
Abstract
Background
Right aortic arch (RAA) is a common congenital aortic arch abnormality. Fetuses with RAA frequently have good outcomes after birth. However, chromosomal abnormalities and genetic syndromes suggest poor prognosis for these patients. So far the underlying genetic etiology is still not identified in most RAA patients based on traditional genetic techniques and a problem is still debated whether fetuses with isolated RAA should be referred for CMA. Our study aims to investigate the genetic etiology of fetuses with right aortic arch (RAA) by chromosomal microarray analysis (CMA) and whole exome sequencing (WES) and evaluate the efficacy of CMA in fetal isolated RAA.
Results
Among these 153 fetuses, 99 (64.7%) with isolated RAA and 54 (35.3%) with non-isolated RAA; 25.5% (39/153) with additional intracardiac anomalies (ICA), and 19.0% (29/153) with extracardiac anomalies (ECA). Tetralogy of Fallot (n = 10) and persistent left superior vena cava (n = 11) are the most common ICA and ECA, respectively. CMA detected 15 clinically significant copy number variations (CNVs) in 14 cases (9.2%); microdeletion of 22q11.21 was the most common pathogenic CNVs (7.8%). The chromosomal abnormalities rate was higher in non-isolated RAA and RAA with ICA groups than in isolated RAA group (16.7% vs. 5.1%; 20% vs. 5.1%, both p < 0.05). From five cases further undergoing WES, a diagnostic variant in MTOR gene (c.7255G > A, de novo) was first reported in prenatal, extending the prenatal manifestation of Smith–Kingsmore syndrome (OMIM: 616638); a clinically relevant variant c.3407A > T in STAG2 was identified, being inherited from the healthy mother. Moreover, the premature birth and termination rates were higher in non-isolated RAA group than in isolated RAA group (11.1% vs. 1.0%; 37.0% vs. 2.0%, both p < 0.01).
Conclusions
We demonstrate that CMA and WES are useful diagnostic tools for fetal RAA, particularly non-isolated RAA, and all fetuses with RAA should be referred for CMA. The data probably aids in prenatal diagnosis and prenatal counseling of fetal RAA.
Similar content being viewed by others

Background
Right aortic arch (RAA) is a kind of congenital aortic arch abnormality (AAA), arising from unusual regression and progression of the double aortic arch system during embryogenesis [1]. RAA is characterized by the transverse aortic arch on the right side of the trachea, with an incidence of about 0.1% in low-risk pregnancies [2]. According to the branching pattern of the head and neck vessels, RAA is divided into two main categories, mirror-image pattern (MI) and aberrant left subclavian artery (ALSA). Fetuses with RAA may be detected with vascular rings encircling the trachea and/or esophagus, which probably affects the development and function of these organs and triggers numerous compression symptoms after birth from mild clinical manifestations to severe respiratory distress, and even to operation and death [3, 4]. In prenatal setting, other sonographic anomalies are frequently recognized in pregnancies with RAA, involving the cardiovascular system, the central nervous system, and the gastrointestinal system, etc [5,6,7,8,9,10,11,12,13,14,15,16,17]. It reveals that conotruncal defects, particularly Tetralogy of Fallot (TOF), constitute the most common intracardiac abnormality (ICA) in fetal RAA [5, 6, 8,9,10,11,12, 14, 16]. Genetic etiology identification is considerably essential for making deliberated decisions regarding pregnancy continuation and corresponding prenatal and postnatal management. Based on conventional karyotype (CK) and fluorescence in situ hybridization (FISH) for 22q11.2 locus, the traditional genetic techniques have detected chromosomal abnormalities, mostly 22q11.2 deletion syndrome (22q11DS) in 12.5 to 15.3% of fetal RAA [10, 11]. However, the underlying genetic etiology is still not identified in most RAA patients.
Chromosomal microarray analysis (CMA) has been recommended as the primary test for fetuses with structural abnormalities in prenatal and can improve approximately 6% additional diagnostic yield in normal CK [18, 19]. Recently, several studies have been carried out on fetal RAA using CMA in prenatal period [5, 6, 9, 20,21,22,23]. The largest cohort study [15] until now discovered 9.7% (11/113) of fetal RAA had chromosome anomalies, including trisomy 21, trisomy 18, microdeletion of 22q11.21, 6p22.2-p22.1 duplication, etc. As the subtype of congenital heart disease (CHD) or AAA, copy number variations (CNVs) involving fetal RAA were also reported in some large prospective or retrospective cohort studies [24,25,26,27,28,29]. Nonetheless, information is still limited about the clinical practice of CMA in pregnancies with RAA, and the problem is still debated whether fetuses with isolated RAA should be referred for CMA. Notably, the definition of isolated RAA varies in published literature. A recent meta-analysis displayed that the chromosomal abnormalities detection rate was 8.2% (95% CI, 5.0%–12.1%) in fetuses with isolated RAA (with extracardiac abnormalities(ECA)) by CMA, higher than that by CK (5.1%, (95% CI, 2.5%–8.4%)) [30]. In contrast, isolated RAA also refers to RAA without additional sonographic findings and the chromosomal abnormalities detection rate in those ranges from 0 to 11.1% by CMA [5, 6, 15, 20, 22]. For fetuses with structure abnormalities, whole exome sequencing (WES), as a candidate testing technology, can help to ascertain the underlying monogenic disorders and has improved approximately 10% diagnostic yield [31]. However, there are still few reports on fetuses with RAA using WES, which probably brings about challenges and difficulties in genetic counseling and pregnancy management for fetal RAA.
Our study aims to investigate the underlying genetic etiology and pregnancy outcome of fetuses with RAA by CMA and WES and evaluate the efficacy of CMA in fetal isolated RAA.
Results
Cohort characteristics
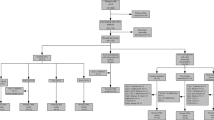
A total of 153 singleton fetuses diagnosed with RAA were included for the invasive procedure from December 2013 to August 2022. These fetuses were examined with or without other ultrasound anomalies but had negative QF-PCR results. The study flowchart was shown in Fig. 1 and the clinical characteristics of the study cohort are listed in Table 1. The median maternal age at RAA diagnosis was 29.8 ± 4.4 years. The median gestational age was 25+6 (24+3, 28+0) weeks. Of those, 75.2% (115/153) were diagnosed in the second trimester, and the remaining 24.8% (38/153) during the third trimester. 32.0% (49/153) were nulliparous women. The branching patterns of RAA in 63 patients were obtained in prenatal or postnatal, including 8 with MI, 54 with ALSA, and 1 with the left subclavian artery (LSA) originating from the pulmonary artery. The additional anomalies rate, especially ICA, was higher in the RAA-MI group than in the RAA-ALSA group (75% vs. 27.8%, p = 0.026; 62.5% vs. 7.4%, p < 0.001). What’s more, 99 (64.7%) had isolated RAA, and 54 (35.3%) had non-isolated RAA.

Flowchart of the cohort in fetuses with RAA for CMA and Trio-WES. RAA, right aortic arch; QF-PCR, quantitative fluorescence polymerase chain reaction; CMA, chromosomal microarray analysis; WES, whole exome sequencing; P/LP CNVS, (likely) pathogenic copy number variants; VUS, variants of uncertain significance; LFU, lost to follow-up; TOP, termination of pregnancy
As shown in Table 2, ICA was identified in 39 (25.5%) patients. The rate of ECA including structure anomalies and soft markers was calculated to be 19.0% (29/153). TOF (n = 10) and PLSVC (n = 11) constitute the most common ICA or ECA, respectively. Remarkedly, 6 patients were detected with TOF, whose DAs were not present or visualized. One of those was found with RAA, ventricular septal defect, crossed pulmonary artery, and small pulmonary arteries in prenatal screening, and then diagnosed with TOF after birth. In the other case, TOF was demonstrated but DA did exist on postnatal ultrasound.
CMA results
In total, amniocentesis was provided for 106 (69.3%) pregnancies and percutaneous umbilical blood sampling was executed for the remaining (30.7%). The detailed CMA results showed in Table 3. CMA produced a 9.2% (14/153) detection rate of clinically significant CNVs. LP CNV was identified in one case, 22q11.21 deletion partially overlapping the pathogenic segment of recurrent 22q11DS (n = 1). There were 14 pCNVs diagnosed in thirteen patients, consisting of microdeletion of 22q11.21 (n = 12), Xp22.33deletion (del) (n = 1), and 17p12del (n = 1). The incidence of classical microdeletion of 22q11.21 was 7.8% (12/153). Of note, the segment of Xp22.33del was reclassified as P from VUS. Additionally, the thymus was absent in patient 11 with 22q11DS.
Patients were classified into three groups, the RAA-MI group, the RAA-ALSA group, and the other RAAs group. The genetic abnormalities rate in the RAA-MI group, the RAA-ALSA group, and the other RAAs group were 12.5% (1/8), 13.0% (7/54), and 6.6% (6/91), respectively. The detection rate was similar in the RAA-MI group and RAA-ALSA group (p = 1.000) (Table 1). In addition, the chromosomal aberrations incidence in cases with isolated RAA, non-isolated RAA, RAA with ICA, RAA with ECA, and RAA with ICA and ECA were 5.1% (5/99), 16.7% (9/54), 20% (5/25), 20% (3/15), and 7.1% (1/14) (Table 1). By comparison, the statistical difference was observed between the non-isolated RAA group or RAA with ICA group and isolated RAA group (p < 0.05 for both). Conversely, the statistical difference was not observed in RAA with ECA group or RAA with ICA and ECA group and isolated RAA group (p > 0.05 for both). Likewise, the detection rate in cases with RAA with other structural anomalies (2/6, 33.3%), RAA with soft markers group (0/8, 0), and RAA with soft markers and other structural anomalies (1/1, 100%) were markedly higher than that in isolated RAA group, but the difference was not statistically significant (p > 0.05 for all).
Furthermore, the uncertain significant CNVs rate was 5.9% (9/153) by CMA. Of those, 12q23.2del and 6q16.3 duplication (dup) were detected respectively in patients 42 and 134, both inherited from their healthy mothers. Notably, the 6q16.3dup was reclassified as VUS from LB. In addition, two duplications of 8p23.2 and 8q13.3 were identified in the same case 53, which was inherited from the phenotypically normal father.
WES results
Following informed consent, trio-WES was performed further for five fetuses with normal CMA, including one with isolated RAA (#85), and four with non-isolated RAA (#18, #22, #23, and #35). LP and VUS variants were detected in cases 22 and 23 respectively (Table 4). In case 22 with non-isolated RAA, WES detected a de novo heterozygous variant in the MTOR gene, NM_004958.3, c.7255G > A, p.(Glu2419Lys), causing Smith–Kingsmore Syndrome (OMIM: 616638) with an autosomal dominant inheritance mode. For case 23 with heterotaxy, a hemizygous variant, NM_001042749.2, c.3407A > T, p.(Asp1136Val) inherited from the healthy mother, was identified, leading to X-linked Mullegama-Klein-Martinez syndrome (OMIM: 301022). In short, trio-WES can yield a 20% (1/5) diagnostic rate in fetuses with RAA over CMA.
Pregnancy outcomes
As shown in Table 1, the pregnancy outcomes included 22 terminations of pregnancy, 32 lost for follow-ups, 99 live births including 92 full-term deliveries, and 7 premature births. As to the 14 cases with 22q11DS, 3 live births (full-term deliveries), 10 terminations of pregnancy, and 1 lost for follow-ups. By calculation, the live birth rate in isolated RAA groups was significantly higher than that in non-isolated RAA groups (73.7% vs. 48.1%, p = 0.002). Instead, the premature birth/terminations rates in non-isolated RAA group were strikingly higher than that in isolated RAA group (11.1% vs. 1.0%, p = 0.008; 37.0% vs. 2.0% p < 0.001).
Discussion
This study investigated the risk of chromosomal abnormalities and monogenic disorders (variants) in pregnancies with fetal RAA by combined CMA and WES testing for the first time and confirmed the diagnostic value of CMA and WES in fetuses with RAA, particularly non-isolated RAA in prenatal setting. CMA and WES improved 9.1% (14/153) and 20% (1/5) additional diagnostic yield, respectively. Herein, a controversial problem was discussed whether these fetuses with isolated RAA should be referred for microarray testing. We summarized all clinical-significant CNVs in reported articles and our study on the utilization of CMA in fetuses only with RAA in prenatal (Table 5). Additionally, we proposed that DA not present or visualized in antenatal scanning may be an indication of TOF. Ultimately, a diagnostic variant in the MTOR gene, NM_004958.3, c.7255G > A, p. (Glu2419Lys) was first reported in prenatal, which extends our knowledge of the prenatal manifestation of Smith–Kingsmore syndrome (SKS, OMIM: 616638). These findings probably contribute to prenatal diagnosis and prenatal counseling of the pregnancy of fetal RAA.
With the introduction and extensive utilization of CMA in prenatal diagnosis, it reveals that CNVs play an essential role in the pathogenesis of fetal structure abnormalities. So far, the largest cohort study showed 9.7% (11/113) of fetal RAA were diagnosed with chromosome anomalies by CK and SNP array [15]. Previous studies found the clinically significant CNV detection yield was 5.2 to 12.1% in fetal RAA [9, 23]. In our cohort, CMA could identify 9.2% additional diagnostic yield in fetuses with RAA, parallel to the data above. Generally, those fetuses with multiple abnormalities tend to suffer from chromosomal disorders, which was proved in the current study. Our data implied that the chromosomal abnormalities rates in non-isolated RAA group, especially in RAA with ICA group were significantly higher than that in isolated RAA group (p < 0.05 for both). By calculations, the clinically significant CNVs detection yield was remarkedly higher in RAA with ICA, RAA with ECA, RAA with ICA and ECA, RAA with structural anomalies, and RAA with structural anomalies and soft markers, except for RAA with soft markers than that in isolated RAA group, while the difference was all not statistically significant (p > 0.05). In a word, coexistent structure malformations, particularly ICA, remarkedly increase the underlying risk of cytogenetic abnormalities in pregnancies of fetuses with RAA.
Fetuses combined with RAA have a highly coexistent prevalence of other structure defects. According to the published studies [5,6,7,8,9,10,11,12,13], additional ultrasound abnormalities are frequently documented in fetal RAA, with an incidence of 26.5%–72% in ICA and 2%–44.44% in ECA. In this study, the ICA and ECA rates were 25.5% and 19.0% separately, corresponding with the previous data. Most studies revealed that the most common ICA of fetuses with RAA in prenatal is conotruncal defects, particularly TOF (14.7%–26.7%) [5, 6, 8,9,10,11]. Equally, TOF was also the most common ICA in our population, but its incidence was 6.5% in the whole cohort, lower than previous reports. What is noteworthy is that six cases were screened with DA not present or visualized in prenatal and all were found with TOF in prenatal or postnatal settings, which correlated well with the discovery in a published study [9]. These findings provide more evidence for the close association between TOF and RAA. Some mechanisms probably count for it that early in embryogenesis neural crest cells migrate into the region of the heart and contribute to the formation of the heart and great vessels [9, 32, 33]. Therefore, we speculate that DA not present or visualized may be an indication of TOF in antenatal scanning, helping in the identification of a group at high risk for TOF. However, the most common ECA associated with fetal RAA fluctuates among the reported articles. Thymus hypoplasia/aplasia, fetal growth restriction, and gastrointestinal malformations, especially esophageal atresia were all found as the most common ECA in different studies[5, 6, 8, 16]. Compared to that, PLSVC was found as the most common ECA from our patients, which is in line with the study by Galindo et al., as a small cohort of 48 fetuses including 3 DAA demonstrated there were seven PLSVC discovered in 45 RAA[11]. This difference above may result from different sizes of samples, distinguished subjects incorporated, various definitions and classifications of associated ECA, as well as advanced ultrasonic technology, and more cases and studies are needed to evaluate the association between RAA and ECA. Based on our data, RAA-ALSA is the most common branching pattern of the head and neck vessels in RAA (35.3%), while RAA-MI is frequently accompanied by CHD (62.5%). The additional anomalies rate and the ICA rate in the RAA-MI group were significantly higher than that in the RAA-ALSA group (p < 0.05 for both). But the detection rate of clinically significant CNVs was similar in the group with RAA-MI and the group with RAA-ALSA (12.5% vs. 13.0%, p > 0.05). In total, when RAAs are detected in prenatal, it is exceedingly necessary for fetuses to perform a systemic scanning including echocardiographic examination in detail, to provide more useful information for counseling.
To date, the problem is still controversial whether CMA testing should be offered for fetuses only with RAA. A paper reviewed and found the overall risk for clinically significant CMA findings was 6.62% (10/151) in pregnancies involving isolated RAA [20]. In the other systematic review [30] including 670 fetuses, the detection rate is 4.7% (95% CI, 1.1%–10.8%), similar to our positive rate (5.1%, 95%CI, 1.7%–11.4%). It indicates that the pregnancies with isolated RAA are combined with an increased risk of P/LP CNVs. In our patients, 22q11DS was the only chromosomal aberration in the isolated RAA group. However, as shown in Table 5, various P/LP CNVs were reported in pregnancies with isolated RAA including 10p15.3deletion, 16p11.2duplication, 22q11.21 deletion, 6p21.31p21.2 deletion, and 17p11.2 duplication [5, 20,21,22,23]. Furthermore, two research groups agreed that CMA should be recommended for isolated RAA [20, 23]. Despite our results not being as expected, we also suggest that CMA should be offered for those with isolated RAA due to the underlying P/LP CNVs. To sum up, a diagnostic yield of 5.1% warrants the application of CMA in pregnancies with isolated RAA.
Microdeletion of 22q11.2 is the most common cause of RAA. DiGeorge syndrome (OMIM: 611867), as known as 22q11DS is the most common chromosomal syndrome, combined with a remarkedly heterogeneous spectrum of phenotypes. The most prevalent manifestation includes CHD, typical facial features, palatal anomalies, and thymic aplasia/hypoplasia [34]. In a study of prenatal features of 22q11DS, 21.7% (16/78) of fetuses had RAA [35]. Additionally, a systematic review including sixteen studies (312 fetuses) displayed that 22q11.2 deletion could count for 6.1% (95% CI, 3.6–9.3%) of fetuses with RAA [17]. In the current study, 12 cases were accompanied by 22q11DS and the overall rate of 22q11DS is 7.8% (12/153), occupying 85.7% of the total chromosomal abnormalities detected. TBX1 gene, a member of the T-box transcription factors family, is the major determinant for the phenotypic features of the 22q11DS [36,37,38,39,40,41]. The TBX1 gene located in chromosome 22q11.2 plays a crucial role in the normal development of the pharyngeal arches [36, 37]. The haploinsufficiency of TBX1 would disturb the development of the fourth branchial arch artery, leading to the various cardiac defects of 22q11DS [40,41,42]. Coincidentally, increased dosage or intragenic pathogenic variants of TBX1 also results in similar cardiac defects [43,44,45]. This suggests that the dosages expression of the TBX1 gene requires precise regulation in heart development. However, the molecular mechanisms how the TBX1 gene causes the production of CHD step by step still need more research to elucidate.
A diagnostic variant in MTOR gene (NM_004958.3, c.7255G > A, p. (Glu2419Lys)) was first reported in prenatal, which extends the prenatal manifestation of Smith–Kingsmore syndrome (SKS, OMIM: 616638). SKS is a rare autosomal dominant neurodevelopmental disorder caused by heterozygous variants in the MTOR genes (OMIM:601231) on chromosome 1p36, described and named firstly by Smith et al. [46] SKS is characterized by megalencephaly/macrocephaly, developmental delay, seizures, and intellectual disability, associated with autism spectrum disorders, hypotonia, dysmorphic facial features, ventriculomegaly, etc. [47]. Case 22 was accompanied with bilateral ventriculomegaly, one of the common phenotypes above. Nevertheless, CHD was not reported in most SKS literature and there’s no clear proof about the correlation between SKS and CHD. Aortic sinus to right atrial fistula was only found in an SKS patient [48]. It could not be determined yet whether the heart defect observed in case 22 was caused by the variants in the MTOR gene, or it was just a coincidence. Based on some reports [47,48,49,50,51], parental germ-cell chimerism was a recurrent finding in SKS patients. The same MTOR variant was identified in two or more sibships from a family but not in parental DNAs, therefore, it was assumed to have been inherited through germline mosaicism [49]. Even though our patients had no siblings and the variant in MTOR gene was not detected from parental samples, the possibility that germ-cell chimerism in at least one parent should not be discounted. For the genetic counseling of this couple, chorionic villus or amniocentesis can be performed for the next pregnancy to rule out the risk of chromosomal disorders without considering the possibility of parental germline mosaicism.
Additional abnormalities and genetic etiology could affect the pregnancy decision and improve the risk of premature births. In the study, these pregnancies in fetuses with RAA were terminated due to the additional sonographic findings, cytogenetic abnormalities, and single gene variants. The rate of terminations of pregnancy and premature birth in non-isolated RAA group were strikingly higher than that in isolated RAA group (p < 0.01), while the livebirth rate in isolated RAA groups was remarkably higher than that in non-isolated RAA groups (p < 0.01).
There are several limitations in the cohort study. It was a retrospective study so the recall bias may be present. There were only five cases further undergoing WES testing so the results may be biased. Due to the limited follow-up time, postnatal conditions related to RAA were unavailable. Therefore, more cases and more research are needed to provide more detailed information for the prenatal and postnatal management of RAA fetuses.
Conclusion
In summary, this study investigated the risk of chromosomal abnormalities and monogenic variants in fetuses with RAA fetuses by combined CMA and WES testing for the first time and demonstrate the diagnostic value of CMA and WES for fetal RAA, particularly non-isolated RAA, in prenatal period. We suggest that CMA can be offered for all fetuses with RAA, including isolated RAA. This data probably assists in prenatal counseling and pregnancy management of the pregnancy with fetal RAA.
Methods
Participants
This retrospective study was conducted at the Center of Prenatal Diagnosis of Guangzhou Women and Children’s Medical Center and approved by the Institutional Review Board of the Ethics Committee. From the clinical record database, a total of 186 pregnancies suspected with RAA were referred to our center for prenatal diagnosis from December 2013 to August 2022. The main inclusion criteria are (1) singleton pregnancy, (2) fetuses diagnosed with RAA with or without additional abnormalities including soft markers in ultrasonography, (3) available and complete clinical information about this pregnancy, (4) negative results in quantitative fluorescence polymerase chain reaction (QF-PCR), (5) available DNA sample for trio WES.
Overall, 153 pregnancies were included in the current study. All cases were performed a systemic examination including echocardiography and the diagnosis was confirmed by two experienced expert sonographers, based on the three vessels and trachea view in which the aortic arch is located on the right side of the trachea [2]. Fetuses were divided into two groups, the isolated RAA group without further sonographic abnormalities and the non-isolated group with ICA and/or ECA. Soft markers observed in our patients included persistent left superior vena cava (PLSVC), single umbilical artery (SUA), ventriculomegaly, nasal bone absence or nasal hypoplasia, short femur length, and enlarged cisterna magna. Patients underwent QF-PCR and CMA after accepting pretest counseling and signing informed consent documents. When these tests were negative, trio-WES was recommended to check the potential monogenic risk. Clinical and follow-up information was obtained by reviewing the medical record database, on occasion via a telephone call. Records included maternal age, gestational age at diagnosis, nullipara or multipara, branching pattern of the head and neck vessels, the position of the DA, ICA and/or ECA, genetic abnormalities, and the pregnancy outcome, etc.
QF-PCR
DNA was extracted from amniotic fluid or umbilical cord blood, using the Qiagen DNA Blood Midi/Mini Kit (Qiagen GmbH, Hilden, Germany) and firstly accomplished for QF-PCR to eliminate maternal cell contamination and check out 13, 18, 21, X, and Y aneuploidies quickly, by the multiplex ligation-dependent probe amplification (MLPA) kit (Guangzhou Darui Biotechnology Co., Ltd, Guangdong, China).
CMA
DNA samples with normal QF-PCR were implemented for CMA by utilizing CytoScan HD Array (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s protocols. Data were analyzed on the Affymetrix Chromosome Analysis Suite software with genome version GRCh37 (hg19). Based on the American College of Medical Genetics and Genomics guidelines [52], CNVs were classified as pathogenic (P), likely pathogenic (LP), variants of uncertain significance (VUS), likely benign (LB), and benign (B). All segments detected were also reclassified. If necessary, numerous databases will be used such as: Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), University of California Santa Cruz (http://genome.ucsc.edu/hg19), DECIPHER (https://www.deciphergenomics.org/), ClinGen resource (https://www.clinicalgenome.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), PubMed (https://pubmed.ncbi.nlm.nih.gov/).
WES
DNA samples for WES were enriched with Agilent SureSelect human exome capture probes (V6, Life Technologies, United States) following the manufacturer’s protocols. The DNA library was sequenced on Hiseq XTen (Illumina, Inc., San Diego, CA, United States) for pair-end 150-bp read. Clean reads were generated by using Trimmomatic to remove adapter-contaminated reads and low-quality reads and then compared with the human reference genome (hg19/GRCh37) with BWA. The BWA originated from SNP analysis, duplication marking, indel realignment, and recalibration by GATK and SAMtools. These variants were annotated according to dbSNP, the 1000 Genome Project, ExAC, EVS, gnomAD, OMIM, ClinVar, the Human Gene Mutation Database, and our in-house database. A series of computational algorithms were used to predict the protein’s effect like the structure of the protein, the conservation domain, and the function domain. These computational algorithms include SIFT, MutationTaster, PolyPhen2, PROVEAN, CADD, Human Splicing Finder, MaxEntScan, and NNSplice. All variants were classified as pathogenic (P), likely pathogenic (LP), variants of uncertain significance (VUS), likely benign (LB), and benign (B) based on the standards and guidelines for the interpretation of sequence variants [53,54,55]. Positive variants including P and LP variants were confirmed by Sanger sequencing.
Statistical analyses
Statistical analyses were fulfilled by using SPSS 25.0. Mean ± standard deviations or median (Q1–Q3) were used for descriptive statistics. The independent samples T-test or the Mann-Whitney U test was used for assessing the significance of difference on continuous variable. The Chi-square test, Continuity correction, or Fisher’s exact test was used for pairwise comparison on categorical data. A value of p < 0.05 was considered statistically significant.
Availability of data and materials
The data that support the findings of this study are not publicly available because the information contained could compromise the privacy of research participants. Further inquiries can be directed to the corresponding author.
Abbreviations
- RAA:
-
Right aortic arch
- AAA:
-
Aortic arch abnormality
- MI:
-
Mirror-image pattern
- ALSA:
-
Aberrant left subclavian artery
- TOF:
-
Tetralogy of fallot
- ICA:
-
Additional intracardiac abnormality
- CK:
-
Conventional karyotype
- FISH:
-
Fluorescence in situ hybridization
- 22q11DS:
-
22Q11.2 deletion syndrome
- CMA:
-
Chromosomal microarray analysis
- CHD:
-
Congenital heart disease
- CNVs:
-
Copy number variations
- ECA:
-
Extracardiac abnormalities
- WES:
-
Whole exome sequencing
- QF-PCR:
-
Quantitative fluorescence polymerase chain reaction
- PLSVC:
-
Persistent left superior vena cava
- SUA:
-
Single umbilical artery
- MLPA:
-
Multiplex ligation-dependent probe amplification
- P:
-
Pathogenic
- LP:
-
Likely pathogenic
- VUS:
-
Variants of uncertain significance
- LB:
-
Likely benign
- B:
-
Benign
- LSA:
-
Left subclavian artery
- del:
-
Deletion
- dup:
-
Duplication
References
Edwards JE. Anomalies of the derivatives of the aortic arch system. Med Clin North Am. 1948;32:925–49.
Achiron R, Rotstein Z, Heggesh J, Bronshtein M, Zimand S, Lipitz S, et al. Anomalies of the fetal aortic arch: a novel sonographic approach to in-utero diagnosis. Ultrasound Obstet Gynecol. 2002;20(6):553–7.
Worhunsky DJ, Levy BE, Stephens EH, Backer CL. Vascular rings. Semin Pediatr Surg. 2021;30(6):151128.
Biermann D, Holst T, Huners I, Rickers C, Kehl T, Ruffer A, et al. Right aortic arch forming a true vascular ring: a clinical review. Eur J Cardiothorac Surg. 2021;60(5):1014–21.
Topbas Selcuki NF, Senol G, Esin D, Ozkose ZG, Caypinar SS, Bornaun H, et al. Prenatal diagnosis and postnatal outcomes of right aortic arch anomalies. Arch Gynecol Obstet. 2022;306(3):745–52.
Petrescu AM, Ruican D, Patru CL, Zorila GL, Tudorache S, Comanescu AC, et al. Prenatal findings and pregnancy outcome in fetuses with right and double aortic arch. A 10-year experience at a tertiary center. Rom J Morphol Embryol. 2020;61(4):1173–84.
Campanale CM, Pasquini L, Santangelo TP, Iorio FS, Bagolan P, Sanders SP, et al. Prenatal echocardiographic assessment of right aortic arch. Ultrasound Obstet Gynecol. 2019;54(1):96–102.
Wojtowicz A, Respondek-Liberska M, Slodki M, Kordjalik P, Pluzanska J, Knafel A, et al. The significance of a prenatal diagnosis of right aortic arch. Prenat Diagn. 2017;37(4):365–74.
Peng R, Xie HN, Zheng J, Zhou Y, Lin MF. Fetal right aortic arch: associated anomalies, genetic anomalies with chromosomal microarray analysis, and postnatal outcome. Prenat Diagn. 2017;37(4):329–35.
Miranda JO, Callaghan N, Miller O, Simpson J, Sharland G. Right aortic arch diagnosed antenatally: associations and outcome in 98 fetuses. Heart. 2014;100(1):54–9.
Galindo A, Nieto O, Nieto MT, Rodriguez-Martin MO, Herraiz I, Escribano D, et al. Prenatal diagnosis of right aortic arch: associated findings, pregnancy outcome, and clinical significance of vascular rings. Prenat Diagn. 2009;29(10):975–81.
Berg C, Bender F, Soukup M, Geipel A, Axt-Fliedner R, Breuer J, et al. Right aortic arch detected in fetal life. Ultrasound Obstet Gynecol. 2006;28(7):882–9.
Zidere V, Tsapakis EG, Huggon IC, Allan LD. Right aortic arch in the fetus. Ultrasound Obstet Gynecol. 2006;28(7):876–81.
Babaoglu K, Dogan Y, Basar EZ, Uzun O. Prenatal diagnosis of the right aortic arch: change in detection rate, the status of associated anomalies, and perinatal outcomes in 137 fetuses. Pediatr Cardiol. 2022;43(8):1888–97.
Yan Y, Yang Z, Li Y, Pei Q, Zhang X, Wang Y, et al. The prenatal diagnosis and prognosis of fetal right aortic arch and double aortic arch malformation: a single-center study. J Obstet Gynaecol Res. 2023;49:2273–82.
Tidrenczel Z, Tardy EP, Ladányi A, Hajdú J, Böjtös I, Sarkadi E, et al. Praenatalisan felismert magzati aortaív-rendellenességek és megszületés utáni következményeik. Orvosi Hetil. 2023;164(28):1111–20.
D’Antonio F, Khalil A, Zidere V, Carvalho JS. Fetuses with right aortic arch: a multicenter cohort study and meta-analysis. Ultrasound Obstet Gynecol Off J Int Soc Ultrasound Obstet Gynecol. 2016;47(4):423–32.
Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175–84.
American College of Obstetricians and Gynecologists Committee on Genetics. Committee opinion no. 581: the use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol. 2013;122(6):1374–7.
Maya I, Singer A, Baris HN, Goldberg Y, Shalata A, Khayat M, et al. Prenatal microarray analysis in right aortic arch–a retrospective cohort study and review of the literature. J Perinatol. 2018;38(5):468–73.
O’Mahony EF, Hutchinson DP, McGillivray G, Nisbet DL, Palma-Dias R. Right-sided aortic arch in the age of microarray. Prenat Diagn. 2017;37(5):440–5.
Vigneswaran TV, Allan L, Charakida M, Durward A, Simpson JM, Nicolaides KH, et al. Prenatal diagnosis and clinical implications of an apparently isolated right aortic arch. Prenat Diagn. 2018;38(13):1055–61.
Wu X, Li Y, Su L, Xie X, Cai M, Lin N, et al. Chromosomal microarray analysis for the fetuses with aortic arch abnormalities and normal karyotype. Mol Diagn Ther. 2020;24(5):611–9.
Turan S, Asoglu MR, Gabbay-Benziv R, Doyle L, Harman C, Turan OM. Yield rate of chromosomal microarray analysis in fetuses with congenital heart defects. Eur J Obstet Gynecol Reprod Biol. 2018;221:172–6.
Wang Y, Cao L, Liang D, Meng L, Wu Y, Qiao F, et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: a prospective cohort study. Am J Obstet Gynecol. 2018;218(2):244.
Qiao F, Wang Y, Zhang C, Zhou R, Wu Y, Wang C, et al. Comprehensive evaluation of genetic variants using chromosomal microarray analysis and exome sequencing in fetuses with congenital heart defect. Ultrasound Obstet Gynecol Off J Int Soc Ultrasound Obstet Gynecol. 2021;58(3):377–87.
Sagi-Dain L, Singer A, Segel R, Berger R, Kanengisser-Pines B, Maya I. The yield of chromosomal microarray in pregnancies with congenital cardiac defects and normal noninvasive prenatal screening. Am J Obstet Gynecol. 2021;225(3):333.
Lu F, Xue P, Zhang B, Wang J, Yu B, Liu J. Estimating the frequency of causal genetic variants in foetuses with congenital heart defects: a Chinese cohort study. Orphanet J Rare Dis. 2022;17(1):2.
Zhang Z, Hu T, Wang J, Hu R, Li Q, Xiao L, et al. Pregnancy outcomes of fetuses with congenital heart disease after a prenatal diagnosis with chromosome microarray. Prenat Diagn. 2022;42(1):79–86.
Luo Q, Chen J, Zhang Y, Li J, Su X, Wang Q, et al. Incidence of chromosomal anomalies in fetuses with isolated right aortic arch: a meta-analysis. Prenat Diagn. 2020;40(3):294–300.
Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet (London, England). 2019;393(10173):758–67.
Erhardt S, Wang J. Cardiac neural crest and cardiac regeneration. Cells. 2022;12(1):111.
McElhinney DB, Clark BJ 3rd, Weinberg PM, Kenton ML, McDonald-McGinn D, Driscoll DA, et al. Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J Am Coll Cardiol. 2001;37(8):2114–9.
Cirillo A, Lioncino M, Maratea A, Passariello A, Fusco A, Fratta F, et al. Clinical manifestations of 22q11.2 deletion syndrome. Heart Fail Clin. 2022;18(1):155–64.
Noel AC, Pelluard F, Delezoide AL, Devisme L, Loeuillet L, Leroy B, et al. Fetal phenotype associated with the 22q11 deletion. Am J Med Genet A. 2014;164A(11):2724–31.
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362(9393):1366–73.
Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006;133(18):3587–95.
Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401(6751):379–83.
Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27(3):286–91.
Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101.
Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–29.
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet (London, England). 2003;362(9393):1366–73.
Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, et al. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38(12):E45.
Zweier C, Sticht H, Aydin-Yaylagül I, Campbell CE, Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet. 2007;80(3):510–7.
Hasten E, McDonald-McGinn DM, Crowley TB, Zackai E, Emanuel BS, Morrow BE, et al. Dysregulation of TBX1 dosage in the anterior heart field results in congenital heart disease resembling the 22q11.2 duplication syndrome. Hum Mol Genet. 2018;27(11):1847–57.
Smith LD, Saunders CJ, Dinwiddie DL, Atherton AM, Miller NA, Soden SE, Farrow EG, Abdelmoity ATG, Kingsmore SF. Exome sequencing reveals de novo germline mutation of the mammalian target of rapamycin (MTOR) in a patient with megalencephaly and intractable seizures. J Genomes Exomes. 2013;2:63–72.
Gordo G, Tenorio J, Arias P, Santos-Simarro F, Garcia-Minaur S, Moreno JC, et al. mTOR mutations in Smith–Kingsmore syndrome: four additional patients and a review. Clin Genet. 2018;93(4):762–75.
Baynam G, Overkov A, Davis M, Mina K, Schofield L, Allcock R, et al. A germline MTOR mutation in aboriginal Australian siblings with intellectual disability, dysmorphism, macrocephaly, and small thoraces. Am J Med Genet A. 2015;167(7):1659–67.
Poole RL, Curry PDK, Marcinkute R, Brewer C, Coman D, Hobson E, et al. Delineating the Smith–Kingsmore syndrome phenotype: investigation of 16 patients with the MTOR c.5395G > A p.(Glu1799Lys) missense variant. Am J Med Genet A. 2021;185(8):2445–54.
Moosa S, Bohrer-Rabel H, Altmuller J, Beleggia F, Nurnberg P, Li Y, et al. Smith–Kingsmore syndrome: a third family with the MTOR mutation c.5395G > A p.(Glu1799Lys) and evidence for paternal gonadal mosaicism. Am J Med Genet A. 2017;173(1):264–7.
Mroske C, Rasmussen K, Shinde DN, Huether R, Powis Z, Lu HM, et al. Germline activating MTOR mutation arising through gonadal mosaicism in two brothers with megalencephaly and neurodevelopmental abnormalities. BMC Med Genet. 2015;16:102.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American college of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med Off J Am Coll Med Genet. 2011;13(7):680–5.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med Off J Am Coll Med Genet. 2015;17(5):405–24.
Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39(11):1517–24.
Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12(1):3.
Acknowledgements
We thank the support of the plan on enhancing scientific research in Guangzhou Medical University and all the participants in this study.
Funding
This research was funded by the sub-project of the National Key R&D Program (2021YFC2701002), the National Natural Science Foundation of China (82060388, 82101796, 82201884), and the Project of Guangzhou Science and Technology Bureau (20221A011029, 202102020061, 20231A011026), the General Guide Project of Guangzhou Health Commission (20221A011038), the Municipal Universities (Institutes) Joint Funding Program of Guangzhou Province (2023A03J0874).
Author information
Authors and Affiliations
Contributions
Manuscript writing: LZ, and RL; Study design and manuscript editing: LZ, RH, HZ, and RL; Document retrieval: XL, and FG; Copy number variants classification: XJ, YZ, and FL; Exome sequence variants classification: RL, FF, QY, and DW; QF-PCR reports: FL; Sanger sequencing testing: GC; Clinical data statistics: RH, HZ, MP, JH, and DL. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Guangzhou Women and Children’s Medical Center. Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Competing interests
All authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, L., Huang, R., Zhou, H. et al. Prenatal diagnosis in fetal right aortic arch using chromosomal microarray analysis and whole exome sequencing: a Chinese single-center retrospective study. Mol Cytogenet 17, 22 (2024). https://doi.org/10.1186/s13039-024-00691-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-024-00691-3