq protocols (e.g., drop-seq or 10x Genomics Chromium protocol) is the contamination of ambient RNAs (cell-free RNAs), which are released in the cell lysis from dead or apoptotic cells before droplet separation. As ambient mRNAs are ubiquitous, they increase background noise and may significantly confound data quality and biological signal [69]. Several methods have been developed to remove the contribution of the ambient RNAs from each cell to recover the true molecular abundance. For example, the SoupX method estimates the ambient mRNA expression profile from empty droplets and the contamination fraction in each cell by making use of known negative cell markers in an identified cell cluster, and then corrects the expression of each cell using the two paratemers [69]. There are other ambient RNA decontamination methods that do not require prior knowledge of negative cell markers, such as DecontX [70] which uses a Bayesian inference model to estimate and remove the background noise, and CellBender [71] which employs a deep generative model to remove the background noise from ambient RNA. In the mixed-sample multiplexing scRNA-seq design, where multiple samples of different genotypes are pooled, or in the presence of subclones, a method called Souporcell can demultiplex cells, identify doublets, and perform joint genotyping and ambient RNA amount estimation by modeling the allele counts of genetic variants available from the reads [72]. Ambient RNA detection and removal is an emerging area of research and just began to be included in the AD snRNA-seq studies [44, 73]. However, since the leak of cytoplasmic RNA by ruptured cells to the cell suspension is unavoidable by the isolation protocols, especially for the case of nuclei isolation from fresh frozen tissues, we expect incorporating the ambient RNA decontamination into the single cell data analysis pipeline will provide much cleaner downstream analysis in future applications of AD.
Quality control and filtering on genes
Genes with low abundance should be removed since they do not contain sufficient information for reliable downstream statistical analysis [74]. Thresholds can be set for the number of cells expressing a gene or the mean expression of a gene [52]. The cell number threshold could be very liberal (e.g., 2 cells in some of the published brain disease studies [8, 68]). Still, it is critical not to exceed the minimal cell cluster size that one may reasonably expect [75].
Further, depending on the downstream analysis, some feature categories such as non-coding genes may not be of interest and hence could be removed to reduce the data complexity [8]. Mitochondrially expressed genes can be also discarded after cell QC in snRNA-seq data to avoid biases introduced during the nuclei isolation since mitochondrial transcripts are not expressed inside a nucleus [8, 67, 68].
Normalization
The observed single-cell read count data could be impacted by many biological and technical factors, including but not limited to sequencing depth, capture efficiency, and cell composition. Between-sample normalization can remove these sample-specific biases and mitigate the batch effect. The simplest method is scaling normalization, which corrects for sequencing depth difference by dividing the feature-level read counts by the library size (i.e., total read counts within each sample) and multiplying a constant value (e.g., 10,000). The library size corrected data is usually log-transformed after adding value 1 to prevent the logarithm of 0. This normalization strategy is implemented in popular tools like scanpy [50] and Seurat [76]. However, similar to bulk RNA-seq normalization, library size as a scaling factor is likely to bias towards highly expressed transcripts. In the context of bulk RNA-seq, there are three most popular methods for robust scaling normalization, including 1) the trimmed mean of M-values (TMM) [77], which calculates scaling factors by trimming away genes with extreme fold changes between samples; 2) the upper-quartile (UQ) method [78], which uses per-sample upper-quartile (75-th percentile) to scale counts; 3) the relative log-expression (RLE) [79], which scales to a pseudo-reference derived from the geometric mean of gene counts across cells.
While these bulk-based methods are still widely used in scRNA-seq data [74, 80], the excessive zeros in the scRNA-seq data jeopardize their effectiveness in calculating proper scaling factors. For example, the TMM method tends to overcorrect for the scaling factors [80] and the upper-quartile could be zero for many cells with low sequencing depth. Moreover, calculating the pseudo-reference sample from the geometric mean across cells can be applied to only the potentially minimal number of genes with non-zero reads in every single cell [80]. Alternatively, the dropout reads can be imputed by assuming a mixture model that includes two latent probability distributions: the probability of the true expressed reads and dropout reads among the true expressed reads. These model-based methods include SAVER [81] (Poisson-Gamma mixture model) and scImpute (Normal-Gamma mixture model) [82]. Markov Affinity-based Graph Imputation of Cells (MAGIC), on the other hand, utilizes a diffusion kernel to identify similar cells in reduced dimension, and infer the dropout reads from the similar cells [83].
Several scRNA-seq-specific normalization methods have been developed and they can be primarily classified into two categories: 1) cell-based normalization by estimating a cell-specific global size factor to normalize all the genes in the same cell, and 2) gene-based normalization by parametric modeling of individual genes. The scran package adopts the cell-based normalization approach by pooling the cells to estimate more robust size factors and avoid the impact of excessive zeros. Then pool-based size factors are “deconvolved” to yield cell-specific factors [84]. In contrast, the gene-based normalization methods, such as the SCnorm [85] and the Pearson residuals method SCTransform in the Seurat package [76], perform adjustments individually for each group of genes with different sequencing depths or different ranges of abundance levels. In addition to the correction for sequencing depth bias for different groups of genes, parametric modeling of count data can account for more complex technical or biological variations, such as batch effect, mitochondrial transcript fraction, cell cycle effect, cellular detection rate (fraction of detected genes), and the average number of counts per detected genes [86,87,88,89]. Moreover, cell-level and gene-level variations can be jointly modeled in a unified framework. For instance, for better separation of unwanted variation from biological signals in noisy, zero-inflated scRNA-seq data, Risso et al. proposed a Zero-Inflated Negative Binomial-based Wanted Variation Extraction (ZINB-WaVE) method, which incorporates not only observed and unobserved sample-level but also gene-level covariates (e.g., sequence length and GC content) [90].
Unwanted sources of variations such as batch effects should be adjusted using single-cell dedicated tools (e.g., MNN [91], CCA [92]), or general linear regression modeling tool (e.g., limma [93], ComBat [94]). Deep learning-based data denoising tools such as deep count autoencoder (DCA) [95] and single-cell variational inference (scVI) [96] are also attractive alternatives to handle unwanted variations in scRNA-seq.
Recommended workflow and application to AD
Figure 2 illustrates a workflow of preprocessing sc/snRNA-seq data, i.e., data QC and normalization. For data QC, we recommend to first inspect the distribution of cell-level read count statistics, such as the total number of UMI counts, the number of detected genes, and the percentage of mitochondrial reads. If no sample presents dramatically different data quality, we expect to see similar cell-level data distribution across donors. Otherwise, we should check if the sample data quality difference is associated with any biological or technical variable. Then confounding technical variables could be taken into account in the data normalization. For example, Seurat’s SCTransform normalization approach has an option “vars.to.regress” to regress out confounding factors. To investigate if any particular variables play significant contribution to the cell-level gene expression variation, mixed model variance component analysis (such as implementation by the R package variancePartition [97]) can be used to quantify the variance attributable to individual factors. For data normalization, we recommend evaluating several different methods. For example, start with the simplest approach of the global scaling by sequencing depth, and proceed to clustering and differential expression analysis (Sections C, D, and E). Then compare the clustering results of the simple method with those from more advanced/complex methods. We favor the methods that lead to better separation of cell clusters with clear cell type annotations and biological meaningful signatures. In cases of combining multiple batches (or conditions) of single cell data where there is batch (or condition) specific cell clustering, an elegant integration method such as Seurat/CCA [92] and harmony [98] should be used after normalization of individual datasets to minimize the batch difference.

A workflow of sc/snRNA-seq data preprocessing. After obtaining the single cell or single nucleus sequencing count data, a series of quality control processes are conducted to filter low quality cells with unusually high or low gene coverage or sequencing depth, unusually high mitochondrial content, ambient RNA, and doublets etc. QCed count data is normalized by either a global scaling approach or advanced parametric modeling of the zero-inflated count data distribution. If there exists batch or condition-specific clustering of the cells, a data integration method like MNN and CCA can be used to correct the batch difference to ensure that cells of the same cell type cluster together
In the first snRNA-seq analysis of control and AD brains by Mathys et al. [8], nuclei with fewer than 200 detected genes or an abnormally high ratio of mitochondrial reads were removed. Mitochondrially encoded genes were removed and only protein-coding genes detected in at least 2 nuclei were selected. In AD snRNA-seq study by Zhou et al. [57], they selected the nuclei with no more than 5% mitochondrial reads, 400–20,000 UMIs and 400–7000 genes as determined by UMI/gene distribution. In another AD snRNA-seq study by Grubman et al., the nuclei with more than 10% of their UMIs assigned to mitochondrial genes or nuclei outside the 5th and 95th percentiles in the number of detected genes or the number of UMIs were filtered out [41]. Similarly, AD snRNA-seq data analyses by Nguyen et al. [43], Lau et al. [99], and Gerrits et al. [44] also QCed their data by mitochondrial content and read count cutoffs, albeit with slightly different threshold values. In addition, Gerrits et al. conducted ambient RNA and cytoplasmic RNA identification to recover more cells from the raw data. In all these AD snRNA-seq studies, QCed data were normalized by the total library size multiplied by a factor of 10,000, except in the Zhou et al. study where they further regressed the total number of UMIs by a negative binomial model.
Feature selection and dimension reduction
Dimension reduction is to identify a few latent variables that explain the most variance in data. Selecting the most informative gene features can improve the detection efficiency and quality of the latent variables. The criteria for selecting informative genes include high biological variances with which the technical variance is modeled by the fitted relationship between mean and variance or spike-ins [74, 76, 84], and strong correlations with different cell types [17, 92] or known pathways (PAGODA) [100].
Then, the latent variables underlying the informative genes are identified by various techniques. Principal Component Analysis (PCA) is an efficient linear algorithm applicable to large-scale matrices and preserves both local and long-range structures. Each principal component is an orthogonal vector to the rest, and their linear combinations can reconstruct the global transcriptome. The PCA dimension can be determined by selecting top PCs accounting for 80 ~ 90% of total variances, PCs with significantly higher loading than bootstrapped data, or detecting an elbow point in the PC loading plot [92]. Several PCA variants have emerged to handle drop-out reads via zero-inflated negative binomial distribution (ZINB) [90].
t-distributed stochastic neighbor embedding (t-SNE) is a non-linear approach to preserve the local structures in the high-dimensional data [101]. Due to the emphasis on the local structure, t-SNE has gained popularity for effectively segregating clusters, but loses long-range structures [34]. Diffusion map (DM) is another popular non-linear method that projects both local and long-range structures to a lower dimension and is optimized to trace gradual changes in a transcriptome [102]. However, DM and t-SNE are computationally expensive. Recently, a computationally more scalable method, uniform manifold approximation, and projection (UMAP), has been proposed to include more long-range structures than t-SNE [103]. UMAP shows superior performances in segregating local clusters than t-SNE while recovering some of the global structures in scRNA-seq data [103]. The non-linear projection methods (DM, t-SNE, UMAP) can be applied directly to the transcriptome or the PCs of interest to compress the data in 2 or 3 dimensions. It is worth noting that, however, they may distort the overall data structure and introduce non-biological artifacts [104] and are often recommended for visualization purposes explicitly. Overall, dimension reduction is useful for visual inspections of cell-level patterns in AD. After data quality control and selection of highly variable genes with, for instance, significant dispersions (FDR < 0.05), cells often aggregate into clusters with distinct molecular characteristics. Such patterns define not only cell types with respective marker gene expressions in the reduced dimensions but also heterogeneous distributions of cells in AD samples in contrast with those from healthy control ones.
Unsupervised cell clustering analysis
Unsupervised cell clustering is a data-driven process to group cells that share similar molecular patterns in scRNA-seq reads. As this is an “unsupervised” approach, it minimizes the impact of external bias and serves to provide biological insights to understand distinct cell populations in the tissue of interest [34, 105].
The cell clustering approaches can be categorized into gene expression-based and genotype-based approaches. Gene expression-based approaches regard each cluster as a unique cell type or sub-population of a known cell type with a distinct expression pattern. However, the high-dimensionality of single-cell transcriptome incurs the “curse of dimensionality”, enforcing distances among the homogeneous cells and making it impossible to distinguish distinct cell populations [34]. This often necessitates dimension reduction before the clustering analysis. Once clusters are identified, the uniquely expressed genes in each cluster can serve as de novo markers to pinpoint, if any, the associated cell type and identify pathways underlying them [74, 92]. Further, gradual expression changes among these clusters may indicate temporal cellular dynamics to infer cell trajectory [106] or sub-clonal evolution [107].
On the other hand, genotype-based cell clustering approaches utilize sequencing reads to identify single nucleotide variants (SNVs) in individual cells and group the cells bearing a similar set of SNVs. The resulting cell clusters can be utilized to demultiplex the reads for individuals with distinct genotypes [108,109,110], identify clonal populations [107], or screen doublets and ambient RNA contamination [72]. We have curated the overview of the single-cell clustering tools in Table 2.
Expression-based clustering approaches
Clustering analysis is performed to infer coherent structures, often from the reduced dimensions. This involves evaluating cell-cell similarity and applying a suitable clustering algorithm to detect a certain number of segregated clusters at some resolution(s). Traditional metrics and clustering algorithms from bulk RNA-sequencing data analysis have been readily adopted in scRNA-seq analysis [105]. For example, SIMLR (Single-cell Interpretation via Multi-kernel LeaRning) utilizes Euclidean distance, Pearson’s correlation and Spearman’s correlation jointly to learn a consensus Gaussian kernel to detect diagonal block structures in these matrices [112]. Similarly, SC3 performs consensus clustering by iteratively performing PCA and k-means on a small subset of principal components, where Euclidean, Pearson, and Spearman correlations jointly evaluate the cell distances [111]. While these consensus methods over multiple similarity matrices identify robust clusters, their scalability is limited to ~ 10,000 to ~ 20,000 cells as calculation of global similarity, and consensus search are computationally expensive [111]. Density-based clustering (e.g. DBSCAN [113]) is a computationally affordable approach that searches for evenly distributed cells in lower dimension space by t-SNE or DM [9, 119]. However, these approaches may suffer stochasticity or distorted data structure due to the dimension reduction.
Graph-theoretic approach
Graph-theoretic approaches do not require dimension reduction and can retain both local and long-range structures in the form of cell-cell networks. k-nearest neighbor (kNN) network has been a popular method to construct these cell-cell networks, linking a cell with k most similar or closest cells [74, 92, 114,115,116]. In scRNA-seq settings, detection of kNN cells requires additional post-processing to account for drop-out reads and sparse expressions. The optimal partition of kNN-graph is computed through quality metrics such as Girvan-Newman (GN) modularity [120] or edge density measures. PhenoGraph constructs kNN network in Euclidean space, prunes spurious links by Jaccard index, then detects the coherent subnetworks by optimizing GN modularity with Louvain’s algorithm [114]. Louvain’s algorithm iteratively merges nodes to improve the global GN modularity, while the modularity measure acts as a scale parameter to capture from scattered and subnetworks (low modularity) to coherent and large subnetworks (large modularity) [121]. Seurat’s popular scRNA-seq analysis workflow adopts a similar strategy to PhenoGraph and allows the users to specify the resolution of resulting clusters [92]. MetaCell, on the other hand, first utilizes Spearman’s correlation in z-score transformed expression data, then applies a series of regularization steps to the adjacency matrix to remove spurious interactions, and finally identifies subnetworks with high edge densities [116]. SNN-Cliq detects dense subnetworks via quasi-clique detection in the mutual nearest neighbor network [115].
Overall, the kNN approach has become popular as it does not make assumptions about the underlying geometry. However, the choice of the kNN parameter has not reached a consensus in the field. Correlation between link weights and shared neighbors [114], global network connectivity [74] and convergence upon iterative regularizations [116] are the imporant criteria adopted by the aforementioned clustering analysis approaches.
Deep neural network approach
Deep neural networks consist of several layers of encoders mapping the input data into a low-dimensional manifold, from which the following decoder layers can reconstruct denoised, full-rank data. The applications in scRNA-seq include denoising single-cell transcriptome [95, 122], batch effect removal [118], probabilistic modeling of gene expressions or cell types [95, 96, 123] or dimension reduction [96, 117, 118, 124]. In cell clustering, these versatile functions of deep neural networks have become an attractive avenue to unveil complex cell architectures in scRNA-seq. Recent releases of TensorFlow [125, 126] with massive GPU parallelization have boosted the application of deep neural network learning to dissect complex patterns in in high-dimensional scRNA-seq data.
Classically, compared to the original input, these deep neural network models are trained by minimizing the reconstructed data loss. However, naïve model learning in this way could lead to over-fitting where non-biological sources of errors (e.g., drop-out reads, low coverage) in scRNA-seq contribute differently to the data noises [122]. Deep count autoencoder (DCA) and single-cell variational inference (scVI) define the reconstruction error as the log-likelihood of the noise model such as ZINB to denoise and impute the drop-out reads. The denoised data are utilized to infer cell clusters. scVI performs k-means clustering in the denoised low-dimensional latent space [96]. Similarly, scVI uses deep generative, variational autoencoder [127] with Gaussian mixture model to identify cell clusters and offer a statistically interpretable framework for downstream analyses [117]. On the contrary, DESC is a model-free approach in which a neural network generates a low-dimensional representation of the input data by minimizing the reconstruction loss [118]. An iterative clustering approach is to combine Louvain’s algorithm and cluster refinement to improve cluster purity [118].
Overall, deep neural network-based approaches offer a promising avenue to model non-linear patterns in single-cell transcriptomes, with computational scalability and flexibility to adapt different single-cell transcriptome models. However, they also face similar challenges as other approaches, such as adequate feature selections and choice of the ‘right’ models for single-cell transcriptome.
Genotype-based approaches
RNA reads from scRNA-seq provide a unique opportunity to infer SNVs per cell to demultiplex for individual samples [108,109,110], or cluster cells to trace clonal evolution [107] or genotype distributions [72]. However, challenges in scRNA-seq variant calling lurk from RNA-splicing, low transcript abundance, allelic drop-out, higher error rate from reverse transcription, incomplete transcript coverage, and 3′- or 5′-end bias in coverages [128, 129]. To handle these challenges, the pre-processing involves splice-aware alignment (e.g., STAR, minimap2), in conjunction with mpileup in samtools to detect variants present in low-coverage regions [72, 129]. To further enhance the confidence in the detected variants, pre-compiled variants from external data sets such as whole-genome sequencing (WGS) from bulk samples are used to detect the reads bearing the alternate alleles with VarTrix [72, 129].
scSplit [109], demuxlet [110] and Vireo [108] are tools dedicated to demultiplex mixed reads from individuals with known (demuxlet) or unknown genotypes (scSplit, Vireo). They are capable of detecting the doublets as outliers by the allele fraction model, which specifies the expected range of observed alternate alleles in singlet cells. On the other hand, Souporcell [72] and DENDRO [107] are specialized in clustering cells with the variant matrix to identify subclones and heterogeneity in the cell populations. Souporcell leverages mixture models to infer centroids in the alternate allele fraction space [72]. DENDRO is tuned more specifically for identifying sub-clones by measuring genetic divergence between the cells. ZINB models the allelic expressions to account for drop-out reads, and different degrees of genetic differences are utilized to construct a phylogeny tree across the cells, where each branching point characterizes sub-clonal expansion [107].
Evaluation of cell clustering quality
Unsupervised cell clustering is an essential component of single-cell transcriptome data analysis, and has been increasingly applied to single-cell transcriptomes [130]. However, there is no consensus on evaluating the cluster qualities to identify the best set of clusters reflecting the underlying geometry and biology in scRNA-seq. In the clustering analysis, the quality of clusters is evaluated by comparing with external gold-standard information (external validation) or the internal geometry in the data (internal validation) [131]. Internal validation evaluates intra-cluster compactness and inter-cluster separability using various indices such as Dunn’s index [132] and Davies-Bouldin index [133] that define different aspects of the underlying data geometry [134], and then determines the optimal number of clusters [134]. On the other hand, external validation evaluates how well the clusters capture relevant information outside the analyzed data. External data can be gold-standard clusters that a clustering algorithm must reproduce (e.g., known subtypes, simulated data with known clusters) [135, 136]. Their concordances can be evaluated by mutual information [137] or adjusted Rand index [134, 138].
As a rule of thumb, a good clustering analysis for scRNA-seq data in AD should reflect major cell populations with robust over-expression of the markers [26, 42, 74, 139], mix cells from different batches of samples [92], and capture key pathways associated with AD pathologies [8, 140] such as immune response, synaptic transmissions and myelination. Furthermore, the reproducibility of the identified clusters should be examined by cross-validation or bootstrapping approaches [7], concordant cell populations in animal models [140,141,142] or respective bulk cohorts [7, 9, 140].
Applications to AD
Several early scRNA-seq studies leveraged brain cells from preclinical disease models to understand cell architectures in neurodegenerative brains under controlled environments. In these studies, cell clustering analysis identified catalogs of distinct cell populations in mouse brains [119, 141], microglial subpopulations from brains undergoing neurodegeneration in mice and humans [139, 140], and differentially regulated neuronal stem cell subpopulation in AD model zebrafish [142].
Studies on the single-cell transcriptome of neurodegenerative human brains have emerged to pinpoint cell populations associated with AD-associated traits. Darmanis et al. 2015 sequenced 466 cells from healthy adult temporal lobe tissue [26]. Gaussian mixture clustering in t-SNE space revealed major brain cell types and distinct neuronal subpopulations with adult-brain-specific MHC-I expressions compared to fetal brains [26]. Olah et al. 2020 analyzed 16,242 cells from fresh prefrontal cortex samples from AD and healthy controls [7]. The study performed iterative Louvain’s clustering on different combinations of the first 15 PCs to identify robust microglial subpopulations depleted in AD [7].
The first phase of unsupervised clustering may be limited in resolution and overlook underlying fine clustering structures. Several studies biologically guided sub-clustering in major cell types to dissect distinct subpopulations. With this strategy, Lau et al. 2020 identified 43 unique cell clusters from 169,496 nuclei from prefrontal cortical samples of postmortem AD and control brains [42]. These clusters included loss of protective glial cells and enriched angiogenic endothelial cells in AD brains [42]. Similarly, Mathys et al. 2019 performed two-stage Louvain’s clustering on kNN on 80,660 nuclei from post mortem prefrontal cortices of 24 AD patients with varying pathology and 24 control subjects [8], and identified sub-clusters associated with AD-related traits and female over-representation in the AD-associated sub-clusters [8].
Recommended workflow: from feature selection, dimension reduction to clustering
This section illustrates the overall recommended workflow from feature selection to clustering analysis (Fig. 3A) and the scripts for these analyses can be found in the companion GitHub repository (see the section "Availability of data and software code" for details). For feature selection, gene dispersion, the gene-wise deviation from the fitted relationship between mean and variance from log-normalized expressions, can serve as the quality metrics for informative features [52] (Fig. 3A). However, in many single-cell AD studies, the cells are confounded with many ‘undesired’ variables (e.g. batches, varying sample quality, different sample preparation procedures), shadowing the meaningful biological signals, and the effects of these undesired variables should be blocked during the gene dispersion modeling [91]. We analyzed the gene dispersions in the snRNA-seq data from the ROSMAP cohort, consisting of postmortem brain tissues from 48 individuals with varying AD pathology [8] (Fig. 3B). Using scran workflow, individual-wise dispersions were first calculated, then summarized into a combined dispersion per gene. Overall, genes with significant dispersions with FDR < 0.05 exhibit high biological variances compared to the technical variances as modeled by the mean-variance curve. This is exemplified by VCAN, an oligodendrocyte progenitor cell marker [143], and APOE, whose polymorphism is a major genetic risk determinant of AD [144] and a marker for astrocyte and activated microglia [8] (Fig. 3B).

Recommended workflow of feature selection, dimension reduction, and clustering, and applications in AD. A Recommended workflow of dimension reduction and unsupervised clustering analysis of AD scRNA-seq data. Software tools are provided for each step. B Technical variance vs biological variance plot from the ROSMAP snRNA-seq data. The red dots depict genes with significantly greater biological variance than the technical variance (FDR < 0.05) and the top 20 most significant genes are labeled. C PC versus percentage of the variance explained. Vertical lines indicate recommended number of PCs from different workflows (red: PC denoising workflow from scran, blue: elbow point from Seurat, green: default number of PCs in Seurat). D UMAP plot of snRNA-seq from ROSMAP cohort. Clustering by PhenoGraph implemented in Seurat is marked by numeric labels. The cell types identified by marker gene expressions in (E) are highlighted as different border colors with relevant cell type name labels (Ast: astrocyte; End, endothelial; Ex: excitatory neurons; In: inhibitory neurons; Mic, microglia; Oli, oligodendrocytes; Opc: oligodendrocytes progenitor cells), and AD pathology (Healthy - green, early AD – yellow, late AD - red) are highlighted as different point colors. E dot plots of brain cell type markers showing their cluster-wise expressions. Clusters on the y-axis are ordered according to their proximity in the UMAP plot in (D). F Proportions of cells at different AD stages. FET is performed to evaluate whether the cells from each AD stage are enriched in each cell cluster. As significant enrichment is based on a cutoff of 0.05 for corrected FET p-value. In the plot, red dots represent the cases with fold enrichment (FE) > 1.3
Then, log-normalized gene expressions across the genes with significant dispersion should be used to perform dimension reduction by PCA. Alternatively, data integration workflows (e.g. CCA [92], MNN [91] and Harmony [98]) offer adjusted features for undesired batch variables, and PCA can be applied to them. Although PCA is not a prerequisite for several down-stream analyses (e.g. deep learning-based clustering), PCA offers a time-cost effective option to identify a few key variables in high-dimensional data, and have been adopted routinely in popular scRNA-seq workflows such as scran [52], Seurat [92] and scanpy [50]. During PCA, determining the number of PCs is crucial, and several criteria such as the elbow in explained variance curve, correlations to technical variance, or PCs with significant variances when randomly permuted should be examined (see Fig. 3C). Among these criteria, random permutation-based evaluation (e.g., Jackstraw statistics in Seurat) is computationally expensive, and may not be suitable for large-scale scRNA-seq data sets (number of cells > 10,000). Instead, the simple elbow detection in the explained variance curve (blue line in Fig. 3C) can be effective without huge computational burden.
Then, the clustering analysis identifies cells with coherent expression patterns (i.e. expression-based clusters). Depending on the nature of the method, the selected gene expression features may be used directly (e.g., autoencoder-based methods), otherwise, the selected PCs should be utilized for methods relying on cell-cell distance metrics (e.g. kNN-based methods, k-means clustering). While deep learning-based methods can simultaneously handle undesired variables and capture non-linear patterns [118], they often require GPU-enabled parallel computation capacity. Thus, in the absence of such high-computation power, we recommend kNN-based methods which can capture local structures and non-linear patterns via complex network topology. Then, the selected PCs can be embedded on the lower dimensions, usually 2- or 3-dimensional space via UMAP to visualize the resulting clusters (Fig. 3D). To evaluate the clusters, the cell clusters associated with similar brain cell types such as excitatory/inhibitory neurons, astrocytes, oligodendrocytes, and microglia should express the respective cell type markers and be located in proximity as demonstrated in the ROSMAP cohort examples in Fig. 3D-E.
To further assess the biological significance of cell clusters in AD, enrichment of cells from various AD pathology (e.g. Braak staging, CERAD score, cognitive declines, AD pathology diagnosis) can guide pinpointing potential key cell populations underlying AD. We demonstrated enrichments of cells from healthy controls (no pathology), early AD (early pathology) and late AD (late pathology) from analyzing the ROSMAP snRNA-seq data (Fig. 3F) by Fisher’s Exact Test (FET). Here we used the sample pathology status defined in the original study by Mathys et al. [8]. Specifically, AD-pathology means increased AD pathological measurements such as β-amyloid (Aβ) while no-pathology represents no or very low AD pathological measurements. Based upon the degree of amyloid neurofibrillary tangle burdens, AD-pathology is further classified into two subgroups including early (modest burden) and late pathology stage AD pathology (higher burden). With a stringent threshold of 0.05 for corrected FET p-value < 0.05 and enrichment fold change (EFC) > 2, it readily uncovers over-represented cell populations in severe AD such as cluster 3 (an oligodendrocyte subpopulation), cluster 18 (microglial subpopulation), and cluster 16 (inhibitory neuron subpopulation).
In contrast to the expression-based clustering, the genotype-based clustering methods can facilitate several data quality control concerns when raw reads are available. For instance, cell clusters with distinct genotypes represent cells from different individuals and provide a systematic way to evaluate the agreement with the clinical annotations [145]. Further, doublet cells can be discerned via leveraging the allele fraction model (Fig. 3A).
Cell type inference and annotation
An essential goal of clustering analysis is to characterize the identity of the cells within each cluster. Marker genes can characterize a cluster with biologically meaningful functions and inform respective cell types. For example, the cell types in the human brains can be annotated by interrogating the expression patterns of known marker genes: NRGN (excitatory neurons), GAD1 (inhibitory neurons), AQP4 (astrocytes), MBP (oligodendrocytes), CSF1R and CD74 (microglia), VCAN (oligodendrocyte progenitor cells), FLT1 (endothelial cells), and AMBP (pericytes) [8].
The drawback of the marker gene-based method is that the markers are often limited to major cell types, hindering the annotation of novel cell clusters or cell subclusters with unknown biological functions. To overcome this drawback, an alternative approach is to use reference signatures derived from existing single-cell datasets [146, 147]. To find the best-matched cell type, the de novo cluster marker genes can be compared with the signatures from the reference single-cell databases by enrichment test or overlapping statistics. The de novo cluster marker genes can be defined as the up-regulated genes in a cluster of interest against the rest clusters through differential expression (DE) analysis (see the Differential expression for disease gene identification section below). For large single-cell datasets, an iteration of clustering and sub-clustering analyses may be needed to reveal the structure of cell clusters. Various automated cell type annotation tools have been developed to assist with cell type annotation. For example, scQuery is a web server that predicts cell types based on over 500 different scRNA-seq experiments [148]. Garnett and scmap allow users to build their own databases or train new cell classifiers to classify cells of interest [149, 150]. These automated annotation tools can be combined with the marker gene-based methods to facilitate the annotation of large complex single-cell datasets.
Differential expression for disease gene identification
DE analysis is useful to discover unique gene expression profiles in novel cell clusters or under disease conditions. In scRNA-seq experiments, DE analysis is presented with additional challenges such as low read depth per cell, the dropout event [151], and multimodality in gene expression values [152]. As the sequenced tissues consist of cells from different types at different states, the heterogeneity leads to variable distributions of gene expression in different cells. Moreover, the stochastic nature of transcription may introduce variability to gene expression levels [153].
A variety of DE methods have been developed to model the dropout events and the multimodal nature of scRNA-seq data. For example, MAST employs a generalized linear model (GLM) and considers the dropouts with a bimodal distribution [89]. Monocle employs a Tobit model to account for dropout events and fits the data with a generalized additive model (GAM) [89]. SCDE models the gene expression as a mixture of ZINB distributions and applies a Bayesian model to estimate the posterior probability for the DE genes [48]. D3E models gene expression distribution by the bursting model of transcriptional regulation [154]. scDD applies a multimodal Bayesian modeling framework to model the multimodal distributions of single cells [155].
To benchmark the performance of different DE methods, extensive experiments have been performed to evaluate many single-cell-based tools as well as popular bulk-tissue-based approaches. Interestingly, the comparative study showed that the single-cell-based tools did not perform better than the bulk-tissue-based methods such as limma [93], DESeq2 [156], and edgeR [157]. The performance of many tools specially designed for scRNA-seq is even worse than the simple t-test or Wilcoxon rank-sum test [158]. Both scRNA-seq and bulk RNA-seq DE tools need to strike a balance between sensitivity and precision [159, 160]. As bulk RNA-seq tools are not specifically designed to model the gene expression profiles of scRNA-seq data, they may suffer poor performance due to zero inflation or multimodality. Indeed, the performance of bulk RNA-seq tools could be further improved by combining with a weighting strategy to down-weight excess zeros [161].
Recommended workflow and applications to AD
Different scRNA-seq DE methods have been applied to reveal gene signatures associated with AD pathology. The bulk-tissue-based DE methods, which have efficient computational speed and sophisticated pipeline, can be directly used for the general purpose of scRNA-seq studies. For example, Grubman et al. used edgeR to identify cluster marker genes as well as the individual-specific and sex-specific differentially expressed genes (DEGs) from 13,214 nuclei of entorhinal cortex samples [41]. Meanwhile, as no single DE tool is superior in all scenarios, we recommend a combination of different methods to identify the most robust DEGs out of consensus calls. The AD study by Mathys et al. combined Wilcoxon rank-sum test and a Poisson mixed model which accounted for individual variability to identify a consensus list of 1031 DEGs in AD-pathology versus no-pathology individuals across cell types [8]. We applied MAST to the ROSMAP AD snRNA-seq data and shared the script through the companion GitHub repository (see the section "Availability of data and software code" for details).
The aforementioned DE methods depend on pre-defined cell clusters or groups, but the optimal number of cell clusters and/or biologically relevant clusters in scRNA-seq data is often hard to find out. singleCellHaystack addresses this issue by applying the Kullback–Leibler Divergence method to identify genes expressed in subsets of non-randomly positioned cells in a multidimensional space [162]. By comparing gene expression profiles to a reference distribution of all cells, singleCellHaystack can identify differentially expressed genes in an unbiased way without relying on cell clusters. The cluster-independent method may serve as a complementary approach for DE analysis when biologically meaningful clusters are not available for scRNA-seq data.
Trajectory inference
Trajectory inference aims at estimating dynamic changes in a single-cell transcriptome landscape, assuming that the cell-wise transcriptome is a static snapshot at a time point along some cellular process. The cascades of these snapshots compose of a dynamic trajectory of cells undergoing continuous changes in the cell states, known as ‘pseudo-temporal trajectory’. Trajectory inference assigns a one-dimensional coordinate, known as pseudotime [163], per cell to approximate the departure from the beginning of the trajectory. It allows us to reconstruct dynamic biological processes without sampling tissues at different time points, identify critical transition points between distinct cell states, and analyze shifts in cell-type composition and cell synchronization [163, 164].
The inferred pseudotime may not progress uniformly in real-time along a trajectory, as the trajectory inferences are based on inferring tree-like geometry in the data rather than by ‘real world’ clocks [165]. RNA velocity provides an alternative way to time-stamp cells by utilizing RNA kinetics. According to the central dogma of molecular biology, the rate of change in mature mRNA abundance, i.e., RNA velocity, can be described by competition between mature, spliced mRNA produced from unspliced pre-mRNA and degraded mature mRNA [166]. In this framework, a greater abundance of pre-mRNA than the mature mRNA indicates an up-regulation, and a down-regulation in the contrary [166, 167]. The summarized kinetics in the global cell transcriptome can facilitate trajectory inference [168].
Information on cellular dynamics could improve our understanding of AD pathologies, such as identification of marker genes for early diagnosis and prompt intervention of neurodegenerative diseases whose pathogenesis precedes many years before clinical manifestation. Herein, we review different computational approaches in cell trajectory inference and discuss its outlooks in AD scRNA-seq analysis.
Overview of trajectory inference methods
Inspired by the metaphorical epigenetic landscape conceived by Waddington, Trapnell et al. adopted a dynamical systems framework. They described the biological process as cells moving in the “gene regulation space” along a particular “trajectory” to finally reach a stable state that corresponds to a clearly defined cell type or an “attractor” in dynamical systems [169]. In this framework, trajectory inference consists of three components: determination of gene regulation space (dimensionality reduction), identification of the attractors (unsupervised cell clustering), and the inference of the trajectory (graph-based data approximation followed by pathfinding and cell ordering). Here, we will primarily focus on the third component as the first two have been extensively discussed in the prior sections in this review.
Graph-based data approximation is used to extract the geometrical skeletons of a given data point cloud. Such graph types include, for example, principle curves [170], minimum spanning trees (MST) [171], nearest neighbor (NN) graphs [172], and more complex networks. Early trajectory inference methods contemplate the trajectory structures to be non-branching (Wanderlust [173]), bifurcated (Wishbone [174]), or even cyclic (DeepCycle on single-cell imaging data [175]), and require prior biological knowledge or user-provided input. Emerging methods, some of which will be covered subsequently, allow unbiased inference of trajectory structures from transcriptomic data at the cost of increased computational complexity, which would impact their scalability and usability.
MST is a tree-graph which spans the entire data points with the minimum overall distance. While each node in the MST represents a single cell, the edge can be the similarity between gene expression profiles or transition probability between neighboring cells. Monocle, one of the pioneer algorithms for trajectory inference, applies the independent component analysis (ICA) and constructs an MST over all the cells [163]. SCOUP models the probability of a cell differentiating into a neighboring cell in a PCA-reduced space based on the Ornestain-Uhlenbeck (OU) process and assumes that a mixture of OU processes represents multiple cell fates during differentiation defined by the shortest paths in the MST [176].
As MST is often sensitive to noise and outliers, Waterfall [177] and TSCAN [178] construct a cluster-based MST to improve the robustness. Slingshot takes one step further by implementing simultaneous principal curves compatible with any dimensionality reduction method to infer multiple fates that individual cells may take during development [106].
Some algorithms use the kNN graphs to overcome the impact of noise and outliers. SLICER takes the shorted path on a kNN graph in a reduced space by locally-linear-embedding (LLE) and determines the branching location by geodesic entropy [179]. Diffusion Pseudotime (DPT) takes a random walk in the nearest neighbor graph in the high-dimensional space. The pseudotime is inferred by Euclidean distance between the probability vectors, rather than gene expression, of any two cells differentiating into all possible fates [102]. However, it might be inappropriate to use a fixed neighborhood size in some cases, as the data are not evenly distributed across the defined space. Moreover, the computational cost of kNN increases drastically with the number of cells.
Others construct complex networks for cell projection to allow assumption-free inference of trajectory topologies. For instance, scEpath [180] builds an energy landscape and infer transition probabilities and lineage relationships between cell stages. Hopland [181] maps cells onto Waddington’s epigenetic landscape and infer pseudotime sequences by geodesic distance.
Another attention-drawing question is whether a continuous transition process is presumed in the trajectory inference algorithms. While the answer is yes in most cases, some would argue that due to limited sampling rate/depth, the experimental data do not always conform to such assumptions. Several methods have been developed to tackle this issue. For example, PArtition-based Graph Abstraction (PAGA) [182] models the connectivity of cell groups and reconstructs both continuous and disconnected topologies at multiple resolutions [183]. Monocle 3 [184] adopts a similar idea to PAGA. It first projects the cells onto a lower-dimensional manifold by UMAP and merged adjacent groups of cells identified by the Louvain community detection algorithm into “supergroups” to resolve the developmental trajectories. Another example is TinGa, a growing neural graph-based algorithm that also allows disconnected topologies [185].
Overview of RNA velocity
The balance between spliced and unspliced mRNA, termed RNA velocity, measures the transcriptional dynamics in the cells and facilitates trajectory inference. In scRNA-seq, Manno et al. 2018 first utilized the relative abundances of exonic and intronic reads to infer the cell-level RNA velocity with a simplified model assuming the same rate of pre-mRNA processing for all genes [166,167,168, 186, 187]. The cell-level RNA velocity inference was applied to scRNA-seq data of mammalian embryo brains and captured dynamic changes in developmental trajectories [168, 187]. Bergen et al. 2020 developed scVelo to implement a more generalized kinetic model with gene-specific pre-mRNA processing rate and infer the kinetics-based cell trajectories in scRNA-seq [187]. While RNA velocity was analyzed mostly in developmental processes, these have not been applied in AD single-cell transcriptome. Potentially, RNA velocity underlying AD-specific microglial or neuronal subpopulations may shed light on key dynamical splicing activities contributing to these AD-specific cell fates.
Recommended workflow and application of trajectory inference to AD
A typical workflow may involve the following steps: 1) conduct data QC, normalization, dimension reduction, and clustering as described above or according to the trajectory inference software package; 2) choose genes that are informative of the cell state progress, such as cell type markers and highly variable genes; 3) conduct data dimension reduction; 4) infer the trajectory and order cells by pseudotime in the trajectory; 5) identify genes regulated over the course of trajectory, such as genes that correlate with the pseudotime or distinguish between cell state along the trajectory branches; and 6) perform additional analyses, such as constructing Granger’s causality network using pseudotime information and identifying trajectory path that correlate with covariates of interest such as AD pathology traits; and 7) generate biologically meaningful hypothesis for experimental validation. As an example, we conducted a trajectory inference for an excitatory neuron cluster of the ROSMAP AD snRNA-seq data using Monocle (Fig. 4). A pipeline implementing this trajectory analysis is provided in the companion GitHub repository (see the section "Availability of data and software code" for details).

Trajectory inference of an excitatory neuron cluster of the ROSMAP AD snRNA-seq data. A Numbered cell states in different colors and two potential trajectory paths as indicated by curved arrows. B Pseudotime indicated by gradient color intensity
scRNA-seq-based trajectory inference methods have been extensively utilized to study the developmental processes and immune systems where cells undergo active transitions from one state to another [188,189,190,191,192]. A unique disease-associated microglia subtype was identified in AD transgenic mouse brains by trajectory inference [139]. Several AD cohort studies [193,194,195] generalized this concept and aligned the individual subjects along the disease trajectory. The inferred models successfully predicted the clinical deterioration and conversion to advanced disease stage from baseline gene expression and disease subtype stratification.
With the development of high-throughput single-cell sequencing techniques, multi-omics data can be simultaneously measured in the same cell. G&Tseq sequences both genome and transcriptome [196], REAPseq measures protein and transcripts [197], scTrio-seq [198] quantifies genetic, epigenetic, and transcriptomic changes, and paired-seq [199] jointly examines the chromatin accessibility with transcriptomic heterogeneity. Despite the modest coverage, such methods add comprehensive information to help infer the trajectory of cells. Algorithms have been developed to compare cross-experiment, cell-type-specific differences and integrate multi-omics at the single-cell scale [200]. Recently, a microarray-based spatial scRNA-seq further resolves the spatial distribution of cell subpopulations in pancreatic tumors [201]. Such progress will prime the ground for novel findings in complex diseases, including AD.
Trajectory inference opens new venues to capture biologically critical dynamic changes that were considered as noises, and enables additional insights via trajectory-based differential analysis [202,203,204], latent variables-pseudotime interactions [205], pseudotime-based gene co-expression network analysis [206], and gene regulatory network inference [207], which will be discussed in the section “Single cell gene network analysis”.
Copy number variation detection
While scRNA-seq is primarily designed to quantify the cell-level expression abundance, the sequencing data contains a substantial portion of the information about the genomic variations, including SNPs and Copy number variations (CNVs). CNVs are one major type of genetic variation. Based on the CNV map of the human genome [208], CNVs occupy 4.8 to 9.5% of the human genome in healthy individuals [208,209,210]. Copy number aberration is involved in the pathogenic process of many diseases, for example, a variety of cancers [9, 211,212,213,214], Parkinson’s disease [215, 216], schizophrenia [217], mental retardation [218], and AD [218,219,220,221,222]. Our recent study found thousands of AD-specific CNVs based on bulk-tissue based whole-genome sequencing data of postmortem brains from AD cases. Whether there are cell- or cell cluster-specific de novo CNVs in AD remains unclear.
Compared with many previous successful CNV calling methods based on bulk tissue sequencing data [223,224,225,226,227,228,229,230,231,232,233], CNV detection from scRNA-seq is challenging due to several technical limitations, including low and non-uniform genome coverage, amplification biases [234, 235] and prevalent monoallelic detection due to transcriptional stochasticity [234, 236,237,238]. The monoallelic bias is more pronounced for lowly expressed genes than highly expressed genes. The monoallelic bias is still high for polymorphic loci with good coverage [237, 238]. Further, 3′-ended scRNA-seq have poor coverage in the 5′-end. Thus, the mutations in the 5′ end may not be sufficiently covered. Together, these limitations reduce the reliability of CNV calling at the gene level in scRNA-seq. Instead, previous studies have suggested large-scale CNVs can be reliably inferred from scRNA-seq at full chromosome-level or chromosome-arm-level [212, 239, 240].
Despite these challenges, several methods, including InferCNV [241], HoneyBADGER [236], CONICS [239], CONICSmat [239], and CaSpER [242] have been developed to detect CNVs from scRNA-seq data (Table 3). InferCNV [241], as a part of the TrinityCTAT toolkit, is the first and the most popular scRNA-seq CNV detection method to predict chromosome-scale CNVs. It calculates residual transcriptomic expression profiles of target cells using a given set of normal reference cells as the baseline. It identifies potential copy number alteration (CNA) regions using the Hidden Markov Model (HMM). To reduce the false-positive rate, a Bayesian mixture model is further implemented to estimate the copy number status of each CNA region in each cell based on the maximized posterior probability [241]. It can work with and without normal-cell reference. If there are no reference cells, the average signal of all target cells will be used as the baseline. It should be kept in mind, though, CNVs shared by all target cells are indistinguishable without reference cells [241]. HoneyBADGER [236] was developed based on a similar algorithm framework as InferCNV, which integrates the HMM and Bayesian inference. To improve the CNV calling accuracy and sensitivity, HoneyBADGER takes continuous allelic imbalance patterns at common SNP loci into consideration. The monoallelic bias rate is also adjusted in their posterior probability model. Based on in-silico simulation, HoneyBADGER can identify sub-clonal CNVs as small as 10 Mb, and chromosome-arm-level CNV events with cell frequency as low as 30%. The inference resolution of the method is higher than the solely normalized expression profile-based method [236]. Like InferCNV, HoneyBADGER also needs expression profiles of normal cells as a reference to calculate residual expression magnitude. Besides the normal cell reference, it also needs whole-genome sequencing or whole-exome sequencing (WES) data from the same sample to get heterozygous SNP positions. If WES data from the same sample is not available, the common SNPs (population frequency ≥ 10%) from natural populations can be used as a location reference. Furthermore, another scRNA-seq CNV calling method CONICS (COpy-Number analysis In single-Cell RNA-Sequencing), released in 2018 [239], infers the CNV status of given cells based on pre-inferred CNV regions from additional bulk-tissue DNA sequencing data (for example, WES data). If the extra DNA-sequencing data or control scRNA-seq data is not available, it also provides an extra caller named CONICSmat. CONICSmat is based on Bayesian inference from averaged gene expression profiling of target scRNA-seq data. HMM is not used to infer potential CNV region in CONICS/CONICSmat. Potential CNV location is inferred from additional bulk-tissue DNA sequencing data. As a result, its resolution depends on the resolution of the bulk-tissue DNA CNV calling method. Without extra bulk-tissue DNA data, the CNV inference is chromosome-arm level [239]. CaSpER [242], which adopted a strategy very similar to HoneyBADGER, integrates allele frequency shift information and normalized expression profiles to predict CNV regions using hierarchical HMM and Bayesian algorithms. It does not need prior variant calling. To speed up the whole CNV calling process, CaSpER takes aligned bam files as input to generate allele and expression profiles.
Here, we present the pros and cons of all methods, with consideration for budget and experiment design (Table 3). While methods utilizing both allelic-imbalance and gene expressions could improve the accuracy and sensitivity of CNV calls, the additional DNA-sequencing data required to generate allelic-imbalance profiles need additional budgets and sample materials. For example, some tools such as HoneyBADGER bypass such issues by leveraging common SNPs from public population datasets instead of the matched WES data in exchange for reduced sensitivity. On the other hand, the performance of expression-based methods is dependent on reference cells of choice [236]. Ideally, matched normal cells from the same individual could be used as references, or germline cell populations may serve as the alternative [9].
An independent normalization procedure of diverse cell types should be done based on the corresponding references [236]. All scRNA-seq CNV methods are highly coverage-dependent [236]. The true positive rate (TPR) of scRNA-seq CNV calling methods is not high in the current stage. For example, the TPR of CaSpER is around 60 to 80% based on in-silico simulation [242]. Additional experiment methods should be used as cross-validation [9], such as fluorescence in situ hybridization (FISH), cytogenetics and bulk WES or WGS. Matched scDNA-seq and scRNA-seq data [196, 243] can be used as ground truth to measure the performances of current scRNA-seq CNV tools, and be valuable for future software development.
All the above scRNA-seq CNV calling tools provide some downstream analysis, such as cell clustering [241], inter-clone differential expression analysis [239], phylogeny analysis [239], intra-clone co-expression network analysis [239], infer clonal evolution [242], and identify gene expression signature of clones [242].
Recommended workflow and applications to AD: copy number variation detection
A recommended workflow for identifying CNVs in single cell RNA-seq data based on the InferCNV tool is illustrated in Fig. 5A. Note that the data preprocessing step will follow prior discussions in Quality control and normalization, Feature selection and dimension reduction, Unsupervised cell clustering analysis and Cell type inference and annotation sections. There can be two kinds of reference cells in AD. First, for each cell cluster, cells from normal controls can be used as the reference cells and can be compared with cells from AD cases in the same cell cluster. Second, brain tissue-based cells can be compared with matched non-brain tissue cells from the same individual, for example, blood, to detect brain-specific somatic CNVs. Further omics data, such as genomic data, are needed to validate the inferred CNVs in both cases.

CNV identification from AD single cell data. A Recommended workflow of CNV detection. B Inference of CNVs in excitatory neurons of the ROSMAP cohort. The red box highlights an amplification region on chromosome 19
We applied this workflow to the sn-RNAseq data from the ROSMAP AD cohort [8]. Figure 5B shows the CNV calling result from the AD cells in one excitatory neuron cluster when using the cells from normal controls in the same cluster as the reference cells. The only predicted CNV region is located at chr19: 571,277-55,403,250 with an extra copy in late AD (Supplementary Table S1). This amplification region contains 66 genes (Supplementary Table S2), including PPP2R1A. PPP2R1A is known AD risk factor [244] and dephosphorylates tau protein [245, 246]. The script for this CNV analysis can be found in the companion GitHub repository (see the section "Availability of data and software code" for details).
Expression associated quantitative trait locus (eQTL) analysis
Expression quantitative trait loci (eQTLs) analysis links single nucleotide polymorphisms (SNPs) with their potential transcriptional effects on downstream genes [247] and has been utilized to pinpoint disease-risk SNPs [248]. Previous studies have shown that disease-risk SNPs are enriched for cis-eQTLs with modest effects [249, 250]. Many large-scale eQTL consortiums have emerged in recent years, such as ImmVar [251], BLUEPRINT [252], GTEx [253], CAGE [254], PsychENCODE [255], and eQTLGen [256]. Although bulk-tissue-based eQTL analysis is still valuable to understand the functional consequences of genetic variations, it has limited power to decipher the context-specific eQTLs, such as the tissue-specificity [247, 253, 257], cell type-specificity [247, 248, 258,259,260], and developmental stage-specificity [251, 261]. These are further complicated by transient eQTLs and those conditional on cell status [247, 261]. Single-cell eQTLs (sc-eQTLs) analysis can shed light on these issues.
Detection of eQTLs from scRNA-seq data
In sc-eQTL analysis, the number of cells should be large enough for sufficient statistical power [247]. At least three parameters need to be considered in the experimental design stage: the sequencing depth per cell, the number of cells per individual, and the number of individuals. The sequencing depth per cell affects the accuracy of gene expression measurement, the total number of cells affects the cell type number, and the number of individuals affects the effective SNP number [247]. Under a fixed budget, sequencing more cells rather than sequencing more reads per cell in fewer cells can increase the power to detect more sc-eQTLs [247].
The sparsity of scRNA-seq makes it less powerful to detect sc-eQTLs than bulk-tissue data [247, 262]. Although the analytic procedure is straightforward, scRNA-seq data may not meet the underlying assumptions for bulk tissue-eQTL methods [247]. For instance, bulk-based methods assume that log-transformed gene expressions follow a particular probability distribution (normal distribution, Poisson distribution, or negative binomial distribution), which may not be valid in scRNA-seq [247]. Further, drop-out events introduce a bias towards highly expressed transcripts [262], and the sparse transcriptome decreases the number of genes with detectable eQTLs [247, 261,262,263,264]. Previous studies show a 6.9-fold difference in the eQTL detection power between single-cell data and bulk RNA-seq data [247, 249, 260].
Several softwares have been developed to address the above challenges in sc-eQTL detection, such as SCeQTL [262] and scReQTL [265]. While dropout reads can be imputed to mitigate the sparsity [81,82,83, 260], SCeQTL (Single Cell expression Quantitative Trait Locus) [262] incorporates the excess of zero expressed genes into the statistic inference framework. SCeQTL separates genes with zero and non-zero expression, and uses zero-inflated negative binomial regression [266].
In addition to sparsity in scRNA-seq, other factors like cell lineages and variant allele frequency can also be incorporated into the inference framework [262]. Inferred single-cell pseudo-time can be utilized to capture eQTLs related to cell differentiation [261]. scReQTL [265] calculates the correlation between variant allele fraction at biallelic polymorphism loci (VAFRNA) and gene expression level in single cells, using a linear regression model. VAFRNA is derived from allele mapping of scRNA-seq data and is sensitive to allele mapping bias. SNP-aware alignment is preferred in the preprocessing step [265]. Prevalent monoallelic expression and single-cell sequencing technique bias towards 3′-end (for example, 10X Genome platform [37]) limit the detection power of scReQTL. scReQTL can only detect a subset of expressed SNPs from the genome-wide SNP profiles. This approach is suitable for single-cell data without matched DNA sequencing information.
Single-cell-based eQTL analysis is still in its infancy. Even though the proof of concept started in 2013 [267], the real application of sc-eQTL analysis started just recently [110, 260, 261, 268]. As one major type of molecular marker like CNV, sc-eQTLs can be used to infer cell type [247, 260], study cell-to-cell expression variability [261], cell type heterogeneity [269], and cell lineage development [247, 260]. It has been shown that the heritability of diseases and complex traits can be explained partially by cell type-specific eQTLs [248]. Other unique advantages of sc-eQTLs are inferring the cell activation states [247, 260] and studying the dynamic process of genetic variations regulating gene expression [260, 261, 268]. Integration of sc-eQTLs and other omics data, for example, scATAC-seq data, can help better understand the genetic mechanism of gene expression regulation at cell type level [247, 261]. Matched scDNA-seq and scRNA-seq datasets can provide higher resolution in sc-eQTL analysis. Unfortunately, current public datasets with paralleled scDNA-seq and scRNA-seq are still rare [262]. Single-cell eQTLGen consortium (sc-eQTLGen) spearheaded the efforts to link disease-related genetic variations with downstream transcriptional consequences in immune cells [147].
Recommended workflow and applications to AD: eQTL detection
We recommend inferring sc-eQTL by summarizing gene expressions by distinct cell populations [247, 260, 261]. The normalized gene expression matrix is averaged per gene, cell type, and individual to derive robust expression values per group to overcome cell-wise sparsity. The summarized gene expression is further integrated with the genotype matrix to identify sc-eQTLs via Spearman rank correlation [270, 271] or linear regression [260, 261, 272, 273]. As an example, we applied this workflow in the ROSMAP AD snRNA-seq cohort and identified cis- and trans-eQTLs in the excitatory neurons of AD cases (Fig. 6). The script for this eQTL analysis can be found in the companion GitHub repository (see the section "Availability of data and software code" for details).

Single cell based eQTL detected in the excitatory neurons in the AD cases of the ROSMAP cohort. The outer track shows eQTL locations. Red bars indicate eQTL locations. The middle track shows chromosome ideogram. The inner track shows correlated genes. The colorful lines in the inner circle link the eQTLs to the target genes. A, cis-eQTLs. B, trans-eQTLs
Single-cell ATAC-seq data analysis
Although scRNA-seq improves our ability to study gene expression variations and interactions among different cell types in the brain, the fundamental mechanisms that regulate the variability with chromatin structure variations remain unclear. The scATAC-seq (Fig. 7A) technology has been developed to study these regulatory elements [20, 21]. Compared to scRNA-seq data, the scATAC-seq feature matrix data is sparser and hence more challenging to analyze [21, 274]. Many computational tools (Table 4) have been developed to analyze scATAC-seq data alone [275,276,277] or integrate scATAC-seq with other single-cell omics data, including scRNA-seq [278, 279], protein profiling [280] and genome variants [281].

Overview of the procedures for analyzing scATAC-seq data. scATAC-seq raw data are collected from sequencing machines (A). Sequencing adaptors are trimmed and reads are then aligned to the reference genome (B). Peaks are called for each cell and merged into a set of unique features (peaks); reads are then counted for each feature in each cell to obtain a feature-by-cell matrix (C). Features and cells go through quality control (D) to remove low-quality features and cells (E). Filtered data are then normalized (F). Top variable features are extracted to perform linear and non-linear dimension reduction (G) that are further utilized for clustering analysis to identify cell clusters (H). Features (peaks) are annotated to gene (I) and reads are counted for each annotated gene in each cell to obtain a gene activity matrix (J). Cell-cluster-specific accessible chromatin regions and cell-cluster-specific activated genes are identified for each cell cluster to identify cell type (K). Genes with differential activity and differentially accessible chromatin regions are identified between conditions in each cell type (L). Transcription factor motifs are identified in each cell type (M). Co-accessibility analysis can be performed to infer cell-type-specific interactions between different genomic elements (N). Function of disease-associated genetic variant can be inferred by integrating them with scATAC-seq data (O). Trajectory analysis can be performed to infer cellular dynamics during developmental or disease progression (P). When available, scRNA-seq can be integrated with scATAC-seq data (Q) to infer cis-regulatory network
The preprocessing steps of scATAC-seq data analysis include data demultiplexing if multiple samples are sequenced simultaneously. This is followed by adaptor trimming, alignment to the reference genome [21, 296], peak calling and merging, read counting, QC, data normalization and transformation, dimension reduction, clustering, and cell identity annotation (Fig. 7B-K). Generally, peaks are called using the MACS2 [297] and then merged to generate a list of potential regulatory elements, termed features herein for simplicity. Reads of each cell are then counted for those features to obtain a feature-by-cell matrix (Fig. 7C). Next, QC is performed on the cells and features to remove low-quality cells and features [282] (Fig. 7D). The filtered feature-by-cell matrix (Fig. 7E) is usually normalized using the term-frequency inverse-document-frequency (TF-IDF) method to normalize the matrix across cells to correct for differences in sequencing depth and give more weight to rare and more variable peaks [277, 293, 294] (Fig. 7F). To reduce redundant information, potential noise and computational time for downstream analysis, dimension reduction is performed after selecting the features (Fig. 7G). Typically, Latent Semantic Indexing (LSI) is applied on the TF-IDF normalized matrix, followed by singular value decomposition (SVD) [282, 293]. Alternative dimension reduction methods include Multi-dimensional scaling (MDS) [289], Diffusion map (DM) [284], and Latent Dirichlet allocation (LDA) [277]. After feature selection and dimension reduction, samples from multiple conditions are integrated, and adjusted for batch effect [282, 293]. Then, non-linear dimension reduction approaches like t-SNE [298] and UMAP [103] are performed to visualize cells in a 2-D or 3-D space. Cell clustering is then performed in the reduced dimensions (Fig. 7H).
After clustering analysis, annotating cell identity for each cluster is a critical step. As lack of cell-type-specific chromatin accessibility features, peaks at promoters and transcription start sites (TSSs) are used in cell cluster annotation by taking advantage of the extensive cell-type-specific genes. For this purpose, peaks associated with regulatory regions and genic regions are annotated (Fig. 7I). Then, a gene activity matrix is created from the scATAC-seq data by summing the reads intersecting peaks associated with regulatory regions and genic regions for each annotated gene (Fig. 7J). Based on the gene activity matrix, two strategies are used to annotate the cell identity of the clusters (Fig. 7K). In the first strategy, gene activity markers are identified and compared to cell-type-specific marker genes [293]. In the second strategy, scATAC-seq gene activity matrix can be projected to the matched scRNA-seq gene expression matrix for the same cell types. Cell type labels can be transferred from the scRNA-seq data to the scATAC-seq data using mutual nearest neighbors (MNN) algorithm [92, 282, 293].
Then, chromatin accessibility can be investigated for individual cell types with or without integrating other omics data [21, 92, 278]. For example, brain regional and cell-type-specific chromatin accessibility dynamics in AD can elucidate the chromatin regulation mechanisms of gene expression changes underlying AD etiology (Fig. 7L). Specifically, such analyses can identify cell-type-specific peaks, differentially accessible regions [282, 299], enriched motifs (Fig. 7M) and co-accessibility [276, 299] (Fig. 7N), and infer cell trajectory [276, 300] (Fig. 7P). In addition, changes in transcription factor (TF) activity can be inferred through calculating the correlations between TF motif-related chromatin accessibility [275] and gene expression levels of AD-associated genes. Furthermore, the function of disease-associated genetic variants identified in the genome-wide association studies (GWAS) studies can be inferred by predicting genetic variants in chromatin accessible peaks that affect regulatory interactions and TF binding (Fig. 7O). For example, Corces et al. identified the SNPs that drive the association of AD with BIN1, PICALM, SLC24A4, and MS4A6A in specific brain cell types using the scATAC-seq data and AD-associated SNPs [281]. Finally, cis-regulatory network can be inferred by integrating scATAC-seq and sc-RNA-seq data (Fig. 7Q).
We applied the Signac pipeline to snATAC-Seq samples (an AD and a control) from the Swarup lab [PMID: 34239132]. The two samples were normalized and integrated into a dataset (Fig. 8A). Gene activities were extracted from fragments and then 11 cell types were identified from the gene activity markers (Fig. 8B and C). The normalized accessibility, peaks, and co-accessible links of the gene SNAP25 are shown in Fig. 8D. The script for this analysis can be found in the companion GitHub repository (see the section "Availability of data and software code" for details)

snATAC-Seq analysis for AD and control. A Sample 47 (AD) and sample 101 (Control) from the Swarup study are integrated and projected onto UMAP. B Cells are clustered into 11 cell clusters. Gene activity matrix is extracted from fragment file. Marker genes are then identified for each cluster and cell types are identified according to the marker genes of each cluster. C Gene activity of SLC1A2, SNAP25 and MBP are shown in AD (left) and control (right), respectively. D Accessibility and co-accessibility of SNAP25. The top panel shows normalized accessibility of 11 cell types. The two panels in the middle show the peaks around SNAP25. The bottom panel shows the co-accessibility scores among the peaks
Single cell gene network analysis
Gene regulatory network (GRN) inference hypothesizes that the etiology of a complex genetic disease is driven by complex signaling cascades [301] and aims to disentangle such signaling maps from molecular data and identify dysregulated subnetworks putative regulators underlying the diseased tissues [302]. GRN inference has been successfully applied to understand complex diseases, including asthma, cancer, flu infection, and neurodegenerative diseases [303,304,305,306,307,308]. However, GRNs from widely populated bulk sequencing data are limited in resolving inter-wined signaling across and within mixed cell populations, although diseased tissues are composed of heterogeneous cell populations with different morphology and functions [8, 302, 309, 310].
To this end, scRNA-seq has recently emerged to uncover cell-level signaling pathways associated with neurodegenerative diseases and their markers [7, 8, 26, 42, 106, 168, 187, 194], and identify key signaling pathways encompassing distinct cell types [7, 8, 130]. However, data-driven network models of the AD cell types and their regulatory mechanisms have been relatively under-explored. To mitigate this gap, we review the current GRN inference methods in scRNA-seq to evaluate their applicability in different contexts, and recommend workflows to construct robust and accurate network models in AD. While we acknowledge several excellent reviews in the literature [302, 311,312,313], we notice that the repertoire of reported methods and categories, including existing bulk-based methods, are still evolving to address arising challenges in the field. Thus, we have structured this review section to list the challenges in scRNA-seq GRN inference, the applicability of established bulk-based methods to infer single-cell GRNs, and currently available scRNA-seq-based GRN inference methods adopting different statistical frameworks. We will further address GRN evaluations and recommend workflows to ensure the discovery of robust network models.
Challenges in scRNA-seq GRN inference
GRN inference in scRNA-seq is presented with challenges to construct robust and reproducible network models. Drop-out reads present false zero-expressions in addition to the sparse cell-wise transcriptome. These noises in scRNA-seq underscored near-random performances of gene-gene similarity measures to uncover meaningful gene interactions in benchmark scRNA-seq data [314]. Model-based dropout imputation methods such as SAVER [81] have shown outstanding imputation performances with the least false-positives [315], and improved the GRN performances [316]. The high-dimensionality of scRNA-seq with thousands to hundreds of thousands of cells can incur ‘curse-of-dimensionality’ and increase the computational complexity to evaluate gene-gene interactions. Careful feature selection by gene dispersion [74] and low dropout rate [317], and cell selection by low mitochondrial rate, read depth, and number of expressed genes [74, 92] can improve network performances. Further, adjustments for technical variations such as batch effects by the mutual nearest neighbor (MNN) [91] and canonical correlation analysis (CCA) [92] are beneficial for GRN inference [316].
scRNA-seq application of bulk-based gene network construction methods
Established bulk-based GRN inference tools have been applied in scRNA-seq as these tools constructed robust network models in complex diseases and are immediately accessible. While there are several excellent reviews on bulk-based GRN inference methods [318,319,320], we review several established methods applied in the scRNA-seq domain.
Co-expression networks identify gene interactions by gene pairwise association measures such as correlations or information-theoretic measures [321]. Weighted gene co-expression network analysis (WGCNA) is the most popular correlation-based method to construct a scale-free gene interaction network model, and identify co-expressed gene modules as putative interactomes [322]. Multiscale Embedded Gene Co-expression Network Analysis (MEGENA) embeds most correlated gene pairs on a topological sphere to construct a sparse co-expression network and detect multi-scale gene modules [323]. While these correlation-based methods capture linear patterns, information-theoretic measures can capture non-linear patterns. Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNE) is an information entropy-based network inference method and prune false-positive interactions by testing all trios with data processing inequality [324]. CLR is based on mutual information to handle gene-gene interactions and controls false-positives by using the global network as the background [325]. MRNet combines both criteria in CLR and ARACNE to screen the false-positives to improve the prediction acuracy [326].
Several statistical frameworks infer directed interactions between causal and effector genes in contrast to undirected interactions. Bayesian Network (BN) inference provides a flexible framework for identifying directed interactions in causal cascades and integrating upstream regulations such as genetic variants as prior network [327, 328]. Well-established BN tools include RIMBANet [328] and bnlearn [329]. GENIE3 is a random forest (RF) regression method to infer directed causal relationships and has won the DREAM4 challenge as the best performing network inference method [330, 331].
Applications in scRNA-seq have discovered key pathways and markers of heterogeneous cell populations underlying human disease tissues. WGCNA has been applied to identify pathways to activate dormant neural stem cells [332], regulators of chemotherapy resistance in esophageal squamous cell carcinoma [333], and prognostic markers for prostate cancer [334]. MEGENA has been applied to identify enriched pathways in different astrocytic subpopulations in Huntington disease [335], viral infection-regulated pathways in lung epithelium [336].
On the other hand, naïve applications of bulk-based GRN methods involve several shortcomings. They show lower retrieval of known functional links than those inferred from bulk RNA-seq data [316] and primarily associate the modules to cell types that the intricate pathways within the cells [337]. GENIE3, BN, ARACNE, and CLR in in silico simulated and experimental scRNA-seq data showed poor performance in retrieving true interactions from reference sets (e.g., known protein-protein interactions) with little overlaps across them [338]. Rigorous QC to remove unintended co-variations such as batch effects and lowly expressed genes have improved the bulk-based co-expression networks [316].
Single-cell-based network analysis tools
Boolean
The Boolean network model simplifies the complex biological pathways into a switch-like process that transits the network change from one state to another [339]. In Boolean networks, a node (i.e., gene) is denoted by two possible states, ON (1) or OFF (0), and the interacting relationship between nodes is characterized by a target-node-specific function f, which formulates the state of a target gene based on the states of some other genes through clauses consisting of only Boolean operators AND (∧), OR (∨) and NOT (¬) [340]. Several Boolean network methods have been proposed for analyzing scRNA-seq data. Single Cell Network Synthesis (SCNS) [341, 342] is a web-graphic-based tool and uses discretized time series snRNA-seq expression data to infer logical rules driving from early phase to late phase transitions, with single gene change at each transition. The resulting logical model predicts the effects of gene perturbations (e.g., knockout or overexpression) on specific lineages by design. A similar Boolean network method that uses cell trajectory lineage tree information was developed by Chen et al. [343]. Unlike SCNS, BTR (BoolTraineR) [344] does not assume trajectories through cell states. Instead, BTR learns the network structure through iteratively modifying existing Boolean models to explore predictions with an improved match to the observed expression data state via a novel Boolean state space scoring function [344].
Differential equation
GRN inference from time-stamped scRNA-seq data can also be facilitated by ordinary differential equation (ODE) models. In these models, a set of ODEs from potential regulators describe temporal changes in the target genes. This model can be expressed in the form of \(\frac{dx}{dt}= Ax\), where x is a time-labeled vector of C single-cell transcriptomic profiles, x1, x2, …, xC ∈ ℝG in which xi represents the expression, for the ith cell, of G genes, and A is a square matrix that characterizes the regulatory network among the genes [345, 346]. One such method, SCODE [346], infers the TF-regulated network by estimating the coefficients of linear ODEs via linear regression in transformed variables. With dimension reduction, this approach leads to a considerable reduction in the time complexity of the algorithm. A similar approach, GRISLI (Gene Regulation Inference for Single-cell with LInear differential equations and velocity inference), was developed [345]. It first estimates each cell’s velocity (i.e., how each gene’s expression value changes as each cell undergoes a dynamical process), then constructs a GRN by solving a sparse regression problem that relates the gene expression and velocity profiles of each cell.
Bayesian
A BN inference approach, AR1MA1-VBEM (Variational Bayesian Expectation-Maximization) [347], uses a first-order autoregressive moving-average (AR1MA1) model to fit the fold change of a gene at a specific time with a linear model that combines the data at the previous timepoint and a noise term. Under a Bayesian framework, the likelihood function for the AR1MA1 model is a multivariate Gaussian with mean expressed as a function of the network structure. For ease of computation, conjugate priors are used, and the unknown network structure is modeled as a hidden latent Gaussian variable while a Normal scaled Inverse-Gamma distribution models the parameters of the AR1MA1 model. For actual network inference, it uses a VBEM framework using variational calculus to optimize the network models’ marginal likelihood and posterior distributions. In a different method, HBFM (Hierarchical Bayesian Factor Model) uses a sparse hierarchical Bayesian factor model to formulate the impact of gene expression by various factors associated with each cell, and a gene regulatory network structure is constructed by examining the shared factors between pairs of genes [348].
Pseudo-temporal dynamics-based regressions
scRNA-seq data provides an opportunity to estimate the cell-level temporal dynamics by assuming gradual changes in the cellwise transcriptome occurs over time and constitute a trajectory. While cell trajectory inference is an active research area in the scRNA-seq domain [106, 163, 194], the inferred ‘pseudo-time’ on individual cells opens the doors to identify causal expressions in cells from preceding time points to explain downstream changes in the later time points, thus enables inference of causal networks. Granger’s causality is a regression-based framework to explain variations at a lagged time point with several precedent time point data [349] and has been adopted in several scRNA-seq GRN inference methods. SINCERITIES (SINgle CEll Regularized Inference using TIme-stamped Expression profiles) assumes such time-stamped cell transcriptome. Sufficient temporal changes between two ‘snapshots’ of single-cell transcriptome are evaluated by Kolmogorov–Smirnov (KS) statistic, and Granger’s causality infers the causal TF activities to the target genes’ expression changes [350]. To address irregularities in inferred pseudo-time that the underlying dynamical process is not uniform and hence hinders correct causal inference, SCINGE uses kernel-based Granger Causality regression to alleviate irregularities in pseudotime values [351].
Different statistical frameworks have also been adopted to evaluate gene-gene relationships across different time windows. LEAP (Lag-based Expression Association for Pseudotime-series) utilizes Pearson’s correlation of normalized expressions at a time window with those expressions from lagged time windows to establish time-lagged associations between the genes [206]. SCENIC (Single-cell regulatory network inference and clustering) couples co-expressed target genes with TFs by GENIE3 [330] and overlaps them with cis-regulatory binding motifs enrichments within each cell trajectory [352]. SCRIBE uses an information-theoretic measure, restricted directed information (RDI), to quantify the information transferred from the potential regulator to the target in a lagged time point. Qiu et al. showed RNA-velocity, a pseudo-dynamic measure based on transcription kinetics [168], best estimates the real time-series and improves GRN performance over pseudotime [165].
Association-based approaches
In contrast to other model-based methods, association-based networks objectively evaluate the likelihood of gene-gene interactions by ‘guilt-by-association’ [320]. Several gene association measures have recently been devised to handle noises and systematic errors specific to scRNA-seq, and optimized algorithms to efficiently dissect robust interactions.
A popular strategy involves a series of data transformation or modeling steps to handle sparsity and dropout expressions and evaluate significant association by correlations or information-theoretic measures. In bigSCale, gene-expressions are grouped into cell clusters [353], and differential expressions between a pair of clusters are transformed into z-scores to calculate Pearson’s correlations between the clusters [354]. With top 0.1% correlations, the global GRN often yields dense networks [311]. CSN (Cell-specific network) [355], on the other hand, aims to infer co-expression networks for individual cells. CSN develops a statistic for each gene pair to evaluate significant patterns in a cell scatter plot, where the statistic is normally distributed if no pattern is present. The significant gene pairs are then collected to construct a cell-specific co-expression network (CSN), and the summarized gene-wise connectivity across cells can serve as denoised and normalized gene expressions for further analyses [355]. Based on multivariate information theory, PIDC utilizes Partial information decomposition (PID) to quantify gene interaction as the proportion of unique information shared explicitly between two genes, compared to the shared information with the rest [338]. When dropout reads were present, PIDC performed favorably over other mutual-information-based methods and yielded sparse GRNs. But, PIDC suffers from data discretization problem, an inherent problem in information-theoretic measures, and is computationally expensive as it sweeps through gene triplets [338]. scLink aims to infer robust and sparse gene-gene covariance structure by modeling the dropout rates per gene to quantify robust expressions. By fitting a Gamma-Normal mixture model for each gene’s expressions, robustly expressed genes are filtered with a low non-detection rate, and the sparse covariance matrix is inferred with a graphical Gaussian model with penalized likelihood method [317].
Cell-cell communication network
Cell-cell communication is vital for multicellular organisms to coordinate functionally unique cell populations in response to internal and external stimuli. Such communication is primarily mediated by ligand (L)-receptor (R) interactions and can be visualized by networks where each node is a cell type and the edges are L-R interactions [311]. Several algorithms and databases have been established to leverage cell-level resolution in scRNA-seq to infer cell-cell communication networks, for instance, in cancer research to dissect L-R signaling in tumor micro-environment [356].
iTalk relies on a built-in curated database of 2648 non-redundant and known L-R pairs to infer communications across or within distinct cell types [357]. It models the gain or loss of interactions by differential gene expression of each L-R pair across all cell types independently, given that different cells may have distinct receptors for the same ligand and vice versa [357]. Instead of relying on differential expressions, Zhou et al. focus on highly expressed L and R genes in sender and receiver cells and evaluate their communications by pairwise Spearman correlation [356]. As a single L-R pair can function in multiple cell type pairs, scTensor models cell-cell communication as a hypergraph where each node is a cell type, but the edges represent different related L-R pair sets. This “many-to-many” model of communications across multiple cell types is detangled by non-negative Tucker decomposition to estimate contributions from the expression patterns of receptors and ligands as well as L-R interacting pairs [358]. Different from other methods discussed, SoptSC considers the signaling-pathway-wise gene expressions downstream of each L-R interaction to infer the L-R activity. The signaling probability is defined based on weighted co-expression of pathway activity in the sender-receiver cell pairs. Together with pseudotemporal information inferred from scRNAseq, SoptSC allows inference of higher-level communication networks with more complex structures such as feedback/feedforward interactions [359].
Context-based network inference
ACTION attributes functional similarity to genes with relatively weak but preferential expression in specific cell types. It identifies cell clusters, termed archetypes, by low-dimensional geometric constructs in the functional space. Further, it infers cell-type-specific TF regulatory networks (TRNs) by assessing significant TFs with their targeted top-ranked cell type markers. Thus, ACTION provides functional annotation and subsequently the phenotype associated with each cell type [360]. SCINET extends the concept of ACTION to project single-cell transcriptomic data onto a reference interactome and identifies cell-type-specific and disease-associated interactions. By using regression-based imputation and rank-based inverse normal transformation, SCINET infers the likelihood of co-expressed gene-gene interactions, assuming the standard normal distribution of the transformed expression data [361].
Network evaluation
The confidence of the inferred interactions is often crucial in identifying key mechanisms and putative regulators, and ultimately, formulating robust and testable biological hypotheses. One approach is to examine the congruence between the established regulatory pathways or relationships and the inferred networks. Gene perturbation databases in established disease models such as Library of Integrated Network-based Cellular Signatures (LINCS) [362] or CREED [363] curate differentially expressed gene signatures by genetic or chemical perturbations in cell lines, animal models, or disease tissues. These signatures represent the downstream pathways of the exerted perturbation on a network regulator, and the topology of the context-matched, inferred networks should capture these pathways within the perturbed regulator’s proximity. Key Driver Analysis (KDA) examines the enrichments of the perturbation signatures in the network neighborhood of the perturbed regulator by Fisher’s Exact Test [307]. The reproducibility of curated reference networks such as protein-protein interactions (PPI) [364], signaling pathways, and text-mining from published studies provide other measures to evaluate inferred networks’ biological relevance. Although incomplete and high in false positives [318], enrichment for the references is a useful metric for evaluating network performance [314]. Perturb-seq provides a single-cell sequencing platform leveraging Cas9/CRISPR to capture the effects of gene perturbations in a high-throughput manner at a cell-level resolution [365].
On the other hand, simulation studies provide the full controls over the model and noise parameters to generate tailored data, whose underlying reference network is known for objective comparison with inferred networks [331]. GeneNetWeaver is a stochastic differential equation-based simulation tool and has been used to evaluate bulk-based GRNs such as DREAM4 challenge [366]. Chen and Mar generated simulated data to resemble scRNA-seq with a combined approach of GeneNetWeaver and added artificial dropout events to evaluate bulk-based, and single-cell-based GRN approaches [367]. Pratapa et al. introduced BEELINE as a single-cell transcriptome simulation framework leveraging Boolean network models [207]. Unlike GeneNetWeaver, BEELINE can simulate stochastic data with underlying cell trajectory, a hallmark feature in single-cell transcriptomes [207].
Recommended workflow & applications to AD
Inference of robust and accurate network models in single-cell transcriptome requires appropriate modeling of data noises specific to scRNA-seq. These involve low per-cell coverage, doublets, dropout reads as the main sources of noises and should be handled as illustrated in QC guidelines and impute the dropout reads with model-based methods. The model-based imputations infer cellwise dropout rates to facilitate the selection of cells robustly expressing each gene to improve the performance of inferred gene-gene interactions [317]. In addition, unwanted sources of variations such as batch effects should be adjusted before gene-gene similarity calculation for improved prediction accuracy [316].
Depending on the type of an inferred GRN, the expression data should be adequately normalized to optimize the accuracy of inferred gene interactions. The denoised counts should be log-transformed to closely follow Gaussian distributions for constructing regression-based or association-based GRNs. On the contrary, kinetic-based methods such as Boolean or ODE-based networks may perform better in the gene count space.
However, GRN inference with scRNA-seq data is still in its infancy and requires an objective performance evaluation. BEELINE [207] and GeneNetWeaver with artificial dropout events provide accessible platforms to generate simulated data mimicking scRNA-seq characteristics and to build up respective reference networks. The simulated data should closely follow noise models from the real-world scRNA-seq data, including branching trajectories and match the dropout rates, and an inferred GRN should be compared to the respective reference network for reproducing network connectivity. In addition, the inferred GRNs can be evaluated in biological contexts as enrichment for protein-protein interactions (PPI) or known pathways.
To illustrate the application of network analysis in AD single-cell data, we constructed gene co-expression and Bayesian networks for an excitatory neuron cluster identified in the ROSMAP snRNA-seq data using two well-established bulk-tissue gene network inference tools. Figure 9 visualizes the two networks. The scripts for the network analyses are provided in the companion GitHub repository (see the section "Availability of data and software code" for details) .

Excitatory neuron cluster-specific gene networks inferred from the ROSMAP AD snRNA-seq data. A Coexpression network constructed by MEGENA, with node color denoting the module assignment. B Bayesian network constructed by RIMBANet. Node size is proportional to the degree of connectivity
Prioritization of cell clusters
scRNA-seq enables accurate classification of individual cells by gene expression profiles, and cluster-based differential gene expression analysis of scRNA-seq data can help resolve brain-region specific changes in heterogeneous cell populations in AD brains. As loss of neurons and increased microglia activation are characteristic of typical AD brains [368, 369], and many genes show differential expression between AD and control [307, 308, 369, 370], cell clusters from scRNA-seq can be prioritized for their relevance to AD or other diseases by considering the following criteria: 1) the change in the proportion of the cells in a cluster between control and disease (e.g., AD) and 2) the number of DEGs in a cluster between control and disease.
For example, Olah et al. used scRNA-seq to characterize differences in the distribution of distinct microglia subpopulations in human cerebral cortex specimens [7]. The study identified nine distinct sequence profile clusters indicative of 9 distinct microglia subpopulations. Among these, they found reduced frequency of the cluster 7 microglia sub-population in AD brain tissues [7]. Moreover, they found cluster 7 is particularly enriched for genes depleted in the AD cortex [7]. As such, these observations would justify prioritizing microglia cluster 7 as a target for follow-up investigations.
In another single-nucleus transcriptomic study of human prefrontal cortex specimens, Mathys et al. identified transcriptionally distinct sub-populations across six major brain cell types, with many of the top DEGs recurring across multiple cell types [8]. Interestingly, the expression changes of myelination-related genes across major cell types, including oligodendrocytes and oligodendrocyte progenitor cells, are indicative of major perturbations in myelin integrity in AD brains. Thus, combined analysis of specific cell subpopulations and DEGs could help illuminate functional and dynamic changes at the single-cell level between AD and control and provide the basis for prioritizing specific clusters for follow-up functional, mechanistic investigations and therapeutic development.
Integration of snRNA-seq and bulk RNA-seq datasets
In the past decade, bulk RNA-seq datasets have been massively accumulated. However, cell type compositions in the bulk tissues represent a significant confounding factor influencing sample comparisons. scRNA-seq can be utilized to estimate cell fractions of the bulk RNA-seq datasets, thus ‘deconvolving’ the cell compositions. Early deconvolution approaches rely on cell markers and gene expressions from cell sorting experiments. For example, CIBERSORT applies support vector regression to characterize the cell composition of complex tissues from their gene expression profiles [371], and CellMix is an R package that incorporates multiple deconvolution methods (e.g., the Digital Sorting Algorithm and semisupervised non-negative matrix factorization methods (ssKL and ssFrobenius)) to analyze heterogeneous samples [372]. More recently, deconvolution methods have been developed to utilize single-cell transcriptome as the reference directly.
In contrast to traditional methods that mainly use marker genes from cell sorting experiments, single-cell-based deconvolution methods utilize sparse gene expression matrix from scRNA-seq and models gene/sample specific variations unique to single-cell experiments. For example, DeconvSeq utilizes a generalized linear model in feature quantification to construct a projection matrix and resolves cell type fractions by a sequential quadratic programming based solver [373]; MuSiC applies a weighting scheme to prioritize consistent genes across subjects and proposes a weighted non-negative least squares (W-NNLS) regression framework to estimate cell fractions [374]; DWLS designs a weighted least squares approach to adjust the contribution of each gene and solves the constrained dampened weighted least squares problem by quadratic programming [375]; Bisque performs a bulk gene expression transformation to explicitly account for the gene-specific variations and employ the non-negative least-squares (NNLS) regression for cell type fraction inference [376]; and SCDC leverages multiple scRNA-seq reference sets for bulk gene expression deconvolution [377]. Extending from CIBERSORT, CIBERSORTx can also use single-cell expression references to infer cell type abundance and cell-type-specific gene expressions. In AD studies, both traditional and scRNA-seq based methods have been applied to deconvolve the cell fractions of bulk RNA-seq datasets [376, 378]. However, the correlations of the inferred cell fractions with immunohistochemistry (IHC) as ground truth were not high [378]. A more recent study showed that Bisque could reliably estimate cell fractions in subcutaneous adipose and dorsolateral prefrontal cortex expression datasets [376]. When applied to the ROSMAP AD snRNA-seq dataset, Bisque outperformed other deconvolution methods [376].
Recommended workflow and applications to AD
Leveraging the ROSMAP AD cohort with both bulk RNA-seq data for over 600 subjects and snRNA-seq data for a subset of the samples, we performed a mini-benchmarking of the performance of several deconvolution methods. Here we aggregated the cells of each individual in the snRNA-seq data by major cell types to calculate a cell type proportion estimate. As shown in Fig. 10, the single-cell-based methods including Bisque, MuSiC, and SCDC tend to have a higher correlation with the snRNA-seq cell type proportions and a lower error rate than the traditional marker gene-based method CIBERSORT (Fig. 10). Among different single-cell-based methods, Bisque’s estimates of the cell type proportions best resemble the results derived from the snRNA-seq experiments. Therefore, single-cell-based methods, especially Bisque, are recommended for the general purpose of deconvolution studies. The script for this integrative analysis is provided in the companion GitHub repository (see the section "Availability of data and software code" for details).

Cell-type deconvolution of the bulk RNA-seq data in the prefrontal cortex in the ROSMAP AD cohort. A The input of cell-type deconvolution analysis. The cell-type marker gene or scRNA-seq expression matrix was used to infer cell type fractions of the bulk RNA-seq datasets. B Cell-type fractions from the deconvolution analysis of 15 individuals from the ROSMAP database. Four different deconvolution methods were applied, including bulk RNA-seq based method (CIBERSORT) and single-cell-based methods (Bisque, MuSiC, and SCDC). The pink color indicates the cell type fractions estimated by the snRNA-seq experiment. C Evaluation of different deconvolution methods by comparing with the cell-type fractions from the snRNA-seq experiment. Two matrices were used to benchmark the tool performance: the Pearson correlation (top panel) and the root-mean-square deviation (RMSE, bottom panel)
The cell-type proportions estimated from deconvolution methods can be used for a number of downstream analyses for AD. For example, in contrast to traditional DE analysis confounded by cell type proportions, deconvolution results can provide new insights into the cell-intrinsic DEGs by adjusting cell type proportions [379]. The fractions of neuron cells calculated by different deconvolution methods negatively correlated with the cognitive diagnosis [376], consistent with our knowledge of neuronal loss in AD brains. Aneal et al. developed a new deconvolution method CelMod to infer proportions of 35 cell subclusters [380], which were identified by snRNA-seq of 24 prefrontal cortex samples with or without AD pathologies. The deconvolution method revealed that highly correlated cell subclusters form distinct cellular communities across 640 individuals. Although biological functions of the cellular communities need validation, the deconvolution analysis provides intriguing applications in AD studies to analyze interactions among different cell subtypes.
Prioritization of gene subnetworks and key drivers
Established strategies for prioritizing co-expressed modules and key causal drivers from bulk transcriptomic data can be directly applied to cell cluster-based coexpression and causal networks [307, 328, 370, 381,382,383]. Specifically, for each cell cluster from scRNA-seq, the modules in the respective coexpression network will be tested for and sorted by associations with trait phenotypes. The association between module eigengenes (the first principal component of the module gene expression data) and the biological covariates can be utilized to evaluate association [384]. Alternatively, the modules can be tested for enrichment of DEGs in the cluster (see Differential expression for disease gene identification section) between disease (e.g., AD) and control, and then all the modules will be rank-ordered by enrichment score (e.g., FET p-value or fold enrichment) [385]. Similarly, the network neighborhood of a gene conforms to the model of the genes’ downstream pathways, and the enrichment of cluster-specific DEGs from diseased cells (e.g., AD) in the network neighborhood can guide the prediction of key drivers of the disease etiology [386]. Such key driver prediction methods have been widely used in numerous network analyses of human diseases, such as AD, coronary artery disease, inflammatory bowel disease, and allograft rejection [307, 386,387,388,389,390]. Key drivers are thought to provide insights into gene regulatory control and serve as targets for therapeutic discovery. Key drivers then can be prioritized for experimental validation by considering a line of evidence whichever available and appropriate, including but not limited to the degree of connectivity, the status of expression change between disease and control, fold enrichment of disease associated genes in the downstream subnetwork, strength of expression correlation with disease trait variables, genetic association signal, literature support, etc. [387].
Spatial transcriptomics
Cellular spatial location is not well retained in the single-cell transcriptomics methods described above. Given that cellular gene expression patterns change in response to environmental cues maintaining the neighborhood environment allows for a deeper understanding of tissue-wide dynamics and biological function of genes and cells. Many of the current spatial technologies were derived from RNA in situ hybridization (ISH) and/or principles of laser capture microdissection. While ISH, a commonly used technique to histologically visualize mRNA localization in tissues on microscope slides, allows for maintained tissue integrity and entire tissue analyses, a major limitation of traditional ISH is the number of targets that can be analyzed concurrently. On the other hand, laser capture microdissection, developed in the 1990s, uses low-power infra-red laser beams to microdissect cells of interest to overcome this obstacle and allows the whole transcriptome profiling, but the laser capture microdissection is destructive for the cells.
Several advanced approaches have been developed to profile whole transcriptomes while preserving spatial information in the past few years. These include fluorescence in situ hybridization (FISH) [391,392,393,394,395,396], in situ sequencing (ISS) [397,398,399], and spatially-barcoded RNA sequencing [23, 400,401,402]. The methods differ in sequencing depth, the number of transcripts analyzed (dozens to the whole transcriptome), tissue integrity (dissociative versus non-destructive), cellular throughput (10s, 100 s, 1000s of cells), spatial information (image or spatial barcode), cellular resolution (multi, single or subcellular) and starting material (fixed or frozen tissue).
These technologies open an unprecedented opportunity to dissect the cellular complexity and characterize the tissue microenvironment for identifying inter-cellular interactions and signaling pathways in complex diseases such as cancer [201, 403], amyotrophic lateral sclerosis (ALS) [404], and AD [405]. Computational methods have been developed to identify spatial patterns [406,407,408,409] and detect spatial ligand-receptor interactions [406]. Application of spatial transcriptomics (ST) analysis to mouse models of familial AD has identified two co-expression networks in small tissue domains, representing spatially coordinated transcriptomic changes induced by amyloid plaques [405].
Chen et al. [405] applied ST (10X Visium protocol that profiles tissue domains up to ~ 10 cells each) and ISS technologies to study AD using APPNL-G-F mice. They identified two co-expression networks, one of which (referred to as plaque-induced genes, or PIG for short) is particularly interesting since their activities are strongly associated with the Aβ plaque. We applied Giotto [406] to re-analyze a subset of the ST data in their paper, corresponding to two wild-type (WT) and two AD (APPNL-G-F) mice at the 18-month-old age. By using Leiden clustering, we identified 8 distinct clusters (Fig. 11A). The spatial patterns of these clusters are robustly reproduced in the normal mice, but vary significantly in the AD mice, suggesting extensive structural differences (Fig. 11B). To quantify the spatial patterns at the gene-level, we used the binSpect algorithm [406] to identify spatially variable genes. The top 300 genes selected by the algorithm were further divided into 20 distinct modules based on co-expression analysis (Fig. 11C). Module 6 is notable as it strongly overlaps with the PIG network previously identified by Chen et al. but a different analysis procedure that requires the knowledge of plaque locations is not used here. Among the 43 genes contained in Module 6, 20 are from the PIG network. These genes are highly expressed in the AD mice but significantly down-regulated in the WT mice (Fig. 11D). We further investigated the cell-type distributions in the normal and AD mice. Since the spatial resolution of the ST technology is limited to 100um, we applied cell-type enrichment analysis to computationally estimate the spatial distribution of different cell types, using publicly available single-cell RNAseq data [410] as guidance. We found that the spatial distributions of neurons are similar, but the proportion of microglia increased significantly in the AD mice. This is consistent with the original study since microglia have been well-known to be associated with AD. Thus, ST analysis provides a valuable tool to identify distinct structural alterations associated with AD and may lead to new hypotheses for future studies. The script for analyzing the spatial transcriptomics data is provided in the companion GitHub repository (see the section "Availability of data and software code" for details).

Spatial transcriptomics analysis of an AD mouse model. Data from four 18-month-old mice, including 2 AD (AD.1 and AD.2) and 2 WT (WT.1 and WT.2), were analyzed. A Visualization of the Leiden clustering of the tissue domains in the UMAP space; B Spatial distribution of the tissue domains colored by the cluster id in A; C Clustering of spatially correlated genes; D Spatial pattern of a spatially correlated gene cluster #6 which is upregulated in the AD mice
Integrative analysis of human and mouse AD scRNA-seq data
As eventually mouse models will be used to validate key findings from human AD single cell sequencing data, integration of mouse and human single cell data is critical for informing the correspondence between AD mouse models and human AD. There are nine papers about single-cell sequencing analysis of AD mouse models [57, 139, 411,412,413,414,415,416,417]. As summarized in Table 5, 14 mouse models have been analyzed, including the most widely used 5XFAD, 3XTg-AD, and APP/PS1 transgenic mice. The mouse ages range from 3 to 24 months, and the brain regions profiled include the prefrontal cortex, cerebral cortex, hippocampus, subventricular zone, and cerebellum.
So far, few studies have conducted an integrative analysis of human and mouse single-cell transcriptomic data in AD. In a recent single-cell study of TRME2 R62H mutation in AD, Zhou et al. [57] analyzed the human AD snRNA-seq data and mouse scRNA-seq data separately and then compared the cell type-specific disease signatures between the two species by overlapping analysis. However, they fell short of joint learning of single-cell datasets between humans and mice. There are a number of challenges in cross-species single-cell data integration, including but not limited to the partially overlapping gene background, distinct cell populations, and poorly conserved cell type transcriptome or markers that may arise from different species. Nonetheless, some tools have been developed to formally reconcile heterogeneous scRNA-seq data from multiple species to gain robust and insightful comparisons between different species [418, 419]. Assuming that at least a subset of gene-gene correlations should be conserved, thus align cells across species, Butler et al. [49] proposed to first use canonical correlation analysis (CCA) to identify conserved gene-gene correlation structure between datasets and then apply nonlinear warping algorithms to align different datasets for a single integrated clustering analysis. When applying this alignment procedure to single cell datasets from human and mouse pancreatic islets, the integrative analysis was able to identify conserved cell states and cell-type markers, with cluster calls agreed very well (> 94%) with the analyses from the independent data sets [49]. In theory, other batch correction pipelines based on similar assumptions, such as MNN [91] and Harmony [98], can also be used for cross-species joint learning. Different from the joint clustering in CCA, Crow et al. developed a supervised framework to align cell clusters across datasets in their method MetaNeighbor [420]. In this method, each dataset is first analyzed separately to label the cell types. Then a cross-dataset cell correlation network is constructed based on the expression of a given set of genes. Next, the cell-type labels (“identity”) in one dataset are held-out and predicted using a neighbor-voting classification model trained from the remaining cell correlation network in other dataset(s). This training and prediction process iterates for every dataset. This method can rapidly evaluate how well cell types are conserved/replicated in different conditions by comparing the predicted labels with original labels from individual analyses. MetaNeighbor has been successfully applied to compare cell atlases across 7 species [418].
To perform a comprehensive comparison of single cell-based gene signatures between human AD studies and AD mouse models, we collected cell-type-specific disease signatures from six single-cell AD mouse model studies [57, 139, 414,415,416,417] and seven single-cell human AD studies [7, 8, 41, 42, 45, 57, 421]. Mouse genes were converted to human homologs by using BioMart before enrichment analysis by FET. We compared the DEG signature for each cell type. In microglia, the DEGs from 5XFAD, APP/PS1, and the CK-p25 mice show the most significant enrichment with human microglia DEGs (Fig. 12). Yet, no single mouse model can capture all molecular alterations in human AD, and the selection of mouse models for signature validation may be cell type-dependent.

Heatmap showing the overlap of microglia specific DEG signatures between human and mouse AD single cell datasets. Each row represents a mouse study. Each column represents a human study
Challenges in using human postmortem and mouse tissues
There are multiple challenges in using human postmortem and mouse tissues to study AD due to the heterogeneity of human samples and human–mouse discrepancies. First, the existing bulk- or single cell/nucleus-based RNA-seq data from human postmortem brain tissues in AD are from terminal-stage patients with extensive neuronal cell death, a prolonged process starting already at preclinical stages before any clinical symptom emerges [368, 422, 423]. However, terminal-stage tissues may not inform the progression of pathogenic mechanisms of earlier disease stages [424, 425]. Mouse tissues, on the other hand, often encapsulate earlier stages of the disease characterized by accelerated amyloid or tau proteinopathy [424]. Second, molecular changes like stress-induced inflammatory response may occur postmortem should cellular energy stores and temperature permit [425], leading to augmented transcriptomic changes that confound the real biological signal. A recent transcriptomic study of brain tissues found rapid loss of neuronal genes and reciprocal expression of glial genes during a postmortem interval (PMI) period of 24 h [426]. Such postmortem changes may lead to loss of cell identity, which in turn biases the subsequent cell clustering. Cautions need be taken when examining cell clusters, especially, neuronal clusters with weak markers expression. Genome-wide modeling of RNA fidelity as a function of PMI is necessary to alleviate such impact and help determine and correct the gene expression pattern for more robust cell clustering and downstream analyses. Therefore, compared to mouse brain samples, human brain samples likely present more variable and poorer RNA quality due to PMI effects [424, 427]. Finally, differences in tissue collection and processing may also confound sequencing data. For example, there are differences in gene and cell type coverages between scRNA-seq and snRNA-seq data as reviewed above. Given current technological and ethical constraints, snRNA-seq analysis is more practical than scRNA-seq for human postmortem brain tissues. However, we must note that snRNA-seq is limited for detecting cellular activation in microglia in human diseases [31], a potential explanation for inconsistent observation of microglia signatures between human studies and between human and mouse studies.
While high quality mouse biospecimens are more readily available, it remains a challenge to choose appropriate AD mouse models (familial AD versus sporadic AD [428]), with a proper experimental design taking into consideration of critical factors including age, sex, brain regions, sample sizes, etc. AD-like pathology occurs in different brain regions of varying mouse models at different developmental stages [429,430,431]. For example, rTg4510 mice show tauopathies in the cortex at 4 months, while the hippocampus at 5.5 months [430]. The PS19 mice show neuron loss in the hippocampus at 8 months old, which spreads to other regions by 12 months. Existing mouse-based single-cell studies of AD have used the prefrontal cortex, cerebral cortex, hippocampus, subventricular zone, and cerebellum for scRNA-seq analysis (Table 5). Some of the brain regions used in human single-cell sequencing studies have not yet been profiled in mice (e.g., the entorhinal cortex and superior frontal gyrus). Lastly, as the existing AD mice mimic some, but not all the key pathologies of human AD, better preclinical models of AD are urgently required.
Future development for bioinformatics of single-cell sequencing data
Outlook of cell clustering analysis in AD scRNA-seq
Cell clustering in scRNA-seq is mostly performed within widely adopted scRNA-seq workflows such as Scater [74], Seurat [92], SCANPY [50] and monocle [432]. However, cell clustering is typically dependent on dimension reduction in these workflows and offers a limited range of clustering algorithms (e.g., kNN-based Louvain’s algorithm). On the other hand, much of the recent developments in cell clustering remained untapped in scRNA-seq study of AD. For instance, the application of genotype-based clustering approaches to dissect cell populations expressing pathogenic variants exemplifies such untapped potentials. Deep learning-based approaches are also attractive, scalable alternatives to extract non-linear patterns in massive AD single-cell transcriptome while seamlessly handling batch effects and scRNA-seq specific noises. In addition, we anticipate single-cell-based spatial transcriptomics to become available in the near future. Then novel clustering methods are needed to leverage the spatial information loss in the current single cell data to bring in the potentially important topological context in cell clustering analysis. Overall, the advances in cell clustering will expand the repertoire of AD-associated cell populations, thereby enhancing our understanding of underlying cell type-specific mechanisms.
Suggestions for future trajectory inference approaches
While several dimension reduction methods such as PCA, t-SNE [101], DiffusionMap [102] and UMAP [103] have been widely adopted in the scRNA-seq analysis [34], some methods are not optimized for dissecting cell trajectories. Non-linear projection methods such as t-SNE and UMAP are known to distort the underlying data structure while mapping cells to low-dimensional manifolds discards long-range structures, and their stochasticity yields slightly different results if the seed is not properly initialized. On the other hand, Diffusion Map reflects local and long-range structures and is optimized to trace gradual changes in the transcriptome, making it an attractive tool for trajectory inference [102]. However, diffusion maps are computationally expensive, and PCA can serve as an efficient alternative to capturing long-range structures to identify the spanning trajectories in large-scale scRNA-seq data.
The surge in trajectory inference method development urgently needs appropriate scenario-specific methods. A growing effort has been put into this direction [433, 434]. A recent comparative study benchmarks 45 trajectory inference methods over hundreds of simulated and real-world data and offers a set of guidelines to walk users through method selection [313]. Notably, users should be aware of whether the trajectory structures can be pre-defined and fit the current experimental settings. Determination of the starting point often requires manual selection based on prior knowledge or marker gene expression. An unsupervised method using the quantile polarization of a cell’s principal component values has been recently proposed and subsequently validated in several independent studies [435]. The scalability and usability should be considered for the efficient characterization of large-scale single-cell data. Most importantly, trajectory inference results should be seen as hypotheses that need validation regarding their applicable scenarios, robustness, and noise tolerance.
Future directions of sc-CNV and sc-eQTL analysis for AD
Even though single-cell based CNV and sc-eQTL analyses have been primarily performed in cancer researches [9, 211, 212] and human-induced pluripotent stem cell studies [261, 268], single-cell based genetic variation analysis hasn’t been applied to AD. So far, there are about 50 eQTL studies [244, 436,437,438,439,440,441,442,443,444,445,446,447,448,449,450,451,452,453,454,455,456,457,458,459,460,461,462,463,464,465,466,467,468,469,470,471,472,473,474,475,476,477,478,479,480,481,482] and about 30 CNV studies [483,484,485,486,487,488,489,490,491,492,493,494,495,496,497,498,499,500,501,502,503,504,505,506,507,508,509,510,511,512,513,514,515] in AD, based on bulk tissues only. Cell-type specific and brain region-specific genetic mutations have been shown more relevant to the pathology process of AD [244, 308, 450, 452, 457, 470, 516]. For example, some sc-eQTLs are cell-status dependent [247, 260]. Additional future study subjects include the functional differences of eQTLs between the normal aging process and pathologic process in AD, and the relevance of cell type-specific eQTL with respect to ApoE or Tau status. Single-cell-based genetic variation analysis can pinpoint key genetic mutations driving the pathological progress of specific cell populations such as microglia cells and neurons in AD. As AD is heterogeneous at both pathological and transcriptomic levels [6], it would be interesting to understand how genetic variations drive AD heterogeneity. Single-cell genetic variation analysis may offer a novel avenue to understand the genetic heterogeneity of AD using single-cell genetic variation analysis.
Drug development for AD using single-cell sequencing data
Single-cell sequencing can identify key molecular pathways targets from distinct cell types in AD and resolve the mixed signals from bulk tissues. Particularly, cell-type-specific signatures from scRNA-seq can be useful for repositioning FDA-approved drugs for treating AD. Through reversal of the cell-type-specific signatures, Connectivity map (CMap) and LINCS have been used to predict candidate FDA-approved drugs for several diseases, including pulmonary arterial hypertension (PAH) [517], COVID-19 [518] and lymphangioleiomyomatosis [519]. For instance, Hong et al. identified NF-κB signaling upregulation and IFN signaling downregulation in several cell types of PAH using scRNA-seq and applied the signatures to the CMap predicting candidate drugs to reverse the changes. Our group developed a more accurate algorithm [520] to identify the CMap and LINCS compounds that reverse the cell-type-specific signatures precisely.
Experimental validation of novel cell subpopulations associated with AD
As high-throughput sequencing techniques produce numerous data and hypotheses, additional experimental validation is often required to confirm the findings. Several scRNA-seq studies have revealed and experimentally validated AD-associated gene regulation and cell subpopulations with a primary emphasis on the glial cells [7, 57]. Co-immunostaining of a cluster-specific signature gene with a known general cell type marker provides the most straightforward visualization and quantification of each cell subpopulation. However, it often depends on the antibody specificity and availability of the marker genes of interest and is generally low throughput. Alternatively, RNAscope, an in-situ hybridization (ISH)-based multiplexing method with high target-detection specificity, can be applied if no working antibody is available or multiple signature genes are required to define a subset of cells [405]. Other validation strategies include the NanoString nCounter system to verify cluster-specific signature gene expression and cross-validation in independent cohorts with cell cluster alignment [49]. In summary, experimental validations from various angles would greatly enhance our confidence in identifying novel cell subpopulations associated with AD and serve as the basis for targeted therapeutic development.
Conclusions
In conclusion, we comprehensively reviewed the state-of-the-art bioinformatics approaches to analyze single-cell sequencing data and their applications to AD in 14 major directions. The basic analyses include data quality control and normalization, cell cluster identification and cell subpopulation characterization and differential expression while more advanced analyses involve trajectory inference, copy number variation, eQTL identification, and integrative gene network inference. We also reviewed the recent progress on analyzing scATAC-seq and spatial transcriptomics data and integrating single-cell multi-Omics data. We summarized their advantages and disadvantages for multiple methods in each direction to help users select the most appropriate approach for specific applications. More importantly, we have implemented the recommended workflow for each major analytic direction and applied it to an snRNA-seq dataset in AD while the scripts and the data are shared with the research community. We further discussed the potential future development of bioinformatics of single-cell sequencing data. We expect both less experienced and advanced single cell data analysts would be greatly benefited from the review and the accompanied software tools. As such, this review not only provides insights into various methods to analyze scRNA-seq data and guidelines for analyzing AD scRNA-seq data but also serves as an invaluable resource for the AD research community and the single cell sequencing community in general.
Availability of data and materials
The human postmortem sequencing data are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership Alzheimer’s Disease (AMP-AD) Target Discovery Program and other NIA-supported programs to enable open-science practices and accelerate translational learning. The data, analyses, and tools are shared early in the research cycle without a publication embargo on secondary use. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/DataAccess/Instructions). The ROSMAP AD snRNA-seq data analyzed in this study is available at https://www.synapse.org/#!Synapse:syn18681734. The scripts and software tools utilized in this study are available in Github (https://github.com/songw01/AD_scRNAseq_companion).
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-beta
- NFT:
-
Neurofibrillary tangle
- CNS:
-
Central nervous system
- EOAD:
-
Sporadic early-onset AD
- LOAD:
-
Sporadic late-onset AD
- MCI:
-
Mild cognitive impairment
- scRNA-seq:
-
Single-cell RNA sequencing
- snRNA-seq:
-
Single-nucleus RNA sequencing
- scATAC-seq:
-
Single-cell assay for transposase-accessible chromatic sequencing
- TMM:
-
Trimmed mean of M-values
- UQ:
-
Upper-quantile
- RLE:
-
Relative log-expression
- CNV:
-
Copy number variation
- eQTL:
-
Expression associated quantitative trait loci
- eCNV:
-
Expression associated CNV
- ZINB:
-
Zero-inflated negative binomial distribution
- ZINB-WaVE:
-
Zero-Inflated Negative Binomial-based Wanted Variation Extraction
- PCA:
-
Principal Component Analysis
- t-SNE:
-
t-distributed stochastic neighbor embedding
- DM:
-
Diffusion map
- UMAP:
-
Uniform manifold approximation and projection
- SNV:
-
Single nucleotide variant
- kNN:
-
k-nearest neighbor
- DCA:
-
Deep count autoencoder
- scVI:
-
Single-cell variational inference
- WGS:
-
Whole-genome sequencing
- WES:
-
Whole-exome sequencing
- DE:
-
Differential expression
- DEG:
-
Differentially expressed gene
- GLM:
-
Generalized linear model
- MST:
-
Minimum spanning trees
- ICA:
-
Independent component analysis
- OU process:
-
Ornestain-Uhlenbeck process
- LLE:
-
Locally-linear-embedding
- DPT:
-
Diffusion Pseudotime
- SNP:
-
Single nucleotide polymorphism
- sc-eQTL:
-
Single-cell eQTL
- TF-IDF:
-
Term-frequency inverse-document-frequency
- LSI:
-
Latent Semantic indexing
- SVD:
-
Singular value decomposition
- MDS:
-
Multi-dimensional scaling
- LDA:
-
Latent Dirichlet allocation
- VAFRNA :
-
Biallelic polymorphism loci
- GRN:
-
Gene regulatory network
- CSN:
-
Cell-specific network
- PID:
-
Partial information decomposition
- TF:
-
Transcription factor
- TRN:
-
TF regulatory network
- KDA:
-
Key driver analysis
- PPI:
-
Protein-protein interaction
- PMI:
-
Postmortem interval
References
Association As. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 2018;14:367–429.
Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer's and Dementia. 2021;17:327–406.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
Fiandaca MS, Mapstone ME, Cheema AK, Federoff HJ. The critical need for defining preclinical biomarkers in Alzheimer’s disease. Alzheimers Dement. 2014;10:S196–212.
Mehta RI, Schneider JA. What is ‘Alzheimer’s disease’? The neuropathological heterogeneity of clinically defined Alzheimer’s dementia. Curr Opin Neurol. 2021;34:237–45.
Neff RA, Wang M, Vatansever S, Guo L, Ming C, Wang Q, et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv. 2021;7(2):eabb5398.
Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun. 2020;11:6129.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–7.
Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–13.
Holmes G, Gonzalez-Reiche AS, Lu N, Zhou X, Rivera J, Kriti D, et al. Integrated transcriptome and network analysis reveals spatiotemporal dynamics of calvarial suturogenesis. Cell Rep. 2020;32:107871.
Zhao J, Zhang S, Liu Y, He X, Qu M, Xu G, et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 2020;6:22.
Wahane S, Zhou X, Zhou X, Guo L, Friedl MS, Kluge M, et al. Diversified transcriptional responses of myeloid and glial cells in spinal cord injury shaped by HDAC3 activity. Sci Adv. 2021;7(9):eabd8811.
Ramskold D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–82.
Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013;14:R31.
Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–73.
Hayashi T, Ozaki H, Sasagawa Y, Umeda M, Danno H, Nikaido I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat Commun. 2018;9:619.
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–14.
Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–7.
Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33:1165–72.
Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910–4.
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–90.
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4.
Liu Y, Yang M, Deng Y, Su G, Enninful A, Guo CC, et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell. 2020;183:1665–1681.e1618.
Stickels RR, Murray E, Kumar P, Li J, Marshall JL, Di Bella DJ, et al. Highly sensitive spatial transcriptomics at near-cellular resolution with slide-seqV2. Nat Biotechnol. 2021;39:313–9.
Alsema AM, Jiang Q, Kracht L, Gerrits E, Dubbelaar ML, Miedema A, et al. Profiling microglia from Alzheimer’s disease donors and non-demented elderly in acute human postmortem cortical tissue. Front Mol Neurosci. 2020;13:134.
Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A. 2015;112:7285–90.
Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature. 2018;563:72–8.
Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJE, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun. 2016;7:11022.
Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat Protoc. 2016;11:499–524.
Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 2018;13:e0209648.
Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, et al. Single-nucleus RNA-Seq is not suitable for detection of microglial activation genes in humans. Cell Rep. 2020;32:108189.
Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–81.
Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–9.
Andrews TS, Hemberg M. Identifying cell populations with scRNASeq. Mol Asp Med. 2018;59:114–22.
Kolodziejczyk AA, Kim JK, Tsang JC, Ilicic T, Henriksson J, Natarajan KN, et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell. 2015;17:471–85.
Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–201.
Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049.
Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11:163–6.
Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33:155–60.
Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat Neurosci. 2017;20:484–96.
Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer's disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22:2087–97.
Lau SF, Cao H, Fu AKY, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2020;117:25800–9.
Nguyen AT, Wang K, Hu G, Wang X, Miao Z, Azevedo JA, et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020;140:477–93.
Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, et al. Distinct amyloid-beta and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 2021;141:681–96.
Morabito S, Miyoshi E, Michael N, Shahin S, Martini AC, Head E, et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet. 2021;53:1143–55.
Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, Proserpio V, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods. 2013;10:1093–5.
Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24:496–510.
Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods. 2014;11:740–2.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–20.
Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15.
Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, et al. Comparative analysis of single-cell RNA sequencing methods. Mol Cell. 2017;65:631–643.e634.
Lun AT, McCarthy DJ, Marioni JC. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122.
Krentz NAJ, Lee MYY, Xu EE, Sproul SLJ, Maslova A, Sasaki S, et al. Single-cell transcriptome profiling of mouse and hESC-derived pancreatic progenitors. Stem Cell Rep. 2018;11:1551–64.
Xi NM, Li JJ. Benchmarking computational doublet-detection methods for single-cell RNA sequencing data. Cell Syst. 2021;12:176–194.e176.
Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291.e289.
Bais AS, Kostka D. scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics. 2020;36:1150–8.
Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. 2020;26:131–42.
Lun A, McCarthy D, Marioni J. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122.
Gayoso A, Shor J, Carr AJ, Sharma R, Pe'er D. GitHub: DoubletDetection (v2.4.1). Zenodo. 2019. https://doi.org/10.5281/zenodo.2678042
McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e324.
Bernstein NJ, Fong NL, Lam I, Roy MA, Hendrickson DG, Kelley DR. Solo: doublet identification in single-cell RNA-Seq via semi-supervised deep learning. Cell Syst. 2020;11:95–101.e105.
DePasquale EAK, Schnell DJ, Van Camp P-J, Valiente-Alandí Í, Blaxall BC, Grimes HL, et al. DoubletDecon: deconvoluting doublets from single-cell RNA-sequencing data. Cell Rep. 2019;29:1718–1727.e1718.
Ilicic T, Kim JK, Kolodziejczyk AA, Bagger FO, McCarthy DJ, Marioni JC, et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 2016;17:29.
Osorio D, Cai JJ. Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics. 2020;37:963–67.
AlJanahi AA, Danielsen M, Dunbar CE. An introduction to the analysis of single-cell RNA-sequencing data. Mol Ther Methods Clin Dev. 2018;10:189–96.
Mercer Tim R, Neph S, Dinger Marcel E, Crawford J, Smith Martin A, Shearwood AM, et al. The human mitochondrial transcriptome. Cell. 2011;146:645–58.
Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun. 2019;10:2832.
Schirmer L, Velmeshev D, Holmqvist S, Kaufmann M, Werneburg S, Jung D, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature. 2019;573:75–82.
Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 2020;9(12):giaa151.
Yang S, Corbett SE, Koga Y, Wang Z, Johnson WE, Yajima M, et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 2020;21:57.
Fleming SJ, Marioni JC, Babadi M. CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv. 2019;791699.
Heaton H, Talman AM, Knights A, Imaz M, Gaffney DJ, Durbin R, et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat Methods. 2020;17:615–20.
Xi S, Gibilisco L, Kummer M, Biber K, Wachter A, Woodbury M. ABACUS: a flexible UMI counter that leverages intronic reads for single-nucleus RNAseq analysis. bioRxiv. 2020;11.13.381624. https://doi.org/10.1101/2020.11.13.381624.
McCarthy DJ, Campbell KR, Lun AT, Wills QF. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33:1179–86.
Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 2019;15:e8746.
Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25.
Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Vallejos CA, Risso D, Scialdone A, Dudoit S, Marioni JC. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat Methods. 2017;14:565–71.
Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods. 2018;15:539–42.
Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun. 2018;9:997.
van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, et al. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174:716–729.e727.
Lun ATL, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016;17:75.
Bacher R, Chu LF, Leng N, Gasch AP, Thomson JA, Stewart RM, et al. SCnorm: robust normalization of single-cell RNA-seq data. Nat Methods. 2017;14:584–6.
Cole MB, Risso D, Wagner A, DeTomaso D, Ngai J, Purdom E, et al. Performance assessment and selection of normalization procedures for single-cell RNA-Seq. Cell Syst. 2019;8:315–328.e318.
Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, et al. Developmental diversification of cortical inhibitory interneurons. Nature. 2018;555:457–62.
Vallejos CA, Marioni JC, Richardson S. BASiCS: Bayesian analysis of single-cell sequencing data. PLoS Comput Biol. 2015;11:e1004333.
Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278.
Risso D, Perraudeau F, Gribkova S, Dudoit S, Vert JP. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat Commun. 2018;9:284.
Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol. 2018;36:421–7.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e1821.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Eraslan G, Simon LM, Mircea M, Mueller NS, Theis FJ. Single-cell RNA-seq denoising using a deep count autoencoder. Nat Commun. 2019;10:390.
Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. Deep generative modeling for single-cell transcriptomics. Nat Methods. 2018;15:1053–8.
Hoffman GE, Schadt EE. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics. 2016;17:483.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods. 2019;16:1289–96.
Lau S-F, Cao H, Fu AKY, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci. 2020;117:25800–809.
Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods. 2016;13:241–4.
Lvd M. Hinton G: Visualizing data using t-SNE. J Mach Learn Res. 2008;9:2579–605.
Haghverdi L, Buttner M, Wolf FA, Buettner F, Theis FJ. Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods. 2016;13:845–8.
Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2019;37:38-44.
Kiselev VY, Andrews TS, Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet. 2019;20:273–82.
Pirim H, Eksioglu B, Perkins A, Yuceer C. Clustering of high throughput gene expression data. Comput Oper Res. 2012;39:3046–61.
Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018;19:477.
Zhou Z, Xu B, Minn A, Zhang NR. DENDRO: genetic heterogeneity profiling and subclone detection by single-cell RNA sequencing. Genome Biol. 2020;21:10.
Huang Y, McCarthy DJ, Stegle O. Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 2019;20:273.
Xu J, Falconer C, Nguyen Q, Crawford J, McKinnon BD, Mortlock S, et al. Genotype-free demultiplexing of pooled single-cell RNA-seq. Genome Biol. 2019;20:290.
Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol. 2018;36:89–94.
Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods. 2017;14:483–6.
Wang B, Ramazzotti D, De Sano L, Zhu J, Pierson E, Batzoglou S. SIMLR: a tool for large-scale genomic analyses by multi-kernel learning. Proteomics. 2018;18. https://doi.org/10.1002/pmic.201700232.
Ester M, Kriegel H-P, Sander J, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. In: Kdd; 1996. p. 226–31.
Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–97.
Xu C, Su Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics. 2015;31:1974–80.
Baran Y, Bercovich A, Sebe-Pedros A, Lubling Y, Giladi A, Chomsky E, et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 2019;20:206.
Ding J, Condon A, Shah SP. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat Commun. 2018;9:2002.
Li X, Wang K, Lyu Y, Pan H, Zhang J, Stambolian D, et al. Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat Commun. 2020;11:2338.
Chen R, Wu X, Jiang L, Zhang Y. Single-cell RNA-Seq reveals hypothalamic cell diversity. Cell Rep. 2017;18:3227–41.
Newman ME. Modularity and community structure in networks. Proc Natl Acad Sci U S A. 2006;103:8577–82.
Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech Theory Exp. 2008;2008:P10008.
Chi W, Deng M. Sparsity-penalized stacked denoising autoencoders for imputing single-cell RNA-Seq data. Genes (Basel). 2020;11(5):532.
Zhang AW, O'Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods. 2019;16:1007–15.
Wang D, Gu J. VASC: dimension reduction and visualization of single-cell RNA-seq data by deep variational autoencoder. Genomics Proteomics Bioinformatics. 2018;16:320–31.
Abadi M, Barham P, Chen J, Chen Z, Davis A, Dean J, et al. Tensorflow: a system for large-scale machine learning. In: 12th {USENIX} symposium on operating systems design and implementation ({OSDI} 16); 2016. p. 265–83.
Rampasek L, Goldenberg A. TensorFlow: biology’s gateway to deep learning? Cell Syst. 2016;2:12–4.
Kingma DP, Welling M. Auto-encoding variational bayes. arXiv preprint arXiv:13126114; 2013.
Liu F, Zhang Y, Zhang L, Li Z, Fang Q, Gao R, et al. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019;20:242.
Petti AA, Williams SR, Miller CA, Fiddes IT, Srivatsan SN, Chen DY, et al. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun. 2019;10:3660.
Jiang J, Wang C, Qi R, Fu H, Ma Q. scREAD: a single-cell RNA-Seq database for Alzheimer’s disease. iScience. 2020;23:101769.
Jain AK, Dubes RC. Algorithms for clustering data. Englewood Cliffs: Prentice-Hall, Inc.; 1988.
Dunn JC. Well-separated clusters and optimal fuzzy partitions. J Cybern. 1974;4:95–104.
Davies DL, Bouldin DW. A cluster separation measure. IEEE Trans Pattern Anal Mach Intell. 1979;PAMI-1:224–27.
Oyelade J, Isewon I, Oladipupo F, Aromolaran O, Uwoghiren E, Ameh F, et al. Clustering algorithms: their application to gene expression data. Bioinform Biol Insights. 2016;10:237–53.
de Souto MC, Costa IG, de Araujo DS, Ludermir TB, Schliep A. Clustering cancer gene expression data: a comparative study. BMC Bioinformatics. 2008;9:497.
Song WM, Di Matteo T, Aste T. Hierarchical information clustering by means of topologically embedded graphs. PLoS One. 2012;7:e31929.
Vinh NX, Epps J, Bailey J. Information theoretic measures for clusterings comparison: variants, properties, normalization and correction for chance. J Mach Learn Res. 2010;11:2837–54.
Steinley D. Properties of the Hubert-Arable adjusted rand index. Psychol Methods. 2004;9:386.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290.e1217.
Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–92.
Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–42.
Cosacak MI, Bhattarai P, Reinhardt S, Petzold A, Dahl A, Zhang Y, et al. Single-cell transcriptomics analyses of neural stem cell heterogeneity and contextual plasticity in a zebrafish Brain model of amyloid toxicity. Cell Rep. 2019;27:1307–1318.e1303.
McKenzie AT, Wang M, Hauberg ME, Fullard JF, Kozlenkov A, Keenan A, et al. Brain cell type specific gene expression and co-expression network architectures. Sci Rep. 2018;8:8868.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–18.
Tian L, Dong X, Freytag S, Le Cao KA, Su S, JalalAbadi A, et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat Methods. 2019;16:479–87.
Franzen O, Gan LM, Bjorkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019;2019:baz046.
Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The human cell atlas. Elife. 2017;6:e27041.
Alavi A, Ruffalo M, Parvangada A, Huang Z, Bar-Joseph Z. A web server for comparative analysis of single-cell RNA-seq data. Nat Commun. 2018;9:4768.
Kiselev VY, Yiu A, Hemberg M. scmap: projection of single-cell RNA-seq data across data sets. Nat Methods. 2018;15:359–62.
Pliner HA, Shendure J, Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat Methods. 2019;16:983–6.
Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58:610–20.
Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–40.
Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–26.
Delmans M, Hemberg M. Discrete distributional differential expression (D3E)--a tool for gene expression analysis of single-cell RNA-seq data. BMC Bioinformatics. 2016;17:110.
Korthauer KD, Chu LF, Newton MA, Li Y, Thomson J, Stewart R, et al. A statistical approach for identifying differential distributions in single-cell RNA-seq experiments. Genome Biol. 2016;17:222.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Soneson C, Robinson MD. Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods. 2018;15:255–61.
Wang T, Li B, Nelson CE, Nabavi S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinformatics. 2019;20:40.
Dal Molin A, Baruzzo G, Di Camillo B. Single-cell RNA-sequencing: assessment of differential expression analysis methods. Front Genet. 2017;8:62.
Van den Berge K, Perraudeau F, Soneson C, Love MI, Risso D, Vert JP, et al. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 2018;19:24.
Vandenbon A, Diez D. A clustering-independent method for finding differentially expressed genes in single-cell transcriptome data. Nat Commun. 2020;11:4318.
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6.
Bar-Joseph Z, Gitter A, Simon I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat Rev Genet. 2012;13:552–64.
Qiu X, Rahimzamani A, Wang L, Ren B, Mao Q, Durham T, et al. Inferring causal gene regulatory networks from coupled single-cell expression dynamics using scribe. Cell Syst. 2020;10:265–274.e211.
Svensson V, Pachter L. RNA velocity: molecular kinetics from single-cell RNA-Seq. Mol Cell. 2018;72:7–9.
Zeisel A, Kostler WJ, Molotski N, Tsai JM, Krauthgamer R, Jacob-Hirsch J, et al. Coupled pre-mRNA and mRNA dynamics unveil operational strategies underlying transcriptional responses to stimuli. Mol Syst Biol. 2011;7:529.
La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature. 2018;560:494–8.
Trapnell C. Defining cell types and states with single-cell genomics. Genome Res. 2015;25:1491–8.
Hastie T, Stuetzle W. Principal curves. J Am Stat Assoc. 1989;84:502–16.
Kruskal JB. On the shortest spanning sub-tree of a graph and the traveling salesman problem. Proc Am Math Soc. 1956;7:48.
Cover T, Hart P. Nearest neighbor pattern classification. IEEE Trans Inf Theory. 1967;13:21–27. https://doi.org/10.1109/TIT.1967.1053964.
Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–25.
Setty M, Tadmor MD, Reich-Zeliger S, Angel O, Salame TM, Kathail P, et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat Biotechnol. 2016;34:637–45.
Rappez L, Rakhlin A, Rigopoulos A, Nikolenko S, Alexandrov T. DeepCycle reconstructs a cyclic cell cycle trajectory from unsegmented cell images using convolutional neural networks. Mol Syst Biol. 2020;16:e9474.
Matsumoto H, Kiryu H. SCOUP: a probabilistic model based on the Ornstein-Uhlenbeck process to analyze single-cell expression data during differentiation. BMC Bioinformatics. 2016;17:232.
Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, et al. Single-cell RNA-Seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. 2015;17:360–72.
Ji Z, Ji H. Pseudotime reconstruction using TSCAN. Methods Mol Biol (Clifton, NJ). 2019;1935:115–24.
Welch JD, Hartemink AJ, Prins JF. SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol. 2016;17:106.
Jin S, MacLean AL, Peng T, Nie Q. scEpath: energy landscape-based inference of transition probabilities and cellular trajectories from single-cell transcriptomic data. Bioinformatics (Oxford, England). 2018;34:2077–86.
Guo J, Zheng J. HopLand: single-cell pseudotime recovery using continuous Hopfield network-based modeling of Waddington's epigenetic landscape. Bioinformatics (Oxford, England). 2017;33:i102–9.
Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:59.
Plass M, Solana J, Wolf FA, Ayoub S, Misios A, Glažar P, et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. New York: Science; 2018. p. 360.
Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502.
Todorov H, Cannoodt R, Saelens W, Saeys Y. TinGa: fast and flexible trajectory inference with growing neural gas. Bioinformatics (Oxford, England). 2020;36:i66–74.
Gaidatzis D, Burger L, Florescu M, Stadler MB. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotechnol. 2015;33:722–9.
Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38:1408–14.
Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K, Timmermans MCP. Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev Cell. 2019;48:840–852.e845.
Engel JA, Lee HJ, Williams CG, Kuns R, Olver S, Lansink LI, et al. Single-cell transcriptomics of alloreactive CD4+ T cells over time reveals divergent fates during gut graft-versus-host disease. JCI Insight. 2020;5(13):e137990.
Lavaert M, Liang KL, Vandamme N, Park JE, Roels J, Kowalczyk MS, et al. Integrated scRNA-Seq identifies human postnatal thymus seeding progenitors and regulatory dynamics of differentiating immature thymocytes. Immunity. 2020;52:1088–1104.e1086.
Lummertz da Rocha E, Malleshaiah M. Trajectory algorithms to infer stem cell fate decisions. Methods Mol Biol (Clifton, NJ). 2019;1975:193–209.
Peng G, Cui G, Ke J, Jing N. Using single-cell and spatial transcriptomes to understand stem cell lineage specification during early embryo development. Annu Rev Genomics Hum Genet. 2020;21:163–81.
Iturria-Medina Y, Khan AF, Adewale Q, Shirazi AH. Blood and brain gene expression trajectories mirror neuropathology and clinical deterioration in neurodegeneration. Brain. 2020;143:661–73.
Mukherjee S, Heath L, Preuss C, Jayadev S, Garden GA, Greenwood AK, et al. Molecular estimation of neurodegeneration pseudotime in older brains. Nat Commun. 2020;11:5781.
Young AL, Marinescu RV, Oxtoby NP, Bocchetta M, Yong K, Firth NC, et al. Uncovering the heterogeneity and temporal complexity of neurodegenerative diseases with subtype and stage inference. Nat Commun. 2018;9:4273.
Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12:519–22.
Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35:936–9.
Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26:304–19.
Zhu C, Yu M, Huang H, Juric I, Abnousi A, Hu R, et al. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol. 2019;26:1063–70.
Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177:1873–1887.e1817.
Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. 2020;38:333–42.
Campbell KR, Yau C. switchde: inference of switch-like differential expression along single-cell trajectories. Bioinformatics (Oxford, England). 2017;33:1241–2.
Cao EY, Ouyang JF, Rackham OJL. GeneSwitches: ordering gene expression and functional events in single-cell experiments. Bioinformatics (Oxford, England). 2020;36:3273–5.
Van den Berge K, Roux de Bezieux H, Street K, Saelens W, Cannoodt R, Saeys Y, et al. Trajectory-based differential expression analysis for single-cell sequencing data. Nat Commun. 2020;11:1201.
Campbell KR, Yau C. Uncovering pseudotemporal trajectories with covariates from single cell and bulk expression data. Nat Commun. 2018;9:2442.
Specht AT, Li J. LEAP: constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics. 2017;33:764–6.
Pratapa A, Jalihal AP, Law JN, Bharadwaj A, Murali TM. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat Methods. 2020;17:147–54.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16:172–83.
Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
Pang AW, MacDonald JR, Pinto D, Wei J, Rafiq MA, Conrad DF, et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010;11:R52.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355(6332):eaai8478.
Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 2018;360:331–5.
Melchor L, Brioli A, Wardell CP, Murison A, Potter NE, Kaiser MF, et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia. 2014;28:1705–15.
Toft M, Ross OA. Copy number variation in Parkinson’s disease. Genome Med. 2010;2:62.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841.
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35.
Cuccaro D, De Marco EV, Cittadella R, Cavallaro S. Copy number variants in Alzheimer’s disease. J Alzheimers Dis. 2017;55:37–52.
Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, et al. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006;129:2977–83.
Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–6.
Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, et al. App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of Down syndrome. J Neurosci. 2003;23:6788–92.
Decourt B, Mobley W, Reiman E, Shah RJ, Sabbagh MN. Recent perspectives on APP, secretases, endosomal pathways and how they influence Alzheimer’s related pathological changes in Down syndrome. J Alzheimers Dis Parkinsonism. 2013;Suppl 7:002.
Mohiyuddin M, Mu JC, Li J, Bani Asadi N, Gerstein MB, Abyzov A, et al. MetaSV: an accurate and integrative structural-variant caller for next generation sequencing. Bioinformatics. 2015;31:2741–4.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84.
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, et al. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–54.
Zarate S, Carroll A, Mahmoud M, Krasheninina O, Jun G, Salerno WJ, et al. Parliament2: accurate structural variant calling at scale. Gigascience. 2020;9(12):giaa145.
Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, et al. Large multiallelic copy number variations in humans. Nat Genet. 2015;47:296–303.
Fan X, Abbott TE, Larson D, Chen K. BreakDancer: identification of genomic structural variation from paired-end read mapping. Curr Protoc Bioinformatics. 2014;45:15 16 11–11.
Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–6.
Xu B, Cai H, Zhang C, Yang X, Han G. Copy number variants calling for single cell sequencing data by multi-constrained optimization. Comput Biol Chem. 2016;63:15–20.
Fan J, Lee HO, Lee S, Ryu DE, Lee S, Xue C, et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 2018;28:1217–27.
Borel C, Ferreira PG, Santoni F, Delaneau O, Fort A, Popadin KY, et al. Biased allelic expression in human primary fibroblast single cells. Am J Hum Genet. 2015;96:70–80.
Wang L, Fan J, Francis JM, Georghiou G, Hergert S, Li S, et al. Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res. 2017;27:1300–11.
Muller S, Cho A, Liu SJ, Lim DA, Diaz A. CONICS integrates scRNA-seq with DNA sequencing to map gene expression to tumor sub-clones. Bioinformatics. 2018;34:3217–9.
Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18:234.
Tickle T, Tirosh I, Georgescu C, Brown M, Haas B. inferCNV of the Trinity CTAT Project. Cambridge: Klarman Cell Observatory, Broad Institute of MIT and Harvard; 2019. https://github.com/broadinstitute/inferCNV.
Serin Harmanci A, Harmanci AO, Zhou X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat Commun. 2020;11:89.
Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol. 2015;33:285–9.
Miron J, Picard C, Labonte A, Auld D, Breitner J, Poirier J, et al. Association of PPP2R1A with Alzheimer’s disease and specific cognitive domains. Neurobiol Aging. 2019;81:234–43.
Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–7.
Drummond E, Pires G, MacMurray C, Askenazi M, Nayak S, Bourdon M, et al. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain. 2020;143:2803–17.
van der Wijst M, de Vries DH, Groot HE, Trynka G, Hon CC, Bonder MJ, et al. The single-cell eQTLGen consortium. Elife. 2020;9:e52155.
Hormozdiari F, Gazal S, van de Geijn B, Finucane HK, Ju CJ, Loh PR, et al. Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat Genet. 2018;50:1041–7.
Zhernakova DV, Deelen P, Vermaat M, van Iterson M, van Galen M, Arindrarto W, et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat Genet. 2017;49:139–45.
Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB. Abundant associations with gene expression complicate GWAS follow-up. Nat Genet. 2019;51:768–9.
Ye CJ, Feng T, Kwon HK, Raj T, Wilson MT, Asinovski N, et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science. 2014;345:1254665.
Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martin D, et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell. 2016;167:1398–1414.e1324.
Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13.
Lloyd-Jones LR, Holloway A, McRae A, Yang J, Small K, Zhao J, et al. The genetic architecture of gene expression in peripheral blood. Am J Hum Genet. 2017;100:228–37.
Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464.
Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. Nat Genet 2021;53:1300–310.
Fu J, Wolfs MG, Deelen P, Westra HJ, Fehrmann RS, Te Meerman GJ, et al. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet. 2012;8:e1002431.
Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, Dilthey A, et al. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat Genet. 2012;44:502–10.
Brown CD, Mangravite LM, Engelhardt BE. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet. 2013;9:e1003649.
van der Wijst MGP, Brugge H, de Vries DH, Deelen P, Swertz MA, LifeLines Cohort S, et al. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat Genet. 2018;50:493–7.
Cuomo ASE, Seaton DD, McCarthy DJ, Martinez I, Bonder MJ, Garcia-Bernardo J, et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat Commun. 2020;11:810.
Hu Y, Xi X, Yang Q, Zhang X. SCeQTL: an R package for identifying eQTL from single-cell parallel sequencing data. BMC Bioinformatics. 2020;21:184.
Bacher R, Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol. 2016;17:63.
Miao Z, Zhang X. Differential expression analyses for single-cell RNA-Seq: old questions on new data. Quant Biol. 2016;4:243–60.
Liu H, Prashant NM, Spurr LF, Bousounis P, Alomran N, Ibeawuchi H, et al. scReQTL: an approach to correlate SNVs to gene expression from individual scRNA-seq datasets. BMC Genomics. 2021;22:40.
Miao Z, Deng K, Wang X, Zhang X. DEsingle for detecting three types of differential expression in single-cell RNA-seq data. Bioinformatics. 2018;34:3223–4.
Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, Sexton DW, et al. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat Biotechnol. 2013;31:748–52.
Sarkar AK, Tung PY, Blischak JD, Burnett JE, Li YI, Stephens M, et al. Discovery and characterization of variance QTLs in human induced pluripotent stem cells. PLoS Genet. 2019;15:e1008045.
Donovan MKR, D'Antonio-Chronowska A, D'Antonio M, Frazer KA. Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants. Nat Commun. 2020;11:955.
Heap GA, Trynka G, Jansen RC, Bruinenberg M, Swertz MA, Dinesen LC, et al. Complex nature of SNP genotype effects on gene expression in primary human leucocytes. BMC Med Genet. 2009;2:1.
Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–24.
Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, Lyle R, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78.
Michaelson JJ, Loguercio S, Beyer A. Detection and interpretation of expression quantitative trait loci (eQTL). Methods. 2009;48:265–76.
Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020;52:1428–42.
Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods. 2017;14:975–8.
Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol Cell. 2018;71:858–871.e858.
Bravo Gonzalez-Blas C, Minnoye L, Papasokrati D, Aibar S, Hulselmans G, Christiaens V, et al. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat Methods. 2019;16:397–400.
Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell. 2020;183:1103–1116.e1120.
Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol. 2019;37:1458–65.
Chen X, Litzenburger UM, Wei Y, Schep AN, LaGory EL, Choudhry H, et al. Joint single-cell DNA accessibility and protein epitope profiling reveals environmental regulation of epigenomic heterogeneity. Nat Commun. 2018;9:4590.
Corces MR, Shcherbina A, Kundu S, Gloudemans MJ, Fresard L, Granja JM, et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat Genet. 2020;52:1158–68.
Stuart T, Srivastava A, Lareau C, Satija R. Multimodal single-cell chromatin analysis with Signac. Nat Methods. 2021;18:1333–341.
Granja JM, Corces RM, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, et al. ArchR: an integrative and scalable software package for single-cell chromatin accessibility analysis. Nat Genet. 2021;53:403–11.
Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat Commun. 2021;12:1337.
de Boer CG, Regev A. BROCKMAN: deciphering variance in epigenomic regulators by k-mer factorization. BMC Bioinformatics. 2018;19:253.
Lal A, Chiang ZD, Yakovenko N, Duarte FM, Israeli J, Buenrostro JD. Deep learning-based enhancement of epigenomics data with AtacWorks. Nat Commun. 2021;12:507.
Urrutia E, Chen L, Zhou H, Jiang Y. Destin: toolkit for single-cell analysis of chromatin accessibility. Bioinformatics. 2019;35:3818–20.
Danese A, Richter ML, Fischer DS, Theis FJ, Colomé-Tatché M. EpiScanpy: integrated single-cell epigenomic analysis. Nat Commun . 2021;12:5228.
Baker SM, Rogerson C, Hayes A, Sharrocks AD, Rattray M. Classifying cells with Scasat, a single-cell ATAC-seq analysis tool. Nucleic Acids Res. 2019;47:e10.
Ji Z, Zhou W, Ji H. Single-cell regulome data analysis by SCRAT. Bioinformatics. 2017;33:2930–2.
Xiong L, Xu K, Tian K, Shao Y, Tang L, Gao G, et al. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat Commun. 2019;10:4576.
Prompsy P, Kirchmeier P, Marsolier J, Deloger M, Servant N, Vallot C. Interactive analysis of single-cell epigenomic landscapes with ChromSCape. Nat Commun. 2020;11:5702.
Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018;174:1309–1324.e1318.
Zamanighomi M, Lin Z, Daley T, Chen X, Duren Z, Schep A, et al. Unsupervised clustering and epigenetic classification of single cells. Nat Commun. 2018;9:2410.
Yu W, Uzun Y, Zhu Q, Chen C, Tan K. scATAC-pro: a comprehensive workbench for single-cell chromatin accessibility sequencing data. Genome Biol. 2020;21:94.
Chen H, Lareau C, Andreani T, Vinyard ME, Garcia SP, Clement K, et al. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019;20:241.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137.
Kobak D, Berens P. The art of using t-SNE for single-cell transcriptomics. Nat Commun. 2019;10:5416.
Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet. 2021;53:403–11.
Chen H, Albergante L, Hsu JY, Lareau CA, Lo Bosco G, Guan J, et al. Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM. Nat Commun. 1903;2019:10.
Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–13.
Cha J, Lee I. Single-cell network biology for resolving cellular heterogeneity in human diseases. Exp Mol Med. 2020;52:1798–808.
Choi H, Song WM, Wang M, Sram RJ, Zhang B. Benzo[a]pyrene is associated with dysregulated myelo-lymphoid hematopoiesis in asthmatic children. Environ Int. 2019;128:218–32.
Forst CV, Zhou B, Wang M, Chou TW, Mason G, Song WM, et al. Integrative gene network analysis identifies key signatures, intrinsic networks and host factors for influenza virus A infections. NPJ Syst Biol Appl. 2017;3:35.
Katsyv I, Wang M, Song WM, Zhou X, Zhao Y, Park S, et al. EPRS is a critical regulator of cell proliferation and estrogen signaling in ER+ breast cancer. Oncotarget. 2016;7:69592–605.
Nakagawa S, Wei L, Song WM, Higashi T, Ghoshal S, Kim RS, et al. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell. 2016;30:879–90.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–20.
Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 2016;8:104.
Song WM, Lin X, Liao X, Hu D, Lin J, Sarpel U, et al. Multiscale network analysis reveals molecular mechanisms and key regulators of the tumor microenvironment in gastric cancer. Int J Cancer. 2020;146:1268-280.
Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, et al. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A. 2019;116:19619–25.
Blencowe M, Arneson D, Ding J, Chen YW, Saleem Z, Yang X. Network modeling of single-cell omics data: challenges, opportunities, and progresses. Emerg Top Life Sci. 2019;3:379–98.
Nguyen H, Tran D, Tran B, Pehlivan B, Nguyen T. A comprehensive survey of regulatory network inference methods using single-cell RNA sequencing data. Brief Bioinform. 2021;22:bbaa190.
Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol. 2019;37:547–54.
Skinnider MA, Squair JW, Foster LJ. Evaluating measures of association for single-cell transcriptomics. Nat Methods. 2019;16:381–6.
Andrews TS, Hemberg M. False signals induced by single-cell imputation. F1000Res. 2018;7:1740.
Crow M, Paul A, Ballouz S, Huang ZJ, Gillis J. Exploiting single-cell expression to characterize co-expression replicability. Genome Biol. 2016;17:101.
Li WV, Li Y. scLink: inferring sparse gene co-expression networks from single-cell expression data. Genomics, Proteomics Bioinformatics. 2021;19:475–92.
Noor A, Serpedin E, Nounou M, Nounou H, Mohamed N, Chouchane L. An overview of the statistical methods used for inferring gene regulatory networks and protein-protein interaction networks. Adv Bioinforma. 2013;2013:953814.
De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat Rev Microbiol. 2010;8:717–29.
Soranzo N, Bianconi G, Altafini C. Comparing association network algorithms for reverse engineering of large-scale gene regulatory networks: synthetic versus real data. Bioinformatics. 2007;23:1640–7.
Carter SL, Brechbuhler CM, Griffin M, Bond AT. Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics. 2004;20:2242–50.
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17.
Song WM, Zhang B. Multiscale embedded gene co-expression network analysis. PLoS Comput Biol. 2015;11:e1004574.
Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7.
Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8.
Meyer PE, Lafitte F, Bontempi G. minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinformatics. 2008;9:461.
Yan J, Risacher SL, Shen L, Saykin AJ. Network approaches to systems biology analysis of complex disease: integrative methods for multi-omics data. Brief Bioinform. 2018;19:1370–81.
Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet. 2008;40:854–61.
Scutari M. Learning Bayesian networks with the bnlearn R package. J Stat Softw. 2010;35:1–22.
Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5:e12776.
Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, Camacho DM, et al. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9:796–804.
Luo Y, Coskun V, Liang A, Yu J, Cheng L, Ge W, et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell. 2015;161:1175–86.
Wu H, Chen S, Yu J, Li Y, Zhang XY, Yang L, et al. Single-cell transcriptome analyses reveal molecular signals to intrinsic and acquired paclitaxel resistance in esophageal squamous cancer cells. Cancer Lett. 2018;420:156–67.
Chen X, Hu L, Wang Y, Sun W, Yang C. Single cell gene co-expression network reveals FECH/CROT signature as a prognostic marker. Cells. 2019;8(7):698.
Al-Dalahmah O, Sosunov AA, Shaik A, Ofori K, Liu Y, Vonsattel JP, et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun. 2020;8:19.
Wang C, Forst CV, Chou TW, Geber A, Wang M, Hamou W, et al. Cell-to-cell variation in defective virus expression and effects on host responses during influenza virus infection. mBio. 2020;11(1):e02880–19.
Crow M, Gillis J. Co-expression in single-cell analysis: saving grace or original sin? Trends Genet. 2018;34:823–31.
Chan TE, Stumpf MPH, Babtie AC. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 2017;5:251–267.e253.
Chen L, Kulasiri D, Samarasinghe S. A novel data-driven Boolean model for genetic regulatory networks. Front Physiol. 2018;9:1328.
Kauffman S, Peterson C, Samuelsson B, Troein C. Random Boolean network models and the yeast transcriptional network. Proc Natl Acad Sci U S A. 2003;100:14796–9.
Woodhouse S, Piterman N, Wintersteiger CM, Gottgens B, Fisher J. SCNS: a graphical tool for reconstructing executable regulatory networks from single-cell genomic data. BMC Syst Biol. 2018;12:59.
Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, Tanaka Y, Wilkinson AC, et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat Biotechnol. 2015;33:269–76.
Chen H, Guo J, Mishra SK, Robson P, Niranjan M, Zheng J. Single-cell transcriptional analysis to uncover regulatory circuits driving cell fate decisions in early mouse development. Bioinformatics. 2014;31:1060–6.
Lim CY, Wang H, Woodhouse S, Piterman N, Wernisch L, Fisher J, et al. BTR: training asynchronous Boolean models using single-cell expression data. BMC Bioinformatics. 2016;17:355.
Aubin-Frankowski P-C, Vert J-P. Gene regulation inference from single-cell RNA-seq data with linear differential equations and velocity inference. Bioinformatics. 2020;36:4774–80.
Matsumoto H, Kiryu H, Furusawa C, Ko MSH, Ko SBH, Gouda N, et al. SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics. 2017;33:2314–21.
Sanchez-Castillo M, Blanco D, Tienda-Luna IM, Carrion MC, Huang Y. A Bayesian framework for the inference of gene regulatory networks from time and pseudo-time series data. Bioinformatics. 2018;34:964–70.
Sekula M, Gaskins J, Datta S. A sparse Bayesian factor model for the construction of gene co-expression networks from single-cell RNA sequencing count data. BMC Bioinformatics. 2020;21:361.
Granger CW. Investigating causal relations by econometric models and cross-spectral methods. Econometrica. 1969;37:424–38.
Papili Gao N, Ud-Dean SMM, Gandrillon O, Gunawan R. SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics. 2018;34:258–66.
Deshpande A, Chu L-F, Stewart R, Gitter A. Network inference with granger causality ensembles on single-cell transcriptomics. Cell Rep. 2022;38:110333.
Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–6.
Iacono G, Mereu E, Guillaumet-Adkins A, Corominas R, Cusco I, Rodriguez-Esteban G, et al. bigSCale: an analytical framework for big-scale single-cell data. Genome Res. 2018;28:878–90.
Iacono G, Massoni-Badosa R, Heyn H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019;20:110.
Dai H, Li L, Zeng T, Chen L. Cell-specific network constructed by single-cell RNA sequencing data. Nucleic Acids Res. 2019;47:e62.
Zhou JX, Taramelli R, Pedrini E, Knijnenburg T, Huang S. Extracting intercellular signaling network of cancer tissues using ligand-receptor expression patterns from whole-tumor and single-cell transcriptomes. Sci Rep. 2017;7:8815.
Wang Y, Wang R, Zhang S, Song S, Jiang C, Han G, et al. iTALK: an R Package to characterize and illustrate intercellular communication. bioRxiv. 2019;507871. https://doi.org/10.1101/507871.
Tsuyuzaki K, Ishii M, Nikaido I. Uncovering hypergraphs of cell-cell interaction from single cell RNA-sequencing data. bioRxiv. 2019;566182. https://doi.org/10.1101/566182.
Wang S, Karikomi M, MacLean AL, Nie Q. Cell lineage and communication network inference via optimization for single-cell transcriptomics. Nucleic Acids Res. 2019;47:e66.
Mohammadi S, Ravindra V, Gleich DF, Grama A. A geometric approach to characterize the functional identity of single cells. Nat Commun. 2018;9:1516.
Mohammadi S, Davila-Velderrain J, Kellis M. Reconstruction of cell-type-specific interactomes at single-cell resolution. Cell Syst. 2019;9:559–568.e554.
Sansone SA, Rocca-Serra P, Field D, Maguire E, Taylor C, Hofmann O, et al. Toward interoperable bioscience data. Nat Genet. 2012;44:121–6.
Wang Z, Monteiro CD, Jagodnik KM, Fernandez NF, Gundersen GW, Rouillard AD, et al. Extraction and analysis of signatures from the gene expression omnibus by the crowd. Nat Commun. 2016;7:12846.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52.
Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167:1853–1866.e1817.
Schaffter T, Marbach D, Floreano D. GeneNetWeaver: in silico benchmark generation and performance profiling of network inference methods. Bioinformatics. 2011;27:2263–70.
Chen S, Mar JC. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinformatics. 2018;19:232.
Coleman PD, Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer’s disease. Neurobiol Aging. 1987;8:521–45.
Felsky D, Roostaei T, Nho K, Risacher SL, Bradshaw EM, Petyuk V, et al. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun. 2019;10:409.
Wang M, Li A, Sekiya M, Beckmann ND, Quan X, Schrode N, et al. Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer’s disease. Neuron. 2020;S0896-6273:8.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Gaujoux R, Seoighe C. CellMix: a comprehensive toolbox for gene expression deconvolution. Bioinformatics. 2013;29:2211–2.
Du R, Carey V, Weiss ST. deconvSeq: deconvolution of cell mixture distribution in sequencing data. Bioinformatics. 2019;35:5095–102.
Wang X, Park J, Susztak K, Zhang NR, Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun. 2019;10:380.
Tsoucas D, Dong R, Chen H, Zhu Q, Guo G, Yuan GC. Accurate estimation of cell-type composition from gene expression data. Nat Commun. 2019;10:2975.
Jew B, Alvarez M, Rahmani E, Miao Z, Ko A, Garske KM, et al. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat Commun. 2020;11:1971.
Dong M, Thennavan A, Urrutia E, Li Y, Perou CM, Zou F, et al. SCDC: bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief Bioinform. 2021;18:416–27.
Patrick E, Taga M, Ergun A, Ng B, Casazza W, Cimpean M, et al. Deconvolving the contributions of cell-type heterogeneity on cortical gene expression. PLoS Comput Biol. 2020;16:e1008120.
Wang X, Allen M, Li S, Quicksall ZS, Patel TA, Carnwath TP, et al. Deciphering cellular transcriptional alterations in Alzheimer's disease brains. Mol Neurodegener. 2020;15:38.
Cain A, Taga M, McCabe C, Hekselman I, White CC, Green G, et al. Multi-cellular communities are perturbed in the aging human brain and with Alzheimer’s disease. bioRxiv. 2020;2020.2012.2022.424084. https://doi.org/10.1101/2020.12.22.424084.
Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–8.
Song WM, Agrawal P, Von Itter R, Fontanals-Cirera B, Wang M, Zhou X, et al. Network models of primary melanoma microenvironments identify key melanoma regulators underlying prognosis. Nat Commun. 2021;12:1214.
Wang Q, Zhang Y, Wang M, Song WM, Shen Q, McKenzie A, et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat Commun. 2019;10:5234.
Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci. 2006;103:17402–7.
Labonté B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med. 2017;23:1102–11.
Shu L, Zhao Y, Kurt Z, Byars SG, Tukiainen T, Kettunen J, et al. Mergeomics: multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics. 2016;17:874.
Peters LA, Perrigoue J, Mortha A, Iuga A, Song WM, Neiman EM, et al. A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat Genet. 2017;49:1437–49.
Mäkinen V-P, Civelek M, Meng Q, Zhang B, Zhu J, Levian C, et al. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014;10:e1004502.
Yi Z, Keung KL, Li L, Hu M, Lu B, Nicholson L, et al. Key driver genes as potential therapeutic targets in renal allograft rejection. JCI Insight. 2020;5:e136220.
Wang M, Li A, Sekiya M, Beckmann ND, Quan X, Schrode N, et al. Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer’s disease. Neuron. 2021;109:257–272.e214.
Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348:aaa6090.
Codeluppi S, Borm LE, Zeisel A, La Manno G, van Lunteren JA, Svensson CI, et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods. 2018;15:932–5.
Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568:235–9.
Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11:360–1.
Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science. 2018;362(6416):eaau5324.
Shah S, Lubeck E, Zhou W, Cai L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse Hippocampus. Neuron. 2016;92:342–57.
Ke R, Mignardi M, Pacureanu A, Svedlund J, Botling J, Wahlby C, et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013;10:857–60.
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343:1360–3.
Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361(6400):eaat5691.
Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–7.
Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82.
Zollinger DR, Lingle SE, Sorg K, Beechem JM, Merritt CR. GeoMx RNA assay: high multiplex, digital, spatial analysis of RNA in FFPE tissue. Methods Mol Biol. 2020;2148:331–45.
Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstrahle J, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun. 2018;9:2419.
Maniatis S, Aijo T, Vickovic S, Braine C, Kang K, Mollbrink A, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89–93.
Chen W-T, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell. 2020;182:976–991.e919.
Dries R, Zhu Q, Dong R, Eng CL, Li H, Liu K, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021;22:78.
Edsgard D, Johnsson P, Sandberg R. Identification of spatial expression trends in single-cell gene expression data. Nat Methods. 2018;15:339–42.
Sun S, Zhu J, Zhou X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat Methods. 2020;17:193–200.
Svensson V, Teichmann SA, Stegle O. SpatialDE: identification of spatially variable genes. Nat Methods. 2018;15:343–6.
Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–42.
Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron. 2020;105:837–854.e839.
Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med. 2020;217(9):e20200785.
Zhang Y, Fung ITH, Sankar P, Chen X, Robison LS, Ye L, et al. Depletion of NK cells improves cognitive function in the Alzheimer disease mouse model. J Immunol. 2020;205:502–10.
Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–80.
Zhong S, Wang M, Zhan Y, Zhang J, Yang X, Fu S, et al. Single-nucleus RNA sequencing reveals transcriptional changes of hippocampal neurons in APP23 mouse model of Alzheimer’s disease. Biosci Biotechnol Biochem. 2020;84:919–26.
Lau SF, Chen C, Fu WY, Qu JY, Cheung TH, Fu AKY, et al. IL-33-PU.1 transcriptome reprogramming drives functional state transition and clearance activity of microglia in Alzheimer’s disease. Cell Rep. 2020;31:107530.
Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23:701–6.
Wang J, Sun H, Jiang M, Li J, Zhang P, Chen H, et al. Tracing cell-type evolution by cross-species comparison of cell atlases. Cell Rep. 2021;34:108803.
Shafer MER. Cross-species analysis of single-cell transcriptomic data. Front Cell Dev Biol. 2019;7:175.
Crow M, Paul A, Ballouz S, Huang ZJ, Gillis J. Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat Commun. 2018;9:884.
Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci. 2021;24:276–87.
Hof PR, Cox K, Morrison JH. Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer’s disease: I. Superior frontal and inferior temporal cortex. J Comp Neurol. 1990;301:44–54.
Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16:4491–500.
Chen Y, Colonna M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J Exp Med. 2021;218:e20202717.
Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018;22:832–47.
Dachet F, Brown JB, Valyi-Nagy T, Narayan KD, Serafini A, Boley N, et al. Selective time-dependent changes in activity and cell-specific gene expression in human postmortem brain. Sci Rep. 2021;11:6078.
Birdsill AC, Walker DG, Lue L, Sue LI, Beach TG. Postmortem interval effect on RNA and gene expression in human brain tissue. Cell Tissue Bank. 2011;12:311–8.
Preuss C, Pandey R, Piazza E, Fine A, Uyar A, Perumal T, et al. A novel systems biology approach to evaluate mouse models of late-onset Alzheimer’s disease. Mol Neurodegener. 2020;15:67.
Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull. 2012;88:3–12.
Jankowsky JL, Zheng H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener. 2017;12:89.
Drummond E, Wisniewski T. Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 2017;133:155–75.
Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14:979–82.
Cannoodt R, Saelens W, Saeys Y. Computational methods for trajectory inference from single-cell transcriptomics. Eur J Immunol. 2016;46:2496–506.
Chervov A, Bac J, Zinovyev A. Minimum spanning vs. principal trees for structured approximations of multi-dimensional datasets. Entropy (Basel, Switzerland). 2020;22(11):1274.
Zhang F, Li X, Tian W. Unsupervised inference of developmental directions for single cells using VECTOR. Cell Rep. 2020;32:108069.
Zhu M, Tao Y, He Q, Gao H, Song F, Sun YM, et al. Common GSAP promoter variant contributes to Alzheimer’s disease liability. Neurobiol Aging. 2014;35:2656.e2651–7.
Zhang L, Feng XK, Ng YK, Li SC. Reconstructing directed gene regulatory network by only gene expression data. BMC Genomics. 2016;17(Suppl 4):430.
Wang X, Chen Y, Wang X, Lu L. Genetic regulatory network analysis for app based on genetical genomics approach. Exp Aging Res. 2010;36:79–93.
Proitsi P, Lee SH, Lunnon K, Keohane A, Powell J, Troakes C, et al. Alzheimer’s disease susceptibility variants in the MS4A6A gene are associated with altered levels of MS4A6A expression in blood. Neurobiol Aging. 2014;35:279–90.
Nelson PT, Wang WX, Wilfred BR, Wei A, Dimayuga J, Huang Q, et al. Novel human ABCC9/SUR2 brain-expressed transcripts and an eQTL relevant to hippocampal sclerosis of aging. J Neurochem. 2015;134:1026–39.
Myers AJ. AD gene 3-D: moving past single layer genetic information to map novel loci involved in Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S15–22.
Martinelli-Boneschi F, Giacalone G, Magnani G, Biella G, Coppi E, Santangelo R, et al. Pharmacogenomics in Alzheimer’s disease: a genome-wide association study of response to cholinesterase inhibitors. Neurobiol Aging. 2013;34(1711):e1717–3.
Li H, Achour I, Bastarache L, Berghout J, Gardeux V, Li J, et al. Integrative genomics analyses unveil downstream biological effectors of disease-specific polymorphisms buried in intergenic regions. NPJ Genom Med. 2016;1:16006.
Karch CM, Ezerskiy LA, Bertelsen S, Alzheimer’s Disease Genetics C, Goate AM. Alzheimer’s disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 loci. PLoS One. 2016;11:e0148717.
Holton P, Ryten M, Nalls M, Trabzuni D, Weale ME, Hernandez D, et al. Initial assessment of the pathogenic mechanisms of the recently identified Alzheimer risk loci. Ann Hum Genet. 2013;77:85–105.
Guerreiro RJ, Beck J, Gibbs JR, Santana I, Rossor MN, Schott JM, et al. Genetic variability in CLU and its association with Alzheimer’s disease. PLoS One. 2010;5:e9510.
Blauwendraat C, Francescatto M, Gibbs JR, Jansen IE, Simon-Sanchez J, Hernandez DG, et al. Comprehensive promoter level expression quantitative trait loci analysis of the human frontal lobe. Genome Med. 2016;8:65.
Zhang A, Zhao Q, Xu D, Jiang S. Brain APOE expression quantitative trait loci-based association study identified one susceptibility locus for Alzheimer’s disease by interacting with APOE epsilon4. Sci Rep. 2018;8:8068.
Xu Z, Xu G, Pan W, Alzheimer’s Disease Neuroimaging I. Adaptive testing for association between two random vectors in moderate to high dimensions. Genet Epidemiol. 2017;41:599–609.
Sun JY, Hou YJ, Zhang Y, Wang L, Liu L, Sun BL, et al. Genetic variants associated with neurodegenerative diseases regulate gene expression in immune cell CD14+ monocytes. Front Genet. 2018;9:666.
Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, de Leeuw CA, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet. 2018;50:912–9.
Ryan KJ, White CC, Patel K, Xu J, Olah M, Replogle JM, et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci Transl Med. 2017;9(421):eaai7635.
Rao S, Ghani M, Guo Z, Deming Y, Wang K, Sims R, et al. An APOE-independent cis-eSNP on chromosome 19q13.32 influences tau levels and late-onset Alzheimer's disease risk. Neurobiol Aging. 2018;66:178.e171–8.
Picard C, Julien C, Frappier J, Miron J, Theroux L, Dea D, United Kingdom Brain Expression C, for the Alzheimer’s Disease Neuroimaging I, Breitner JCS, Poirier J. Alterations in cholesterol metabolism-related genes in sporadic Alzheimer’s disease. Neurobiol Aging. 2018;66:180.e181-180.e189.
Padhy B, Hayat B, Nanda GG, Mohanty PP, Alone DP. Pseudoexfoliation and Alzheimer’s associated CLU risk variant, rs2279590, lies within an enhancer element and regulates CLU, EPHX2 and PTK2B gene expression. Hum Mol Genet. 2017;26:4519–29.
Montesanto A, Crocco P, Dato S, Geracitano S, Frangipane F, Colao R, et al. Uncoupling protein 4 (UCP4) gene variability in neurodegenerative disorders: further evidence of association in frontotemporal dementia. Aging (Albany NY). 2018;10:3283–93.
Mirza N, Appleton R, Burn S, du Plessis D, Duncan R, Farah JO, et al. Genetic regulation of gene expression in the epileptic human hippocampus. Hum Mol Genet. 2017;26:1759–69.
Malamon JS, Kriete A. Integrated systems approach reveals sphingolipid metabolism pathway dysregulation in association with late-onset Alzheimer’s disease. Biology (Basel). 2018;7(1):16.
Liu G, Wang T, Tian R, Hu Y, Han Z, Wang P, et al. Alzheimer’s disease risk variant rs2373115 regulates GAB2 and NARS2 expression in human brain tissues. J Mol Neurosci. 2018;66:37–43.
Le Guen Y, Philippe C, Riviere D, Lemaitre H, Grigis A, Fischer C, et al. eQTL of KCNK2 regionally influences the brain sulcal widening: evidence from 15,597 UK Biobank participants with neuroimaging data. Brain Struct Funct. 2019;224:847–57.
Katsumata Y, Nelson PT, Estus S, Alzheimer’s Disease Neuroimaging I, Fardo DW. Translating Alzheimer’s disease-associated polymorphisms into functional candidates: a survey of IGAP genes and SNPs. Neurobiol Aging. 2019;74:135–46.
Jawinski P, Kirsten H, Sander C, Spada J, Ulke C, Huang J, et al. Human brain arousal in the resting state: a genome-wide association study. Mol Psychiatry. 2019;24:1599–609.
Hu Y, Zhao T, Zang T, Zhang Y, Cheng L. Identification of Alzheimer’s disease-related genes based on data integration method. Front Genet. 2018;9:703.
Fang X, Tang W, Yang F, Lu W, Cai J, Ni J, et al. A comprehensive analysis of the CaMK2A gene and susceptibility to Alzheimer’s disease in the Han Chinese population. Front Aging Neurosci. 2019;11:84.
Carrasquillo MM, Allen M, Burgess JD, Wang X, Strickland SL, Aryal S, et al. A candidate regulatory variant at the TREM gene cluster associates with decreased Alzheimer’s disease risk and increased TREML1 and TREM2 brain gene expression. Alzheimers Dement. 2017;13:663–73.
Bi R, Zhang W, Zhang DF, Xu M, Fan Y, Hu QX, et al. Genetic association of the cytochrome c oxidase-related genes with Alzheimer’s disease in Han Chinese. Neuropsychopharmacology. 2018;43:2264–76.
Zheng Q, Bi R, Xu M, Zhang DF, Tan LW, Lu YP, et al. Exploring the genetic association of the ABAT gene with Alzheimer’s disease. Mol Neurobiol. 2021;58(5):1894–903.
Zhao T, Hu Y, Zang T, Wang Y. Integrate GWAS, eQTL, and mQTL data to identify Alzheimer’s disease-related genes. Front Genet. 2019;10:1021.
Wang T, Peng Q, Liu B, Liu Y, Wang Y. Disease module identification based on representation learning of complex networks integrated from GWAS, eQTL summaries, and human Interactome. Front Bioeng Biotechnol. 2020;8:418.
Sieberts SK, Perumal TM, Carrasquillo MM, Allen M, Reddy JS, Hoffman GE, et al. Large eQTL meta-analysis reveals differing patterns between cerebral cortical and cerebellar brain regions. Sci Data. 2020;7:340.
Shigemizu D, Akiyama S, Higaki S, Sugimoto T, Sakurai T, Boroevich KA, et al. Prognosis prediction model for conversion from mild cognitive impairment to Alzheimer’s disease created by integrative analysis of multi-omics data. Alzheimers Res Ther. 2020;12:145.
Sanchez-Mut JV, Glauser L, Monk D, Graff J. Comprehensive analysis of PM20D1 QTL in Alzheimer’s disease. Clin Epigenetics. 2020;12:20.
Picard C, Poirier A, Belanger S, Labonte A, Auld D, Poirier J, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) in Alzheimer’s disease: a genetic and proteomic multi-cohort study. PLoS One. 2019;14:e0220254.
Malamon JS, Kriete A. Erosion of gene co-expression networks reveal deregulation of immune system processes in late-onset Alzheimer’s disease. Front Neurosci. 2020;14:228.
Maj C, Azevedo T, Giansanti V, Borisov O, Dimitri GM, Spasov S, et al. Integration of machine learning methods to dissect genetically imputed transcriptomic profiles in Alzheimer’s disease. Front Genet. 2019;10:726.
Ma Y, Jun GR, Chung J, Zhang X, Kunkle BW, Naj AC, et al. CpG-related SNPs in the MS4A region have a dose-dependent effect on risk of late-onset Alzheimer disease. Aging Cell. 2019;18:e12964.
Lin CW, Chang LC, Ma T, Oh H, French B, Puralewski R, et al. Older molecular brain age in severe mental illness. Mol Psychiatry. 2021;26:3646–56.
Kikuchi M, Hara N, Hasegawa M, Miyashita A, Kuwano R, Ikeuchi T, et al. Enhancer variants associated with Alzheimer’s disease affect gene expression via chromatin looping. BMC Med Genet. 2019;12:128.
Gerring ZF, Lupton MK, Edey D, Gamazon ER, Derks EM. An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:43.
Fan KH, Feingold E, Rosenthal SL, Demirci FY, Ganguli M, Lopez OL, et al. Whole-exome sequencing analysis of Alzheimer’s disease in non-APOE*4 carriers. J Alzheimers Dis. 2020;76:1553–65.
Baird DA, Liu JZ, Zheng J, Sieberts SK, Perumal T, Elsworth B, et al. Identifying drug targets for neurological and psychiatric disease via genetics and the brain transcriptome. PLoS Genet. 2021;17:e1009224.
Amlie-Wolf A, Tang M, Way J, Dombroski B, Jiang M, Vrettos N, et al. Inferring the molecular mechanisms of noncoding Alzheimer’s disease-associated genetic variants. J Alzheimers Dis. 2019;72:301–18.
Zappoli R, Versari A, Paganini M, Arnetoli G, Muscas GC, Gangemi PF, et al. Brain electrical activity (quantitative EEG and bit-mapping neurocognitive CNV components), psychometrics and clinical findings in presenile subjects with initial mild cognitive decline or probable Alzheimer-type dementia. Ital J Neurol Sci. 1995;16:341–76.
Zappoli R, Versari A, Arnetoli G, Paganini M, Muscas GC, Arneodo MG, et al. Topographic CNV activity mapping, presenile mild primary cognitive decline and Alzheimer-type dementia. Neurophysiol Clin. 1991;21:473–83.
Swaminathan S, Kim S, Shen L, Risacher SL, Foroud T, Pankratz N, et al. Genomic copy number analysis in Alzheimer’s disease and mild cognitive impairment: an ADNI study. Int J Alzheimers Dis. 2011;2011:729478.
Shaw CA, Li Y, Wiszniewska J, Chasse S, Zaidi SN, Jin W, et al. Olfactory copy number association with age at onset of Alzheimer disease. Neurology. 2011;76:1302–9.
McNaughton D, Knight W, Guerreiro R, Ryan N, Lowe J, Poulter M, et al. Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol Aging. 2012;33(426):e413–21.
Kay DM, Stevens CF, Hamza TH, Montimurro JS, Zabetian CP, Factor SA, et al. A comprehensive analysis of deletions, multiplications, and copy number variations in PARK2. Neurology. 2010;75:1189–94.
Ghani M, Pinto D, Lee JH, Grinberg Y, Sato C, Moreno D, et al. Genome-wide survey of large rare copy number variants in Alzheimer’s disease among Caribbean hispanics. G3 (Bethesda). 2012;2:71–8.
Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, Le Bastard N, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–33.
Borovecki F, Klepac N, Muck-Seler D, Hajnsek S, Mubrin Z, Pivac N. Unraveling the biological mechanisms in Alzheimer’s disease--lessons from genomics. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35:340–7.
Zheng X, Demirci FY, Barmada MM, Richardson GA, Lopez OL, Sweet RA, et al. Genome-wide copy-number variation study of psychosis in Alzheimer’s disease. Transl Psychiatry. 2015;5:e574.
Zheng X, Demirci FY, Barmada MM, Richardson GA, Lopez OL, Sweet RA, et al. A rare duplication on chromosome 16p11.2 is identified in patients with psychosis in Alzheimer’s disease. PLoS One. 2014;9:e111462.
Szigeti K, Lal D, Li Y, Doody RS, Wilhelmsen K, Yan L, et al. Genome-wide scan for copy number variation association with age at onset of Alzheimer’s disease. J Alzheimers Dis. 2013;33:517–23.
Swaminathan S, Shen L, Kim S, Inlow M, West JD, Faber KM, et al. Analysis of copy number variation in Alzheimer’s disease: the NIALOAD/ NCRAD family study. Curr Alzheimer Res. 2012;9:801–14.
Swaminathan S, Huentelman MJ, Corneveaux JJ, Myers AJ, Faber KM, Foroud T, et al. Analysis of copy number variation in Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. PLoS One. 2012;7:e50640.
Rovelet-Lecrux A, Charbonnier C, Wallon D, Nicolas G, Seaman MN, Pottier C, et al. De novo deleterious genetic variations target a biological network centered on Abeta peptide in early-onset Alzheimer disease. Mol Psychiatry. 2015;20:1046–56.
Li Y, Shaw CA, Sheffer I, Sule N, Powell SZ, Dawson B, et al. Integrated copy number and gene expression analysis detects a CREB1 association with Alzheimer’s disease. Transl Psychiatry. 2012;2:e192.
Hooli BV, Mohapatra G, Mattheisen M, Parrado AR, Roehr JT, Shen Y, et al. Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology. 2012;78:1250–7.
Guffanti G, Torri F, Rasmussen J, Clark AP, Lakatos A, Turner JA, et al. Increased CNV-region deletions in mild cognitive impairment (MCI) and Alzheimer’s disease (AD) subjects in the ADNI sample. Genomics. 2013;102:112–22.
Chapman J, Rees E, Harold D, Ivanov D, Gerrish A, Sims R, et al. A genome-wide study shows a limited contribution of rare copy number variants to Alzheimer’s disease risk. Hum Mol Genet. 2013;22:816–24.
Bushman DM, Kaeser GE, Siddoway B, Westra JW, Rivera RR, Rehen SK, et al. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife. 2015;4:e05116.
Yamasaki M, Makino T, Khor SS, Toyoda H, Miyagawa T, Liu X, et al. Sensitivity to gene dosage and gene expression affects genes with copy number variants observed among neuropsychiatric diseases. BMC Med Genet. 2020;13:55.
Szigeti K. New genome-wide methods for elucidation of candidate copy number variations (CNVs) contributing to Alzheimer’s disease heritability. Methods Mol Biol. 2016;1303:315–26.
Shaw PX, Stiles T, Douglas C, Ho D, Fan W, Du H, et al. Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol Sci. 2016;3:196–221.
Sekine M, Makino T. Inference of causative genes for Alzheimer’s disease due to dosage imbalance. Mol Biol Evol. 2017;34:2396–407.
Schroter F, Sleegers K, Van Cauwenberghe C, Bohndorf M, Wruck W, Van Broeckhoven C, et al. Lymphoblast-derived integration-free iPSC lines from a female and male Alzheimer’s disease patient expressing different copy numbers of a coding CNV in the Alzheimer risk gene CR1. Stem Cell Res. 2016;17:560–3.
Saykin AJ, Shen L, Yao X, Kim S, Nho K, Risacher SL, et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: progress, opportunities, and plans. Alzheimers Dement. 2015;11:792–814.
Nelson PT, Fardo DW, Katsumata Y. The MUC6/AP2A2 locus and its relevance to Alzheimer’s disease: a review. J Neuropathol Exp Neurol. 2020;79:568–84.
Lew AR, Kellermayer TR, Sule BP, Szigeti K. Copy number variations in adult-onset neuropsychiatric diseases. Curr Genomics. 2018;19:420–30.
Keogh MJ, Wei W, Wilson I, Coxhead J, Ryan S, Rollinson S, et al. Genetic compendium of 1511 human brains available through the UK Medical Research Council Brain Banks Network Resource. Genome Res. 2017;27:165–73.
Kaeser G, Chun J. Brain cell somatic gene recombination and its phylogenetic foundations. J Biol Chem. 2020;295:12786–95.
Iorio A, Polimanti R, Calandro M, Graziano ME, Piacentini S, Bucossi S, et al. Explorative genetic association study of GSTT2B copy number variant in complex disease risks. Ann Hum Biol. 2016;43:279–84.
Byman E, Nagga K, Gustavsson AM, Netherlands Brain B, Andersson-Assarsson J, Hansson O, et al. Alpha-amylase 1A copy number variants and the association with memory performance and Alzheimer’s dementia. Alzheimers Res Ther. 2020;12:158.
Blauwendraat C, Wilke C, Simon-Sanchez J, Jansen IE, Reifschneider A, Capell A, et al. The wide genetic landscape of clinical frontotemporal dementia: systematic combined sequencing of 121 consecutive subjects. Genet Med. 2018;20:240–9.
Rustenhoven J, Smith AM, Smyth LC, Jansson D, Scotter EL, Swanson MEV, et al. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol Neurodegener. 2018;13:44.
Hong J, Arneson D, Umar S, Ruffenach G, Cunningham CM, Ahn IS, et al. Single-cell study of two rat models of pulmonary arterial hypertension reveals connections to human pathobiology and drug repositioning. Am J Respir Crit Care Med. 2021;203:1006–22.
Alakwaa FM. Repurposing didanosine as a potential treatment for COVID-19 using single-cell RNA sequencing data. mSystems. 2020;5(2):e00297–20.
Al Mahi N, Zhang EY, Sherman S, Yu JJ, Medvedovic M. Connectivity map analysis of a single-cell RNA-sequencing -derived transcriptional signature of mTOR signaling. Int J Mol Sci. 2021;22(9):4371.
Zhou X, Wang M, Katsyv I, Irie H, Zhang B. EMUDRA: Ensemble of Multiple Drug Repositioning Approaches to improve prediction accuracy. Bioinformatics. 2018;34:3151–9.
Acknowledgments
Not applicable.
Funding
This work was supported in parts by grants from the National Institutes of Health (NIH)/National Institute on Aging (U01AG046170, RF1AG054014, RF1AG057440, R01AG057907, U01AG052411, R01AG068030, RF1AG074010, R01DA051191, R01AG063819, R01DE029322, R01AG062355, R21AI149013 and R01AG062661).
Author information
Authors and Affiliations
Contributions
Conceptualization, B.Z.; Investigation, S.W., M.W., X.Z., Q.W., C.M., P.X., L.H., Y.Y. M.O., and G.Y.; Resources, B.Z.; Writing – Original Draft, All Authors.; Writing – Review & Editing, All Authors; Supervision, B.Z.; Funding Acquisition, B.Z., M.O. and G.Y. The author(s) read and approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors are consent to the publication of this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table S1.
The chr19 amplification region inferred in the Cluster 6 of late-pathology AD cases. Supplementary Table S2. Genes in the chr19 amplification region in the Cluster 6 of late pathology ADs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, M., Song, Wm., Ming, C. et al. Guidelines for bioinformatics of single-cell sequencing data analysis in Alzheimer’s disease: review, recommendation, implementation and application. Mol Neurodegeneration 17, 17 (2022). https://doi.org/10.1186/s13024-022-00517-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-022-00517-z