
Abstract
Despite decades of investigation into the genetics of autism spectrum disorder (ASD), a current consensus in the field persists that ASD risk is too heterogeneous to be diagnosed by a single set of genetic variants. As such, ASD research has broadened to include assessment of other molecular biomarkers implicated in the condition that may be reflective of environmental exposures or gene by environment interactions. Epigenetic variance, and specifically differential DNA methylation, have emerged as areas of particularly high interest to ASD, as the epigenetic markers from specific chromatin loci collectively can reflect influences of multiple genetic and environmental factors and can also result in differential gene expression patterns. This review examines recent studies of the ASD epigenome, detailing common gene pathways found to be differentially methylated in people with ASD, and considers how these discoveries may inform our understanding of ASD etiology. We also consider future applications of epigenetics in ASD research and clinical practice, focusing on substratification, biomarker development, and experimental preclinical models of ASD that test causality. In combination with other -omics approaches, epigenomics allows an improved conceptualization of the multifactorial nature of ASD, and opens future lines of inquiry for both basic research and clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Current knowledge of the autism spectrum disorder (ASD) epigenome implicates gene pathways that may contribute to condition development. |
Application of epigenomics to the fields of patient substratification, biomarker identification, and causality determination hold the potential to improve patient outcomes and understanding of heterogeneous ASD etiologies. |
1 Introduction
Autism spectrum disorder (ASD) is a diagnosis that encompasses a group of neurodevelopmental disorders characterized by persistent impairments in social interaction and language, together with repetitive patterns of behavior and interests [1]. ASD has been consistently observed to have a male bias, with ASD being reported as 4.2 times more prevalent in male children than female children [2]. Prevalence of ASD has been increasing in the US since the Centers for Disease Control and Prevention (CDC) first began monitoring in 1996, with the most recent surveillance report observing ASD prevalence at 23.0/1000 children (1 in 44 children) [2]. However, due to increased awareness of ASD and changing criteria for ASD diagnosis over this time frame, debate persists over whether this increase in prevalence is the result of biological or sociological factors [3,4,5]. Nonetheless, the sheer magnitude of the rise in ASD prevalence in recent decades warrants consideration of biological explanations for this increase, including consideration of environmental risk [5,6,7,8].
Parsing how much of the increase in ASD prevalence is attributable to environmental risk is made difficult by ASD being both highly heterogenous in its etiology and dependent on diagnosis through behavioral assessment. While ASD is defined by the presence of both social/language impairments and repetitive behaviors, these two parameters can be particularly difficult to assess in children under 3 years of age, leading to high potential for changing diagnosis even when adhering to ‘gold standard’ diagnostic tools such as the Autism Diagnostic Observation Schedule (ADOS) and the Autism Diagnostic Interview-Revised (ADI-R) [9]. Furthermore, 70% of individuals with ASD also have psychiatric comorbidities (including social anxiety and/or attention deficit and hyperactivity disorders), whose own behavioral traits can mask ASD [9, 10].
Given the difficulties of behavioral diagnosis of ASD, studies of the molecular etiology of the condition have attracted interest for their potential to identify quantifiable biomarkers that can act as metrics of ASD severity. Studies of ASD heritability have firmly established a genetic component of ASD etiology, with a 58–90% concordance rate in monozygotic twins [11, 12]. However, heritability estimates as well as specific genetic risk factors for ASD vary dramatically between studies, with insufficient evidence to designate a singular set of ASD-specific genetic risk factors [13,14,15,16]. Epigenetics, literally meaning ‘on top of genetics’, is defined as alterations to DNA or chromatin that can alter gene expression without a change to the DNA sequence. DNA methylation, which collectively refers to methyl groups added at cytosine bases in DNA, is the most investigated epigenetic mark in human tissues and will be the focus of this review. As a multifactorial disorder, ASD development is often a result of the interplay between genetic and environmental factors, which is what DNA methylation marks are frequently reflecting. Thus, a complete understanding of the molecular etiology of ASD requires knowledge of both genetics and epigenetics associated with the condition.
The genetic components of ASD risk have been thoroughly covered in prior publications [16,17,18,19,20]. As such, this review will focus on recent advances in the comparatively young field of ASD epigenetics, with the specific goal of envisioning future prospects of improved diagnosis and targeted behavioral therapy.
2 Autism Spectrum Disorder Epigenetics
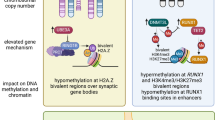
The insufficiency of solely considering genetic risk factors for the purpose of understanding ASD etiology has prompted a plethora of investigations into the environmental factors contributing to ASD development [21]. Various maternal medical conditions, including maternal prepregnancy body mass index (BMI) [22], diabetes [23], and hypertension [24], have been shown to increase the risk of ASD. Perinatal exposure to some common environmental pollutants, such as air pollution [25], and pesticides [26] have also shown an association with increased ASD risk. Some differences in maternal nutrients, including maternal iron supplement consumption [27] and maternal use of folic acid and multivitamin supplements [28], have also been shown to have potential protective effects against ASD development. These environmental exposures are often hypothesized to act at least partially through epigenetic mechanisms [29,30,31,32,33], although the tissue-specific and temporally influenced nature of epigenetic modifications can make repeated observation of epigenetic markers of ASD difficult. In spite of these inherent challenges, some progress has been made in identifying DNA methylation patterns from multiple tissue sources that distinguish ASD from typically developing controls (Fig. 1).

A summary of multiple genetic and environmental risk factors implicated in ASD that can be informed by distinct epigenetic profiles in both postmortem brain and perinatal tissues collected prior to diagnosis. ASD autism spectrum disorder, AP3S2 adaptor-related protein complex 3 subunit sigma 2, ARHGAP15 Rho GTPase-activating protein 15, BRSK2 brain-specific serine/threonine-protein kinase 2, CAMK2A calcium/calmodulin-dependent protein kinase IIα, CCDC1171/C90rf93 coiled coil domain containing 171, CD44 CD44 molecule (Indian blood group), CHST12 carbohydrate sulfotransferase 12, DGKZ diacylglycerol kinase zeta, DLGAP2 DLG-associated protein 2, EN2 engrailed homeobox 2, EPHA6 EPH receptor A6, ETS1 ETS proto-oncogene 1 transcription factor, FMR1 fragile X messenger ribonucleoprotein 1, GRIA3 glutamate ionotropic receptor AMPA type subunit 3, HDAC3 histone deacetylase 3, IL1B interleukin 1β, ISLR4 immunoglobulin superfamily containing leucine rich repeat, NHIP neuronal hypoxia-inducible placenta-associated, SATB2 SATB homeobox 2, SOCS2 suppressor of cytokine signaling 2, STK38L serine/threonine kinase 38 like, TSPAN32 tetraspanin 32, ZIC3 Zic family member 3. a Wong et al. [35]; b Nardone et al. [45]; c Ladd-Acosta et al. [43]; d Cheng et al. [48]; e James et al. [46]; f Corley et al. [37]; g Bakulski et al. [53]; h Mordaunt et al. [3]; i Zhu et al. [54] (full citations provided in the References section)
The neurodevelopmental origin of ASD makes post-mortem human brain tissue the most relevant tissue to provide direct epigenetic links between environmental exposures and altered genes in ASD. Insights gained from epigenetic changes associated with ASD in the brain therefore serve as the standard against which epigenetic data from other tissues are compared. Epigenomic analysis of post-mortem ASD brain DNA has focused largely on the cortex and cerebellum, as these regions of the brain have been implicated in prior examination of neural abnormalities associated with ASD [34,35,36,37,38] and are more commonly available from public brain banks. Among the most common methods for studying ASD epigenetics is the methylation array, which is manufactured to target regions of epigenetic interest in humans and uses hybridization-based fluorescent signaling to determine whether a given target nucleotide is methylated [39]. Despite the relative scarcity of available ASD brain tissue for investigations, methylation array-based analyses of epigenetic changes in the ASD brain have revealed a large number of differences at individual CpGs or differentially methylated regions (DMRs). These analyses rarely identify global differences in DNA methylation between ASD and control brains, regardless of the brain region surveyed.
Cerebral cortex has been the brain tissue of focus for multiple array-based ASD methylome analyses. For instance, Wong et al. extracted DNA from post-mortem prefrontal cortex and temporal cortex to perform region comethylation network analysis, revealing multiple comethylation modules significantly associated with ASD diagnosis [35]. Gene ontology (GO) pathway analysis of these comethylation modules revealed them to be enriched for pathways relating to immune functionality, synaptic signaling and regulation, and postsynaptic density, with these enrichments reflecting the known dual contributions of immune and neuronal dysregulation associated with ASD [40, 41]. Using an alternative approach of whole-genome bisulfite sequencing (WGBS), Vogel Ciernia et al. found similar gene pathway enrichments in ASD cortex, focusing on microglia as the source of the inflammatory signal [42]. Other array-based methylomic studies of ASD cortex have made similar findings. Ladd-Acosta et al. reported three DMRs in ASD temporal cortex, with these DMRs being proximal to genes involved in neuronal and immune pathways as well as maintaining imprinting patterns [43]. Nardone et al. identified differentially methylated CpGs in ASD prefrontal cortex samples, with GO analysis revealing hypomethylated CpGs enriched for immune functions and hypermethylated CpGs enriched for neuron-neuron synaptic activity [44]. Further ASD methylomic analysis of the sorted nuclei from frontal cortex neurons revealed differential regions of comethylation that were enriched for synaptic, neuronal, GABAergic and immune processes [45].
Methylomic analyses of cerebellum has shown similar general findings, with differences in genic locations of ASD differences. Corley et al. performed DNA methylation analysis of the subventricular zone, a component of the cerebellum that serves as a source of neuronal stem cells, and reported differentially methylated loci (DML) in ASD samples [37]. The observed DML were underrepresented at promoter regions and overrepresented at gene bodies and intergenic sequences, with GO analysis of the ASD-related DMLs showing enrichment of genes involved in neuron proliferation, differentiation, and migration. A targeted investigation of the ASD-associated and neurodevelopmentally linked EN2 gene by James et al. identified significant hypermethylation in the EN2 promoter region of ASD cerebellum samples relative to non-ASD samples [46]. In addition to ASD DMRs being identified in the cerebellum, differentially hydroxymethylated regions (DhMRs) have also been observed. Hydroxymethylated cytosines are the result of Tet protein interaction with methylated cytosines, and have been implicated to function in a manner unique from methylated cytosines in some biological functions [47]. Cheng et al. screened for DhMRs between ASD and non-ASD cerebellum and identified 797 DhMRs [48]. GO analysis of the 181 DhMRs that were identified in intragenic regions were found to be significantly enriched for ASD-implicated pathways such as ‘cell-cell communication’ and ‘heparan sulfate biosynthesis’, and cis functionality of the 599 intergenic DhMRs associated with ASD was predicted.
While studies of ASD-specific differential methylation in the brain are limited due to low sample availability, methylomic analysis of peripheral tissues does not suffer this same limitation. A wide range of investigations into DNA methylation patterns associated with ASD in non-neuronal tissue have been performed, with many of these studies using either ASD blood or maternal placenta as sources of DNA. While these peripheral tissue studies are less informative about consequential methylomic changes associated with ASD than studies on brain tissue, blood methylation levels at specific loci have been shown to be correlative with brain tissue methylation in prior studies [49, 50], and placental methylation levels have likewise been shown to associate with child ASD diagnosis [51,52,53,54].
Blood samples from individuals diagnosed with ASD versus matched controls offer an easily accessible sample type for interrogating the ASD epigenome, but questions have been raised about how well alterations in blood reflect the brain. ASD blood samples were demonstrated to have an impaired metabolic capacity for methylation [55], sparking further interest. Investigations of potential associations between blood DNA methylation and ASD have shown modest associations [56,57,58]. Of particular note is the meta-analysis of Andrews et al., where the authors conducted the largest methylome-wide association study on blood DNA ever performed, using samples from 1654 children [56]. No single CpG surveyed in this study was identified as being significantly associated with ASD, although seven CpGs passed the much lower threshold for suggestive association. A study from a fairly small subset of ASD-discordant monozygotic twin pairs showed some significant differentially methylated genes, with the significance of association decreasing when comparing ASD with non-ASD individuals more broadly [59]. Furthermore, the methylation chip used in this study screened for nearly 19x fewer CpG sites than the chips used in future studies, including Andrews et al. [60]. While blood DNA methylation profiles may well contain ASD-associated DMRs in small subsamples of highly genetically similar individuals, contemporary studies have shown them to be ill-suited to examining large and heterogenous groups.
In contrast to the somewhat limited utility of blood methylation studies from ASD patients, recent prospective investigations into the methylome of ASD placental, cord blood, and maternal blood samples have identified some consistent epigenetic changes that exist prior to the diagnosis of ASD. This is not particularly surprising given the importance of the placental in utero environment for fetal neurodevelopment [61, 62]. Mordaunt et al. performed WGBS of cord blood from a high-risk ASD cohort and identified 2299 DMRs across male and female samples, with ASD-associated DMRs being significantly enriched for methyl-sensitive developmental transcription factors that play a role in fetal neurodevelopment [63]. Sex-stratified DMRs associated with ASD were found to be highly enriched for X chromosome location, implying a potential epigenetic etiology to ASD’s male bias. Further implication of the role of the methylome in ASD development was reported by Zhu et al., where the authors performed WGBS of placenta samples and observed an ASD-associated 118 kb hypomethylated region at 22q13.33 [54]. This hypomethylated region was comprised of ASD-associated DMRs that were significantly enriched for multiple fetal brain enhancers and a novel gene named NHIP (neuronal hypoxia-inducible, placenta-associated). Bakulski et al. assessed the methylomes of both cord blood and placenta, finding nominally ASD-associated differentially methylated positions (DMPs) that correlated across both tissues [53]. Additionally, these DMPs were highly enriched for SFARI ASD risk genes. The authors also assessed the methylation of maternal whole blood DNA and found ASD DMPs that overlapped with those identified in placenta and cord blood. These results contribute to emerging evidence of conserved regions of cross-tissue methylomic profiles associated with neuropsychiatric disorders such as ASD [50, 51, 63, 64].
It is noteworthy that much of the existing methylomic analyses of ASD DNA (both in brain and peripheral tissues) are limited by the relatively narrow focus of the commercially available methylation array platforms. The vast majority of contemporary methylomic studies assess methylation with Illumina HumanMethylation450 or Infinium MethylationEPIC platforms, which provide coverage of only approximately 1.5–3% of all CpG sites in the human genome [60]. The targeted regions were determined by expert panels prior to whole methylome maps based on sequencing, introducing additional bias for particular regions of the genome with the least variation in methylation [65, 66]. As an example, > 80% of the ASD-associated DMRs identified in cord blood by WGBS did not overlap with a single probe represented on the Illumina HumanMethylation450 or Infinium MethylationEPIC platforms [63]. While these methylation array platforms do provide broad coverage of Refseq genes and CpG islands, the limited scope of their query should be kept in mind when considering potential ASD-differential methylation patterns outside of these locations.
Nonetheless, the current body of methylomic investigations of ASD already consists of numerous studies describing alterations in DNA methylation in gene pathways relevant to the ASD phenotype (summarized in Fig. 1). As future research deepens our understanding of the methylomic profile of ASD, epigenomic knowledge will be increasingly actionable, both in the clinic and in the laboratory. The remainder of this article discusses three areas of high interest in the future of ASD epigenetics: epigenetic substratification of ASD, predictive epigenetic biomarkers of ASD, and direct mechanistic investigation of epigenetic factors on ASD-related phenotypes in preclinical models.
3 Application of ASD Epigenetics in Substratification
The heterogeneity of symptomatic presentation in ASD poses a challenge to both researchers and clinical practitioners. While all ASD cases are broadly diagnosed by the presence of impairments in social behavior, language, and repetitive behavioral patterns, the manifestations and severity of these symptoms can vary substantially between individuals. Furthermore, the potential for common comorbid psychopathologies produces great difficulty in attempting to generalize studies of ASD individuals to the broader population [67, 68]. In response to this problem, clinicians and scientists have often attempted to stratify ASD individuals into subgroups for the purpose of studying more distinct manifestations of ASD, such as those individuals with or without intellectual disability. The identification of reliable, consistent subtypes of ASD reflective of psychological and biological phenomena has transformative potential, as it would enable more highly targeted studies of specific pathways involved in the manifestation of subphenotypes. Such an approach may also highlight subtype-specific treatment outcomes from behavioral intervention therapy with ASD youth [69, 70].
Unfortunately, review of an assortment of subtype stratification studies reports that many identified ASD subtypes are insufficiently verified by more than two strategies [71]. Furthermore, many employed substratification methods have been found to be over-reliant on symptom scores and cognitive measures, with few adequately including sensory processing and biological variables [72].
The incorporation of ASD-associated methylation patterns into substratification criteria, in combination with other data such as transcriptomics or metabolomics, has the potential to substantially enhance the biological considerations of substratification criteria. This is especially true with regard to discerning subtypes of idiopathic ASD, defined as cases of ASD arising from unknown causes [73]. In contrast to occurrences of ASD where a specific cause can be identified (known as syndromic ASD), idiopathic ASD describes the approximately 85% of ASD cases arising from an unknown combination of genetic and environmental factors. Epigenetic analyses are uniquely well-suited to uncover the mechanics of gene-environment interactions, and have been highlighted in reviews of neurodevelopmental conditions such as ADHD and anxiety disorders as a likely origin of condition risk that cannot be directly explained by genetic variation [74, 75].
While few studies have attempted to incorporate epigenetic data into ASD substratification, preliminary investigations of the epigenetic profiles associated with phenotypic ASD subtypes offer support for an epigenetic component in substratification criteria. Lee and Hu compared methylation profiles across three phenotypically distinct subtypes (severe language impairment, intermediate, and mild) of ASD lymphoblastoid cell lines, and observed that a greater number of significant DMRs were detected when comparing control cell lines with a specific phenotype subtype than were detected when comparing control cell lines to all pooled ASD samples [76]. Furthermore, functional analysis of subtype-specific DMRs revealed those DMRs to be enriched for subtype-specific biological pathways. For example, severe language impairment ASD DMRs were shown to be highly enriched for neuritogenesis genes, while the intermediate ASD DMRs were heavily populated with neuroinflammation genes and the mild ASD DMRs included genes involved in sensory system development. Epigenetic investigation of ASD subtyping holds the potential to uncover subtype-specific insights into ASD etiology that will enable researchers and clinicians alike to better understand the molecular dynamics of ASD’s various manifestations and etiologies (Fig. 2). In reducing the heterogeneity of ASD, these approaches may also empower large-scale ASD differential methylation analyses of peripheral tissue to uncover subtype-specific DMRs, even in the absence of more generalizable ASD DMRs [56, 58].

A hypothetical example showing how epigenetic substratification of ASD patients could facilitate subtype-specific interventions and improve patient outcomes. ASD autism spectrum disorder
4 Epigenetic ASD Biomarkers
ASD’s heterogeneity of presentation can also contribute to difficulties in diagnosis and treatment, especially for younger children. The average age of clinical diagnosis has been found to be 4–5 years [77, 78], although it is fairly common for youth with ASD with poor access to health care services to remain undiagnosed until early adolescence [79]. This is particularly concerning given the body of evidence indicating early behavioral interventions can confer substantial improvements to ASD children’s social, vocal, and adaptive skills that translate into increased thriving later in life [80,81,82]. Such interventions have already been proven to reduce symptom severity, and sometimes even diagnosis rates, when administered at approximately 12 months of age [83, 84].
One strategy to lessen the consequences of delayed ASD diagnosis is to identify ASD biomarkers that are predictive of risk for ASD prior to diagnosis. Infants at a higher risk could be offered behavioral intervention, followed up with behaviorally based ASD diagnoses at 3–5 years of age, a time when diagnosis is more informative (Fig. 3). Extensive studies in this field have identified a broad spectrum of ASD biomarkers across various sample types, but have generally failed to identify a consistent set of genetic differences collectively associated with ASD diagnosis [85,86,87]. In light of this ‘missing heritability problem’ [88], epigenetic biomarkers of ASD have been investigated as alternative predictive features for ASD diagnosis. Studies focusing on biomarkers detectable before the typical ASD diagnosis window hold particularly high value for potential ASD intervention, and some early investigations have shown success in this field.

Hypothetical example of how epigenetic ASD biomarkers can result in earlier ASD behavioral therapy interventions, and the potential reductions to ASD severity resulting from earlier therapy administration. ASD autism spectrum disorder, yrs years
Reports of significant ASD-associated methylation biomarkers detected early in development from accessible peripheral tissues, including buccal swabs [89] and placenta [51, 54], could be implemented in ASD risk factor calculations, thereby giving clinicians additional levels of evidence when attempting to make an early ASD diagnosis. Furthermore, the development of methods for assessing ASD biomarkers prenatally through the assessment of cell-free fetal DNA (cffDNA) would provide a window of opportunity for in utero interventions. CffDNA is derived from the placenta, representing circulating fetal genetic material deposited in the maternal bloodstream throughout gestation [90, 91]. This potential value is highlighted in a preprint by Laufer et al. describing how longitudinal cffDNA DMRs in a macaque model of maternal obesity was associated with behavioral measure and brain methylation patterns of offspring [92]. Additionally, these cffDNA and brain DMRs associated with maternal obesity were found to significantly overlap with DMRs in human ASD brain and placenta, a result that aligns with established knowledge of maternal obesity as an ASD risk factor [22, 93]. Larger studies searching for prenatal cffDNA epigenetic biomarkers of ASD in humans may likewise identify consistent signals associated with the likelihood of ASD development. Working in combination with established postnatal ASD biomarkers, these new tools would empower earlier and more accurate ASD diagnosis, and in turn allow clinicians to begin administering behavioral intervention therapies in the developmental window where they are proven to be most effective.
5 Direct Investigation of the Epigenetic Mechanisms of ASD
While additional investigations of existing associations between epigenetics and ASD will provide actionable results that support clinical practices such as substratification and biomarker development, they are fundamentally limited in the extent to which these correlative findings can uncover the direct mechanisms by which epigenetic alterations influence ASD phenotype. By their very nature, association studies of epigenetic alterations and ASD can only report differential epigenetic layers that associate with ASD phenotype. Even more direct interventions, such as knockout of chromatin-modifying enzymes [94, 95], are complicated due to the broad range of non-explicitly epigenetic functionality observed in such proteins [96,97,98]. Thus, these studies are capable of identifying loci whose epigenetics may play a role in ASD development but are incapable of identifying true causality [99].
However, the rapid proliferation of epigenetic editing technologies has begun to make casual studies between epigenetic alterations and phenotypes feasible [100,101,102]. Many laboratories have generated fusion proteins where a given deactivated DNA-targeting protein such as dCas9 is ligated to an effector domain capable of making chromatin or DNA methylation modifications. These fusion proteins are then introduced to a given organism, such as a mouse model or human cell line model, and researchers directly observe how the engineered modifications result in changes to organism phenotype [103]. Rather than being merely associative, these investigations using direct epigenetic editing of a locus make possible observations of causality (Fig. 4).

Hypothetical experiment investigating epigenetic mechanisms of ASD through phenotype rescue attempt using dCas9 fused to a DNMT. ASD autism spectrum disorder, dCas9-DNMT deactivated CRISPR-associated protein 9-deoxyribose nucleic acid methyltransferase, Me methyl functional group, DNMT DNA methyltransferase
Applied to ASD, these methods can prove helpful in narrowing the scope of epigenetic investigations to loci shown to have direct phenotypic impacts in other models. While human epigenetic editing of loci implicated in ASD remains clinically difficult [104] and ethically dubious [105, 106], epigenetic mechanism experiments in non-human models of ASD can reveal the degree to which a given locus’s chromatin conformation directly contributes to the ASD phenotype. Considering the growing list of epigenetically divergent loci associated with ASD, filtering for proven high-impact epigenetic variants will enable researchers to focus their studies on regions whose epigenetic profile is likely to be most impactful in endeavors such as substratification and biomarker discovery.
For example, a plethora of preclinical mouse models of rare genetic syndromic forms of ASD have been generated that display novel social, communicative, and behavioral symptoms [107]. While investigations of ASD epigenetic mechanism in these models have been undertaken using enzymatic inhibition [108,109,110], an additional degree of mechanistic certainty of the role a particular epigenetic mark plays in ASD development could be acquired by making precise epigenetic edits to loci of interest and observing downstream effects on both the molecular and phenotypic level. Lu et al. describe an early example of dCas9-based epigenetic investigation, with the authors reporting targeted methylation of the MECP2 transcription start site in mice-induced deficits in social behavior [111]. These tools could also be employed in developed ASD mouse models to study the potential for phenotypic rescue. Furthermore, one could imagine a study applying the CRISPRoff tool designed by Nuñez et al. to selectively repress loci of interest in the Myt1l+/- mouse model of ASD developed by Chen et al. [101, 112]. The Myt1l+/- mouse model was shown to replicate ASD-related social impairments in males, and also had greater chromatin accessibility in some regions of cortex than wild-type mice. By using an epigenetic editor such as CRISPRoff to target some of these more accessible regions in the Myt1l+/- mouse model, and observing any changes in ASD-related trait severity, direct hypotheses as to whether the action of the MYT1L gene in mouse ASD-like pathways is explicitly epigenetic could be tested. Such conclusions could then inform future human studies.
Unfortunately, the heterogeneous origins of idiopathic ASD makes it a more challenging target for direct model generation and subsequent mechanistic investigations [113]. Nonetheless, some attempts to generate non-syndromic rodent ASD models have been mildly successful at reproducing ASD phenotypes [114,115,116]. These non-syndromic models exhibit a wide range of neurodevelopmental impacts caused by environmental exposures and accumulation of numerus mutations implicated in altered neurodevelopment. These multifactorial etiologies mirror the etiologies of many idiopathic cases of ASD in the US. While limited in their direct translation to humans, non-syndromic ASD rodent models could offer a valuable tool for refinement for epigenetic variants of interest in future studies.
6 Conclusions
ASD is one of the most common neurodevelopmental conditions in the US, and understanding its etiology requires epigenetic investigations at the interface of genetics and environment. The elucidation of differentially methylated gene pathways in ASD brain and peripheral tissues has substantially improved our knowledge of ASD epigenetics, and future studies will build upon this knowledge in basic research clinical application. While this review focuses on a small handful of potentially high-impact applications for epigenetics in the ASD field, others exist that are not detailed in this article. Incorporated together with other -omics informed technologies and predictive machine learning algorithms, epigenetics can facilitate the next-generation of ASD care, informing healthcare decisions from gestation to adulthood.
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. American Psychiatric Association; 2013. https://doi.org/10.1176/appi.books.9780890425596.
Maenner MJ. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 Sites, United States, 2018. MMWR surveill summ. 2021. p. 70.
Zeidan J, et al. Global prevalence of autism: a systematic review update. Autism Res. 2022;15:778–90.
Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism. 2017;8:13.
Russell G, et al. Time trends in autism diagnosis over 20 years: a UK population-based cohort study. J Child Psychol Psychiatry. 2022;63:674–82.
Hertz-Picciotto I, Delwiche L. The rise in autism and the role of age at diagnosis. Epidemiology. 2009;20:84–90.
Matelski L, Van de Water J. Risk factors in autism: thinking outside the brain. J Autoimmun. 2016;67:1–7.
Ye BS, Leung AOW, Wong MH. The association of environmental toxicants and autism spectrum disorders in children. Environ Pollut. 2017;227:234–42.
Hus Y, Segal O. Challenges surrounding the diagnosis of autism in children. Neuropsychiatr Dis Treat. 2021;17:3509–29.
Simonoff E, et al. Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J Am Acad Child Adolesc Psychiatry. 2008;47:921–9.
Genovese A, Butler MG. Clinical assessment, genetics, and treatment approaches in autism spectrum disorder (ASD). Int J Mol Sci. 2020;21:4726.
Hallmayer J. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095.
Pizzo L, et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med. 2019;21:816–25.
Myers SM, et al. Insufficient evidence for “Autism-Specific” genes. Am J Hum Genet. 2020;106:587–95.
Srivastava S, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413–21.
Ramaswami G, Geschwind DH. Genetics of autism spectrum disorder. Published in Handbook of Clinical Neurology, (eds. Geschwind DH, Paulson HL, Klein C.), Neurogenetics. 147:321–9. (Elsevier Amsterdam 2018).
Vorstman JAS, et al. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. 2017;18:362–76.
de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med. 2016;22:345–61.
Hashem S, et al. Genetics of structural and functional brain changes in autism spectrum disorder. Transl Psychiatry. 2020;10:229.
Turner TN, et al. Genomic patterns of de novo mutation in simplex autism. Cell. 2017;171:710-722.e12.
Saxena R, Babadi M, Namvarhaghighi H, Roullet FI. Role of environmental factors and epigenetics in autism spectrum disorders. Published in Progress in Molecular Biologyand Translational Science, (eds. Ilieva M. and Lau WKW.), Autism. 173:35-60 (Elsevier Amsterdam, 2020).
Sanchez CE, et al. Maternal pre-pregnancy obesity and child neurodevelopmental outcomes: a meta-analysis. Obes Rev. 2018;19:464–84.
Wan H, Zhang C, Li H, Luan S, Liu C. Association of maternal diabetes with autism spectrum disorders in offspring: A systemic review and meta-analysis. Medicine (Baltimore). 2018;97: e9438.
Maher GM, et al. Association of hypertensive disorders of pregnancy with risk of neurodevelopmental disorders in offspring: a systematic review and meta-analysis. JAMA Psychiat. 2018;75:809.
Costa LG, Cole TB, Dao K, Chang Y-C, Garrick JM. Developmental impact of air pollution on brain function. Neurochem Int. 2019;131: 104580.
Bennett DH, et al. Environmental exposures to pesticides, phthalates, phenols and trace elements are associated with neurodevelopment in the CHARGE study. Environ Int. 2022;161: 107075.
Gerges P, et al. Risk and protective factors in autism spectrum disorders: a case control study in the Lebanese population. Int J Environ Res Public Health. 2020;17:6323.
Levine SZ, et al. Association of maternal use of folic acid and multivitamin supplements in the periods before and during pregnancy with the risk of autism spectrum disorder in offspring. JAMA Psychiat. 2018;75:176.
Sharp GC, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet. 2017;26:4067–85.
Ornoy A, Reece EA, Pavlinkova G, Kappen C, Miller RK. Effect of maternal diabetes on the embryo, fetus, and children: congenital anomalies, genetic and epigenetic changes and developmental outcomes: maternal diabetes and pregnancy outcome. Birth Defects Res Part C Embryo Today Rev. 2015;105:53–72.
Wang C, Geng H, Liu W, Zhang G. Prenatal, perinatal, and postnatal factors associated with autism: a meta-analysis. Medicine (Baltimore). 2017;96: e6696.
Raghavan R, et al. Maternal multivitamin intake, plasma folate and vitamin B12 levels and autism spectrum disorder risk in offspring. Paediatr Perinat Epidemiol. 2018;32:100–11.
Lintas C. Linking genetics to epigenetics: the role of folate and folate-related pathways in neurodevelopmental disorders. Clin Genet. 2019;95:241–52.
Bruchhage MMK, Bucci M-P, Becker EBE. Cerebellar involvement in autism and ADHD. In: Handbook of clinical neurology. 2108;155:61–72 (Elsevier, 2018).
Wong CCY, et al. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum Mol Genet. 2019;28:2201–11.
Ramaswami G, et al. Integrative genomics identifies a convergent molecular subtype that links epigenomic with transcriptomic differences in autism. Nat Commun. 2020;11:4873.
Corley MJ, et al. Epigenetic delay in the neurodevelopmental trajectory of DNA methylation states in autism spectrum disorders. Front Genet. 2019;10:907.
Fetit R, Hillary RF, Price DJ, Lawrie SM. The neuropathology of autism: a systematic review of post-mortem studies of autism and related disorders. Neurosci Biobehav Rev. 2021;129:35–62.
Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95.
Gilbert J, Man H-Y. Fundamental elements in autism: from neurogenesis and neurite growth to synaptic plasticity. Front Cell Neurosci. 2017;11:359.
Meltzer A, Van de Water J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacology. 2017;42:284–98.
Vogel Ciernia A, et al. Epigenomic convergence of neural-immune risk factors in neurodevelopmental disorder cortex. Cereb Cortex. 2020;30:640–55.
Ladd-Acosta C, et al. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry. 2014;19:862–71.
Nardone S, et al. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry. 2014;4:e433–e433.
Nardone S, Sams DS, Zito A, Reuveni E, Elliott E. Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb Cortex. 2017;27:5739–54.
James SJ, Shpyleva S, Melnyk S, Pavliv O, Pogribny IP. Complex epigenetic regulation of Engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl Psychiatry. 2013;3: e232.
Rustad SR, Papale LA, Alisch RS. DNA methylation and hydroxymethylation and behavior. Berlin: Springer; 2019. https://doi.org/10.1007/7854_2019_104.
Cheng Y, et al. 5-Hydroxymethylcytosine alterations in the human postmortem brains of autism spectrum disorder. Hum Mol Genet. 2018;27:2955–64.
Tylee DS, Kawaguchi DM, Glatt SJ. On the outside, looking in: a review and evaluation of the comparability of blood and brain “-omes.” Am J Med Genet B Neuropsychiatr Genet. 2013;162:595–603.
Lin D, et al. Characterization of cross-tissue genetic-epigenetic effects and their patterns in schizophrenia. Genome Med. 2018;10:13.
Zhu Y, et al. Placental DNA methylation levels at CYP2E1 and IRS2 are associated with child outcome in a prospective autism study. Hum Mol Genet. 2019;28:2659–74.
Bahado-Singh RO, Vishweswaraiah S, Aydas B, Radhakrishna U. Placental DNA methylation changes and the early prediction of autism in full-term newborns. PLoS ONE. 2021;16: e0253340.
Bakulski KM, et al. Autism-associated DNA methylation at birth from multiple tissues is enriched for autism genes in the early autism risk longitudinal investigation. Front Mol Neurosci. 2021;14: 775390.
Zhu Y, et al. Placental methylome reveals a 22q13.33 brain regulatory gene locus associated with autism. Genome Biol. 2022;23:46.
James SJ, et al. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004;80:1611–7.
Andrews SV, et al. Case-control meta-analysis of blood DNA methylation and autism spectrum disorder. Mol Autism. 2018;9:40.
Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10:1024–32.
Hannon E, et al. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 2018;10:19.
Wong CCY, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19:495–503.
Pidsley R, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208.
Mir IN, Leon R, Chalak LF. Placental origins of neonatal diseases: toward a precision medicine approach. Pediatr Res. 2021;89:377–83.
Rosenfeld CS. The placenta-brain-axis. J Neurosci Res. 2021;99:271–83.
Mordaunt CE, et al. Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Med. 2020;12:88.
Bakulski KM, Halladay A, Hu VW, Mill J, Fallin MD. Epigenetic research in neuropsychiatric disorders: the “Tissue Issue.” Curr Behav Neurosci Rep. 2016;3:264–74.
Zhou L, et al. Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing. Sci Rep. 2019;9:10383.
Gunasekara CJ, et al. A genomic atlas of systemic interindividual epigenetic variation in humans. Genome Biol. 2019;20:105.
Masi A, DeMayo MM, Glozier N, Guastella AJ. An overview of autism spectrum disorder, heterogeneity and treatment options. Neurosci Bull. 2017;33:183–93.
Siegel M, Beaulieu AA. Psychotropic medications in children with autism spectrum disorders: a systematic review and synthesis for evidence-based practice. J Autism Dev Disord. 2012;42:1592–605.
Uljarević M, et al. Exploring social subtypes in autism spectrum disorder: a preliminary study. Autism Res. 2020;13:1335–42.
Bohane L, Maguire N, Richardson T. Resilients, overcontrollers and undercontrollers: a systematic review of the utility of a personality typology method in understanding adult mental health problems. Clin Psychol Rev. 2017;57:75–92.
Agelink van Rentergem JA, Deserno MK, Geurts HM. Validation strategies for subtypes in psychiatry: a systematic review of research on autism spectrum disorder. Clin Psychol Rev. 2021;87:102033.
Wolfers T, et al. From pattern classification to stratification: towards conceptualizing the heterogeneity of autism spectrum disorder. Neurosci Biobehav Rev. 2019;104:240–54.
Casanova MF, et al. Editorial: secondary vs. idiopathic autism. Front Psychiatry. 2020;11:297.
Palladino VS, McNeill R, Reif A, Kittel-Schneider S. Genetic risk factors and gene–environment interactions in adult and childhood attention-deficit/hyperactivity disorder. Psychiatr Genet. 2019;29:63–78.
Schiele MA, Domschke K. Epigenetics at the crossroads between genes, environment and resilience in anxiety disorders: epigenetics in anxiety disorders. Genes Brain Behav. 2018;17: e12423.
Lee EC, Hu VW. Phenotypic subtyping and re-analysis of existing methylation data from autistic probands in simplex families reveal ASD subtype-associated differentially methylated genes and biological functions. Int J Mol Sci. 2020;21:6877.
Brett D, Warnell F, McConachie H, Parr JR. Factors affecting age at ASD diagnosis in UK: no evidence that diagnosis age has decreased between 2004 and 2014. J Autism Dev Disord. 2016;46:1974–84.
Neimy H, Pelaez M, Carrow J, Monlux K, Tarbox J. Infants at risk of autism and developmental disorders: establishing early social skills. Behav Dev Bull. 2017;22:6–22.
Malik-Soni N, et al. Tackling healthcare access barriers for individuals with autism from diagnosis to adulthood. Pediatr Res. 2021;91(5):1028–103. https://doi.org/10.1038/s41390-021-01465-y.
Kodak T, Bergmann S. Autism spectrum disorder. Pediatr Clin N Am. 2020;67:525–35.
Blanc R, et al. Early intervention in severe autism: positive outcome using exchange and development therapy. Front Pediatr. 2021;9: 785762.
Pierce K, et al. Get SET early to identify and treatment refer autism spectrum disorder at 1 year and discover factors that influence early diagnosis. J Pediatr. 2021;236:179–88.
Green J, et al. Randomised trial of a parent-mediated intervention for infants at high risk for autism: longitudinal outcomes to age 3 years. J Child Psychol Psychiatry. 2017;58:1330–40.
Whitehouse AJO, et al. Effect of preemptive intervention on developmental outcomes among infants showing early signs of autism: a randomized clinical trial of outcomes to diagnosis. JAMA Pediatr. 2021;175: e213298.
Janšáková K, Kyselicová K, Ostatníková D, Repiská G. Potential of salivary biomarkers in autism research: a systematic review. Int J Mol Sci. 2021;22:10873.
Wang Y, et al. A proteomic analysis of urine biomarkers in autism spectrum disorder. J Proteom. 2021;242: 104259.
Bjørklund G, et al. Diagnostic and severity-tracking biomarkers for autism spectrum disorder. J Mol Neurosci. 2018;66:492–511.
Génin E. Missing heritability of complex diseases: case solved? Hum Genet. 2020;139:103–13.
Gui A, et al. Leveraging epigenetics to examine differences in developmental trajectories of social attention: a proof-of-principle study of DNA methylation in infants with older siblings with autism. Infant Behav Dev. 2020;60: 101409.
Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science. 2021;372:eaaw3616.
Chiu RWK, Lo YMD. Cell-free fetal DNA coming in all sizes and shapes. Prenat Diagn. 2021;41:1193–201.
Laufer BI, et al. Multi-omic brain and behavioral correlates of cell-free fetal DNA methylation in macaque maternal obesity models. The preprint can be found at bioRxiv, Preprint 2021. Accepted for publication at Nature Communications. https://doi.org/10.1101/2021.08.27.457952.
Lei X-Y, Li Y-J, Ou J-J, Li Y-M. Association between parental body mass index and autism spectrum disorder: a systematic review and meta-analysis. Eur Child Adolesc Psychiatry. 2019;28:933–47.
Grosswendt S, et al. Epigenetic regulator function through mouse gastrulation. Nature. 2020;584:102–8.
Michowski W, et al. Cdk1 controls global epigenetic landscape in embryonic stem cells. Mol Cell. 2020;78:459-476.e13.
Dimitrova E, Turberfield AH, Klose RJ. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015;16:1620–39.
Wu Z, Connolly J, Biggar KK. Beyond histones—the expanding roles of protein lysine methylation. FEBS J. 2017;284:2732–44.
Morgan MAJ, Shilatifard A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat Genet. 2020;52:1271–81.
Stricker SH, Köferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18:51–66.
Liu XS, et al. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172:979-992.e6.
Nuñez JK, et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184:2503-2519.e17.
Xu X, et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat Commun. 2018;9:3509.
Gomez JA, Beitnere U, Segal DJ. Live animal epigenome editing: convergence of novel techniques. Trends Genet. 2019;35527–541.
Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. 2018;17:641–59.
Ripamonti L. Disability, diversity, and autism: philosophical perspectives on health. New Bioeth. 2016;22:56–70.
Schuck RK, et al. Neurodiversity and autism intervention: reconciling perspectives through a naturalistic developmental behavioral intervention framework. J Autism Dev Disord. https://doi.org/10.1007/s10803-021-05316-x(Epub 13 Oct 2021).
Bey AL, Jiang Y. Overview of mouse models of autism spectrum disorders. Curr Protoc Pharmacol. 2014;66:5.66.1–26.
Qin L, et al. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat Neurosci. 2018;21:564–75.
Clark DF, Schmelz R, Rogers N, Smith NE, Shorter KR. Acute high folic acid treatment in SH-SY5Y cells with and without MTHFR function leads to gene expression changes in epigenetic modifying enzymes, changes in epigenetic marks, and changes in dendritic spine densities. PLoS ONE. 2021;16: e0245005.
Shen T, et al. Brain-specific deletion of histone variant H2A.z results in cortical neurogenesis defects and neurodevelopmental disorder. Nucleic Acids Res. 2018;46:2290–2307.
Lu Z, et al. Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death Dis. 2020;11:85.
Chen J, et al. A MYT1L syndrome mouse model recapitulates patient phenotypes and reveals altered brain development due to disrupted neuronal maturation. Neuron. 2021;109:3775-3792.e14.
Varghese M, et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol (Berl). 2017;134:537–66.
Nicolini C, Fahnestock M. The valproic acid-induced rodent model of autism. Exp Neurol. 2018;299:217–27.
Haddad FL, Patel SV, Schmid S. Maternal immune activation by poly I: C as a preclinical model for neurodevelopmental disorders: a focus on autism and schizophrenia. Neurosci Biobehav Rev. 2020;113:546–67.
Meyza KZ, Blanchard DC. The BTBR mouse model of idiopathic autism—current view on mechanisms. Neurosci Biobehav Rev. 2017;76:99–110.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research has been supported by NIH R01 ES029213, a Cultivating Team Science Award from UC Davis School of Medicine, and a fellowship from the Perinatal Origins of Disparities IMPACT Center to LAW.
Conflicts of interest
Logan A. Williams declares no conflicts of interest. Janine M. LaSalle has submitted a patent application on ‘Methods and Compositions for Determining Risk of Autism Spectrum Disorders’.
Author contributions
LAW and JML conceived, planned, and wrote the manuscript, and both authors contributed to the article and approved the submitted version.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Code availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Williams, L.A., LaSalle, J.M. Future Prospects for Epigenetics in Autism Spectrum Disorder. Mol Diagn Ther 26, 569–579 (2022). https://doi.org/10.1007/s40291-022-00608-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-022-00608-z