
Abstract
Background
Aortic arch abnormalities (AAA) are abnormal embryologic developments of the aorta and its branches. Their outcomes often depend on their association with other congenital diseases and genetic testing results.
Objective
This study aimed to evaluate the yield of chromosomal microarray analysis (CMA) in fetuses with different patterns of AAA and normal karyotype.
Methods
Data from 158 pregnancies referred for prenatal CMA testing due to fetal AAA were obtained between April 2016 and April 2019. Fetuses with isolated AAA, AAA accompanied by soft ultrasound markers, and AAA with other ultrasound malformations were classified into groups A, B, and C, respectively. Cases with detectable karyotype aberrations were excluded from the study.
Results
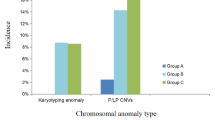
Twenty cases (12.7%) of submicroscopic anomalies were detected in 158 cases with normal karyotype, comprising 16 cases (10.1%) of clinically significant variants, two cases (1.3%) of variants of unknown significance, and two variants (1.3%) that were likely benign. Microdeletion of 22q11.2 accounted for 25% (4/16) of the clinically significant variants. The overall incremental yields by CMA in group A, group B, and group C were 1.8%, 2.3%, and 24.1%, respectively. Except for double aortic arch, the incremental yield of clinical significant findings for each type of AAA in group C was much higher than that in group A and group B. In group A, a clinically significant variant was only detected in one fetus with right aortic arch (RAA) (1.8%, 1/57).
Conclusions
In addition to 22q11.2 microdeletion, many other clinically significant submicroscopic variants are present in fetuses with AAA, especially in fetuses with other ultrasound malformations. Although CMA is always recommended in the presence of any malformation in many countries, our results suggest insufficient evidence to recommend CMA in fetuses with isolated AAA, except for isolated RAA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Chromosomal microarray analysis (CMA) increases the detection rate of clinically significant aberrations in fetuses with aortic arch abnormalities (AAA) compared with karyotyping. |
It is of great importance to perform ultrasound screening for other systems when AAA is encountered. |
There was insufficient evidence to offer CMA in fetuses with isolated aberrant right subclavian artery, coarctation of the aorta, and double aortic arch, but it is advocated in isolated right aortic arch and AAA accompanied with additional ultrasound abnormalities. |
1 Introduction
Fetal aortic arch abnormalities (AAA) refer to position, length, and size abnormalities of the aortic arch and its branching vessels. These aberrations are the results of abnormal embryologic development of branchial arches, with a prevalence of approximately 1–3% [1, 2]. There are diverse patterns of aortic arch malformations. Common AAA patterns include right aortic arch (RAA), left aortic arch with aberrant right subclavian artery (LAA-ARSA), double aortic arch (DAA), coarctation of the aorta (CoA), and aortic arch interruption. They can occur in isolation or in association with cardiac defects and/or non-cardiac abnormalities [3, 4].
Many scholars have analyzed different patterns of AAA separately. Lodeweges et al. [5] analyzed an adult survival cohort with different types of congenital AAA, and provided the following information: most isolated AAA are asymptomatic and require no surgical therapy. Dyspnea was the most common syndrome associated with AAA, followed by gastrointestinal syndrome. They can be relieved by surgery; however, in prenatal settings, the premise is that there are no genetic abnormalities. Published studies on genetic testing for AAA have mainly focused on aberrant right subclavian artery (ARSA) and RAA. Their association with trisomy 21 (T21) as well as with 22q11 microdeletion detected by fluorescent in situ hybridization (FISH) has been reported frequently in both prenatal and postnatal settings [6,7,8,9,10,11,12]. Chromosomal microarray analysis (CMA) has become a front-line method for the genetic detection of fetal heart defects, because of its ability to detect both microscopic and submicroscopic chromosomal aberrations. It has been widely accepted for use in fetuses with AAA accompanied with cardiac anomalies or non-cardiac malformations, but its use in the management of cases with an isolated AAA remains controversial. For fetuses with ARSA as an isolated finding, an invasive procedure for CMA testing is not indicated, unless additional ultrasound abnormalities are detected [10, 13]. Some authors have recommended karyotyping and FISH for chromosome 22q11.2 microdeletion screening in all fetuses with RAA [3, 14]. The present study aimed to evaluate the implications of CMA in fetuses with various patterns of AAA with or without cardiac and/or non-cardiac malformations, and to provide incidence data for pregnancy guidance and fetal prognosis evaluation.
2 Materials and Methods
2.1 Patients and Samples
This retrospective study reviewed 204 consecutive pregnant women referred to our center because of fetal AAA accompanied with or without cardiac/non-cardiac malformations between August 2016 and May 2019. CMA was not performed in 30 cases. The other 174 cases underwent traditional karyotyping and CMA analysis concurrently, among whom, 16 cases with karyotype-detectable CMA findings were excluded from the study. As a result, 158 cases were enrolled in the study. The specimens included 112 cases with amniotic fluid obtained during 19 and 24 gestational weeks as well as 46 cases with umbilical cord blood obtained during 25 and 32 gestational weeks. The demographic characteristics are presented in Table 1. According to the ultrasound findings, the enrolled 158 patients were classified into three groups: isolated AAA (group A), AAA accompanied with soft ultrasound markers (group B), and AAA accompanied with other ultrasound malformations (group C). In group B, AAA with soft ultrasound markers was identified in 43 cases, and the most common soft ultrasound markers were echogenic intracardiac focus (34.9%, 15/43) and heart valve regurgitation (34.9%, 15/43). In group C, 58 fetuses had additional ultrasound malformations; among them, the most frequent cardiac anomaly was ventricular septal defect (VSD) (44.8%, 26/58), followed by persistent left superior vena cava (PLSVC) (17.2%, 10/58). The most frequent non-cardiac anomaly was fetal growth retardation (FGR) (13.8%, 8/58) (assigned when fetal weight, using the formula of Hadlock, was below the 10th percentile for gestational age), followed by urinary system anomalies (8.6%, 5/58) and skeletal system abnormalities (6.9%, 4/58).
A clinical and ultrasound examination was suggested in surviving infants. Follow-up assessments were performed via clinical records or a telephone call, with the age of the infant at follow-up ranging from 5 to 24 months. The study was approved by the Ethics Committee of Fujian Provincial Maternity and Children’s Hospital. Written informed consent to participate in the study was obtained from each patient.
2.2 DNA Extraction and CMA Platforms
Genomic DNA was extracted from uncultured or cultured amniotic fluid and fetal cord blood using a QIAGEN kit (Qiagen, Germany) according to the manufacturer’s instructions. CMA was performed using a whole genome-wide Affymetrix CytoScan 750 K array (Affymetrix Inc., Santa Clara, CA, USA), which includes 200,000 probes for single nucleotide polymorphisms and 550,000 probes for copy number variations distributed across the entire human genome.
2.3 Data Interpretation
To analyze the results, Chromosome Analysis Suite software (Affymetrix) and human genome version GRCh37 (hg19) were used. A resolution was generally applied: gains or losses of ≥ 400 kb and loss of heterozygosity (LOH) ≥ 10 Mb. Uniparental disomy (UPD) was reported based on the identification of the region of homozygosity (ROH) covering the entire chromosome. UPDtool was used for genome-wide detection of UPD within the child–parent trios to confirm maternal or paternal UPD origin. All detected copy number variants (CNVs) were compared with in-house and national public CNV databases as follows: Database of Genomic Variants (DGV), Database of Chromosome Imbalance and Phenotype in Humans Using Ensemble Resources (DECIPHER), International Standards for Cytogenomic Arrays Consortium, and Online Mendelian Inheritance in Man (OMIM).
The incremental yield of CMA was defined as the yield of CMA over traditional karyotyping. The CMA results were classified into five levels according to the American College of Medical Genetics (ACMG) definitions [15]: pathogenic, benign, likely pathogenic, likely benign, and variants of unknown significance (VOUS). All of these results have been reported for patients. Submicroscopic (< 7 Mb) pathogenic/likely pathogenic CNVs were considered clinically significant. Parental CMA was recommended to determine the inheritance of CNVs. In general, CNVs inherited from normal phenotype parents were regarded as likely benign, whereas de novo fetal mutations were regarded as likely pathogenic. If the CNV had been reported to have incomplete penetrance and/or variable expressivity, we considered it a likely pathogenic variant, even though it was inherited from a parent with a normal phenotype.
2.4 Statistical Analysis
The data were analyzed using SPSS software v19.0 (SPSS Inc., Chicago, IL, USA). Statistical comparisons were performed using the Chi squared test, and p < 0.05 was considered statistically significant.
3 Results
Twenty cases (12.7%) of submicroscopic abnormalities were detected in the 158 cases with normal karyotype, and the details are summarized in Table 2. Among them, 1.3% (2/158) were variants of likely benign CNVs, 1.3% (2/158) were variants of VOUS, and 10.1% (16/158) were variants of clinical significance, which included nine cases of pathogenic CNVs, five cases of likely pathogenic CNVs, and two cases of UPD, as shown in Fig. 1. CNVs of clinical significance (n = 14) ranged in size from 507 kb to 7.4 Mb, and ten of them were related to clinical syndromes: DiGeorge syndrome (OMIM # 611867, cases 2, 3, 6, and 13), Potocki-Lupski syndrome (OMIM # 610883, cases 1 and 7), 1q21.1 duplication syndrome (OMIM # 612475, case 4), 8q21.11 deletion syndrome (OMIM # 614230, case 9), Williams-Beuren syndrome (OMIM # 194050, case 10), and 22q11.2 duplication syndrome (OMIM # 608363, case 14). Aberrations derived from 22q11.2, comprising four cases of deletion and one case of duplication, were observed with the highest frequency among the aberrations of clinical significance (25.0%, 4/16). In addition to CNVs of clinical significance, CMA yielded two cases of UPD from fetuses with AAA combined with intra- and non-cardiac abnormalities. Case 15 showed a maternal UPD (2) that affected the entirety of chromosome 2. Case 16 revealed a maternal UPD (16) involving 10.3 Mb and 19.2 Mb in regions 16q23.2q24.3 and 16p13.3p12.3, respectively. Of note, both fetuses manifested as FGR.

Flow chart illustrating the CMA findings of fetuses with AAA. AAA aortic arch abnormalities, CMA chromosomal microarray analysis, CoA coarctation of the aorta, LAA-ARSA left aortic arch with aberrant right subclavian artery, RAA right aortic arch, UPD uniparental disomy, VOUS variants of unknown significance
Of the enrolled 158 cases, the most frequent pattern was LAA-ARSA, accounting for 51.9% (82/158), followed by CoA (24.1%, 38/158), RAA (20.9%, 33/158), which included RAA with aberrant left subclavian artery (RAA-ALSA) as well as RAA with mirror-image branching, and DAA (3.16%, 5/158); their incremental yield of clinically significant findings was 4.9%, 21.1%, 12.1%, and 0%, respectively. Except for DAA, the incremental yield of clinical significant findings for each type of AAA in group C was much higher than that in group A and group B. A detailed summary is presented in Table 3. In group A, one out of 57 cases (1.8%) with isolated AAA yielded clinically significant results; the case was a fetus with RAA with mirror-image branching (Table 2, case 8). In group B, only one case of clinically significant findings was noted in a fetus with ARSA accompanied by echogenic cardiac focus and mild tricuspid regurgitation (Table 2, case 12), contributing to an incremental yield of 2.3%. In group C, 14 aberrations of clinical significance were identified, contributing to an incremental yield of 24.1%, comprising three cases of ARSA, three cases of RAA, and eight cases of CoA. It is worth noting that in group C, 35 patients had additional cardiac anomalies, ten cases had non-cardiac anomalies, and 13 cases had both non-cardiac and cardiac anomalies; their frequencies of clinically significant CMA findings showed no significant difference (17.1% vs. 20.0% vs. 46.2%, p > 0.05).
Follow-up was obtained in 143 (90.5%) of our patients. Thirty-three fetuses were terminated or died in utero. All surviving infants had regularly visited a professional pediatrician. During the follow-up period, three cases with CoA and intracardiac malformations confirmed by postnatal echocardiography underwent cardiac surgery; one infant with ARSA as well as VSD showed developmental delay; one infant with isolated ARSA showed thyroid hypofunction; the rest of the cases showed no obvious phenotypic abnormality. Postnatal echocardiography was performed in only 38 cases; among them, prenatal diagnosis was confirmed in nine out of 13 fetuses (69.2%) with CoA, 13 out of 15 fetuses (86.7%) with RAA, two out of two cases (100%) with DAA, as well as seven out of eight cases (87.5%) with ARSA.
4 Discussion
The traditional classification of AAA was described by Crawford in 1964 [16]. In the present study, the enrolled cases mainly involved four common patterns: RAA, LAA-ARSA, CoA, and DAA.
The 22q11.2 microdeletion was considered to be the most relevant abnormality in previous studies on the correlation between various AAA and submicroscopic chromosomal abnormalities [17]. As isolated findings, the relationship is controversial [3, 8, 10,11,12,13,14, 18, 19]. In the current study, 22q11.2 was the most frequently affected region, with four cases of microdeletion and one case of microduplication. The 22q11.2 microdeletion accounted for 25% of fetuses with clinically significant submicroscopic chromosomal abnormalities. Among them, none was found in the isolated AAA group. One was found in the fetus with ARSA and a soft ultrasound marker (case 5); the fetus had echogenic cardiac focus and mild tricuspid regurgitation, and the pregnancy was terminated. The remaining three fetuses had additional cardiac anomalies. Some studies have suggested that the risk is significantly increased when the thymus is small or cannot be visualized and when extra-cardiac abnormalities are present [4, 19, 20]. In our study, only the fetus with 22q11.2 deletion detected in group C had thymus dysplasia, which was present in two cases of the whole cohort. Another variant that occurred with high frequency was 17p11.2 microduplication, which was present in two fetuses. One fetus with RAA with mirror-image branching was the only case of submicroscopic anomaly detected in group A (case 1). Another case was reported in a fetus with ARSA and strephenopodia (case 7). This aberration has not been reported in previous studies, especially for fetuses with isolated AAA. The duplication of 17q11.2 included the gene RAI1, whose overexpression has been linked to Potocki-Lupski syndrome (OMIM # 610883), resulting in various congenital abnormalities including developmental delays, autism, intellectual disability, cardiovascular anomalies, and certain other structural anomalies [21, 22]. Our findings suggest that, other than 22q11.2 microdeletion, more submicroscopic pathogenic aberrations may occur in isolated AAA.
The risk of genetic abnormalities increased when AAA was combined with cardiac and/or non-cardiac abnormalities. Pathogenic/likely pathogenic submicroscopic variants involved in clinical syndrome were also found in eight fetuses. As shown in Table 2, these fetuses all had additional cardiac anomalies except for case 10. Case 10 revealed Williams-Beuren syndrome, a well-defined multisystem disorder that is caused by a chromosome 7q11.23 deletion. As previously described, congenital cardiovascular defects are the most clinically significant in 80% of patients with Williams-Beuren syndrome, which is caused by haplo insufficiency in the elastin (ELN) [23, 24]. The ultrasound feature of case 10 was CoA and FGR, which was also reported in a study by Yuan et al. [25]. In their study, the researchers found that the most common ultrasound manifestations of William-Beuren syndrome in prenatal cases were FGR and congenital cardiovascular abnormalities mainly involving supravalvular aortic stenosis, VSD, or aortic coarctation. 1q21.1 duplication syndrome was detected in one fetus with RAA, right ventricular hypoplasia, and pulmonary stenosis (case 4). According to previous studies, 1q21.1 microduplication can be found in individuals with normal or abnormal phenotypes. Most patients were reported to be neurologically abnormal, mainly characterized by developmental delay, intellectual disabilities, or autism spectrum disorder [26, 27], contributing to 1q21.1 duplication syndrome; congenital heart defect was less commonly observed [28]. One fetus with CoA, VSD, increased spine curvature, and FGR revealed a 7.3-Mb deletion on 8q21.11, associated with 8q21 deletion syndrome. The syndrome is mainly characterized by intellectual disability and common facial dysmorphic features [29, 30], and congenital heart defects have also been rarely reported [31]. The fetus in our study died in utero at 34 gestational weeks, with no obvious abnormalities in the appearance of the delivered fetus.
Other rare variants of clinical significance, such as CNVs in the region of 2q13 and 18q23, have been reported to contribute to congenital heart disease [32,33,34]. The deletion of 2q13 encompasses the FBLN7 and TMEM87B genes, which account for cardiac defects; loss of FBLN7 may be responsible for impaired branchial arch development [33]. Although the deletion was inherited from the healthy parent, considering the incomplete penetrance and abnormal ultrasound features, the variant was classified as likely pathogenic, and the pregnancy was terminated. As for the deletion of 8q23.1q23.2 and 15q11.2 in this series, there are no previous studies about their association with congenital heart defects, and we considered them to be incidental findings. In addition, UPD was detected in two cases of CoA accompanied by cardiac anomalies and FGR. The pathogenesis of UPD is determined by both epigenetic imprinting and unmasking of autosomal-recessive diseases. Currently, two maternally expressed imprinted genes are located on chromosome 16: ZNF597 and NAA60. Fetuses with maternal UPD (16) were found in most cases to have FGR, and also in a few cases had malformations like cardiac defects [35, 36]. Similar to UPD (16), maternal UPD (2) was mainly reported in fetuses with FGR [37, 38], but fetal heart defects have not been reported previously, and they may also be an incidental finding.
Isolated ARSA is frequently considered to be benign and not associated with T21 or 22q11.2 deletion [18]. Some scholars have even concluded that CMA had no additive value in such cases, but a detailed ultrasound scan should be performed to further evaluate the risk of genetic abnormalities [10, 13]. But some scholars suggested that CMA should be considered in cases of prenatally diagnosed fetal cardiovascular malformations, even if the lesion is apparently isolated based on prenatal imaging [39]. Our findings are compatible with the former viewpoints. Compared to ARSA, the association of 22q11.2 microdeletion and RAA was more frequently documented. Some authors suggested that genetic testing, especially CMA, should be advocated regardless of co-existing malformations [8, 11, 12, 14, 19]. In previous studies, the incidence of clinically significant submicroscopic variants was variable. Ruan et al. reported an incidence of 5.2% in RAA fetuses with normal karyotype, all of which were 22q11.2 microdeletions [14]. In the study by Vigneswaran et al., an incidence of 8.6% was found in 69 apparently isolated RAA with normal karyotype, and the most common was a 22q11.2 microdeletion [40]. In our study, the incidence in isolated RAA fetuses and the total cohort was 7.7% and 12.5%, respectively. It is worth mentioning that various aberrations other than 22q11.2 microdeletion were present in our study, possibly because many additional ultrasound anomalies were also at high risk of genetic abnormalities. As far as we know, there have been few studies about the relationship between genetic abnormalities and fetal CoA, which may partly be because of insufficient accuracy for prenatal diagnosis of CoA, and its occurrence was always accompanied with other cardiac abnormalities. In our study, seven out of the 16 cases of clinically significant abnormalities were fetuses with CoA, accounting for the highest proportion. They all had additional ultrasound anomalies, and only 69.2% of the CoA cases who underwent postnatal echocardiography were confirmed. Therefore, the relationship between genetic abnormalities and CoA could not be assessed. In group C, most cases had both cardiac and non-cardiac anomalies, and the incidence of clinically significant submicroscopic variants was as high as 46.2%, which further emphasizes the importance of ultrasound screening for other systems when AAA is encountered.
Our study has several limitations. Firstly, it is a retrospective study and not all patients with AAA accepted CMA, especially the fetus with apparently isolated AAA. Secondly, not all fetal AAA were confirmed after birth or termination. Many parents were not willing to conduct echocardiography for their infants with no obvious symptom at early age. We should accept that, potentially, there may have been some misdiagnoses. Finally, the inability to detect single gene point mutations associated with AAA is a limitation of CMA; therefore, clinicians should be aware that karyotyping and CMA cannot find all the genetic aberrations.
In summary, our research suggests that CMA increases the diagnostic yield of clinically significant aberrations in fetuses with AAA compared to karyotyping, especially in fetuses with other ultrasound malformations. There was insufficient evidence to offer CMA in fetuses with isolated ARSA, CoA, and DAA, but it is advocated in isolated RAA and AAA accompanied with additional ultrasound abnormalities.
References
Licari A, Manca E, Rispoli GA, Mannarino S, Pelizzo G, Marseglia GL. Congenital vascular rings: a clinical challenge for the pediatrician. Pediatr Pulmonol. 2015;50(5):511–24.
Yoo SJ, Min JY, Lee YH, Roman K, Jaeggi E, Smallhorn J. Fetal sonographic diagnosis of aortic arch anomalies. Ultrasound Obst Gynecol. 2003;22(5):535–46.
Miranda JO, Callaghan N, Miller O, Simpson J, Sharland G. Right aortic arch diagnosed antenatally: associations and outcome in 98 fetuses. Heart. 2014;100(1):54–9.
Razon Y, Berant M, Fogelman R, Amir G, Birk E. Prenatal diagnosis and outcome of right aortic arch without significant intracardiac anomaly. J Am Soc Echocardiogr. 2014;27(12):1352–8.
Lodeweges JE, Dikkers FG, Mulder BJM, Roos-Hesselink JW, Vliegen HW, van Dijk APJ, et al. The natural and unnatural history of congenital aortic arch abnormalities evaluated in an adult survival cohort. Can J Cardiol. 2019;35(4):438–45.
Chaoui R, Heling KS, Sarioglu N, Schwabe M, Dankof A, Bollmann R. Aberrant right subclavian artery as a new cardiac sign in second- and third-trimester fetuses with Down syndrome. Am J Obstet Gynecol. 2005;192(1):257–63.
Rauch R, Rauch A, Koch A, Zink S, Kaulitz R, Girisch M, et al. Laterality of the aortic arch and anomalies of the subclavian artery-reliable indicators for 22q112 deletion syndromes? Eur J Pediatr. 2004;163(11):642–5.
McElhinney DB, Clark BJ 3rd, Weinberg PM, Kenton ML, McDonald-McGinn D, Driscoll DA, et al. Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J Am Coll Cardiol. 2001;37(8):2114–9.
Pico H, Mancini J, Lafouge A, Bault JP, Gorincour G, Quarello E. Prenatal associated features in fetuses diagnosed with an aberrant right subclavian artery. Fetal Diagn Ther. 2016;40(3):187–94.
Maya I, Kahana S, Yeshaya J, Tenne T, Yacobson S, Agmon-Fishman I, et al. Chromosomal microarray analysis in fetuses with aberrant right subclavian artery. Ultrasound Obstet Gynecol. 2017;49(3):337–41.
Berg C, Bender F, Soukup M, Geipel A, Axt-Fliedner R, Breuer J, et al. Right aortic arch detected in fetal life. Ultrasound Obst Gynecol. 2006;28(7):882–9.
Yerlikaya G, Efeturk T, Springer S, Reischer T. Prenatal detection of right aortic arch. Arch Gynecol Obstet. 2019;299(4):933–8.
Sagi-Dain L, Singer A, Josefsberg S, Peleg A, Lev D, Samra NN, et al. Microarray analysis has no additional value in fetal aberrant right subclavian artery: description of 268 pregnancies and systematic literature review. Ultrasound Obst Gynecol. 2019;53(6):810–5.
Peng R, Xie HN, Zheng J, Zhou Y, Lin MF. Fetal right aortic arch: associated anomalies, genetic anomalies with chromosomal microarray analysis, and postnatal outcome. Prenat Diagn. 2017;37(4):329–35.
South ST, Lee C, Lamb AN, Higgins AW, Kearney HM. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet Med. 2013;15(11):901–9.
Crawford T. An atlas of vascular rings and related malformations of the aortic arch system. J Clin Pathol. 1964;17(5):579–80.
Sklansky MS. Fetal cardiology simplified: a practical manual. Future Cardiol. 2013;9(5):611–3.
Ranzini AC, Hyman F, Jamaer E, van Mieghem T. Aberrant right subclavian artery: correlation between fetal and neonatal abnormalities and abnormal genetic screening or testing. J Ultrasound Med. 2017;36(4):785–90.
Perolo A, De Robertis V, Cataneo I, Volpe N, Campobasso G, Frusca T, et al. Risk of 22q11.2 deletion in fetuses with right aortic arch and without intracardiac anomalies. Ultrasound Obst Gynecol. 2016;48(2):200–3.
D’Antonio F, Khalil A, Carvalho JS. OC15.03: Management of fetal right aortic arch without associated intracardiac abnormalities: cohort study and meta analysis. Ultrasound Obst Gynecol. 2013;42(s1):31.
Treadwell-Deering DE, Powell MP, Potocki L. Cognitive and behavioral characterization of the Potocki–Lupski syndrome (duplication 17p11.2). J Dev Behav Pediatr JDBP. 2010;31(2):137–43.
Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80(4):633–49.
Yuan SM. Congenital heart defects in Williams syndrome. Turk J Pediatr. 2017;59(3):225–32.
Collins RT 2nd. Cardiovascular disease in Williams syndrome. Circulation. 2013;127(21):2125–34.
Yuan M, Deng L, Yang Y, Sun L. Intrauterine phenotype features of fetuses with Williams–Beuren syndrome and literature review. Ann Hum Genet. 2020;84(2):169–76.
Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359(16):1685–99.
Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40(12):1466–71.
Christiansen J, Dyck JD, Elyas BG, Lilley M, Bamforth JS, Hicks M, et al. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res. 2004;94(11):1429–35.
Palomares M, Delicado A, Mansilla E, de Torres ML, Vallespin E, Fernandez L, et al. Characterization of a 8q21.11 microdeletion syndrome associated with intellectual disability and a recognizable phenotype. Am J Hum Genet. 2011;89(2):295–301.
Happ H, Schilter KF, Weh E, Reis LM, Semina EV. 8q21.11 microdeletion in two patients with syndromic Peters anomaly. Am J Med Genet Part A. 2016;170(9):2471–5.
Quintela I, Barros F, Castro-Gago M, Carracedo A, Eiris J. Clinical characterization of a male patient with the recently described 8q21.11 microdeletion syndrome. Am J Med Genet Part A. 2015;167(6):1369–73.
Riley KN, Catalano LM, Bernat JA, Adams SD, Martin DM, Lalani SR, et al. Recurrent deletions and duplications of chromosome 2q11.2 and 2q13 are associated with variable outcomes. Am J Med Genet Part A. 2015;167(11):2664–73.
Russell MW, Raeker MO, Geisler SB, Thomas PE, Simmons TA, Bernat JA, et al. Functional analysis of candidate genes in 2q13 deletion syndrome implicates FBLN7 and TMEM87B deficiency in congenital heart defects and FBLN7 in craniofacial malformations. Hum Mol Genet. 2014;23(16):4272–84.
Hernandez LE, Shepard CW, Bamforth SD, Anderson RH. The right subclavian artery arising as the first branch of a left-sided aortic arch. World J Pediatr Congenit Heart Surg. 2014;5(3):456–9.
Yingjun X, Zhiyang H, Linhua L, Fangming S, Linhuan H, Jinfeng T, et al. Chromosomal uniparental disomy 16 and fetal intrauterine growth restriction. Eur J Obstet Gynecol Reprod Biol. 2017;211:1–7.
Kotzot D. Prenatal testing for uniparental disomy: indications and clinical relevance. Ultrasound Obst Gynecol. 2008;31(1):100–5.
Hansen WF, Bernard LE, Langlois S, Rao KW, Chescheir NC, Aylsworth AS, et al. Maternal uniparental disomy of chromosome 2 and confined placental mosaicism for trisomy 2 in a fetus with intrauterine growth restriction, hypospadias, and oligohydramnios. Prenat Diagn. 1997;17(5):443–50.
Kotzot D, Lurie IW, Mehes K, Werder E, Schinzel A. No evidence of uniparental disomy 2, 6, 14, 16, 20, and 22 as a major cause of intrauterine growth retardation. Clin Genet. 2000;58(3):177–80.
Jansen FA, Blumenfeld YJ, Fisher A, Cobben JM, Odibo AO, Borrell A, et al. Array comparative genomic hybridization and fetal congenital heart defects: a systematic review and meta-analysis. Ultrasound Obst Gynecol. 2015;45(1):27–35.
Vigneswaran TV, Allan L, Charakida M, Durward A, Simpson JM, Nicolaides KH, et al. Prenatal diagnosis and clinical implications of an apparently isolated right aortic arch. Prenat Diagn. 2018;38(13):1055–61.
Acknowledgements
The authors want to thank Gang An, Min Zhang, and Yan Wang for their technical support on single nucleotide polymorphism array analysis.
Author information
Authors and Affiliations
Contributions
XW and LX prepared the main manuscript; XX, MC, and LS prepared the experiment. All authors have read and approved the final article.
Corresponding author
Ethics declarations
Funding
The present study and open access was supported by The Fujian Provincial Natural Science Foundation (Grant no. 2017 J01238).
Conflict of Interest
The authors, Xiaoqing Wu, Ying Li, Linjuan Su, Xiaorui Xie, Meiying Cai, Na Lin, Hailong Huang, Yuan Lin, and Liangpu Xu, declare there is no conflict of interest regarding the publication of this paper.
Availability of Data and Material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wu, X., Li, Y., Su, L. et al. Chromosomal Microarray Analysis for the Fetuses with Aortic Arch Abnormalities and Normal Karyotype. Mol Diagn Ther 24, 611–619 (2020). https://doi.org/10.1007/s40291-020-00474-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-020-00474-7