
Abstract
Human papillomaviruses (HPVs) belong to a small spherical virus family and are transmitted through direct contact, most often through sexual behavior. More than 200 types of HPV are known, a dozen or so of which are classified as high-risk viruses (HR HPV) and may contribute to the development of cervical cancer. HPV is a small virus with a capsid composed of L1 and L2 proteins, which are crucial for entry to the cell. The infection begins at the basal cell layer and progresses to involve cells from higher layers of the cervical epithelium. E6 and E7 viral proteins are involved in the process of carcinogenesis. They interact with suppressors of oncogenesis, including p53 and Rb proteins. This leads to DNA replication and intensive cell divisions. The persistent HR HPV infection leads to the development of dysplasia and these changes may progress to invasive cancer. During the initial stage of carcinogenesis, telomeres shorten until telomerase activates. The activation of telomerase, the enzyme necessary to extend chromosome ends (telomeres) is the key step in cell immortalization. Analyzing the expression level of hTERT and hTERC genes encoding telomerase and telomere length measurement may constitute new markers of the early carcinogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Persistent infection of high-risk human papillomaviruses (HR HPVs) may cause cervical cancer |
Molecular mechanism of carcinogenesis induced by HR HPV is partly known but new biomarkers of early carcinogenesis are still required for effective cervical cancer screening |
Results of several studies indicate that telomere length measurements and expression analysis of genes coding telomerase may potentially be useful in early diagnosis of a precancerous state, but those methods are currently under development |
1 Introduction
Human papillomavirus (HPV) is transmitted by direct contact, mostly through sexual behavior [1]. Currently, more than 200 types of HPV are known that belong to the family Papillomaviridae. Differences in the L1 gene sequence are the basis for the latest classification of HPV, according to which five genera of viruses are distinguished: α, β, γ, μ, and ν. Approximately 60 HPVs belonging to the α group show affinity for cervical epithelial cells. HPVs are classified into two groups in relation to their oncogenic potential. The group of high-risk viruses includes 13 types: 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 66, and 68. The group of low-risk viruses includes types 6, 11, 42, 43, and 44, and are responsible for the clinically overt form, genital warts (GW), and other HPVs of lesser clinical significance [2,3,4]. It is believed that about 80% of sexually active women will be infected with HPV before the age of 50 years [5]. Infection with the most oncogenic types, 16 and 18, increases the risk of developing cervical cancer by more than 200-fold [6]. Every year, around 500,000 new cases of this type of cancer are diagnosed in the world [7], over 90% of which is a result of persistent HPV infection, and at least half of which are caused by HPV16 [8]. In addition to latent infection with highly oncogenic HPV types (high risk HPVs [HR HPVs]), epidemiological risk factors include early sexual initiation, large number of partners, numerous births, low socioeconomic status, habitual smoking, coexistence of other infectious agents, and vitamin deficiencies. Genital infections with HR HPV are also associated with squamous cell carcinoma of the anus [9], vagina and vulva [10], and penis [11]. In addition, the presence of HPV is detected in 47% of cases of pharyngeal cancers, 11% of oral cancers [12], and 23% of laryngeal cancers [1]. HPV types 6 and 11, belonging to the so-called ‘low-risk’ HPVs, are responsible for the formation of 95% of GW; they are more common in young people [13].
The HPV infection begins with the basal cells in the stratified squamous epithelium and progresses to involve cells from higher layers. Microabrasions facilitate HPV reaching the basal layer. The balance of growth and differentiation in stratified squamous epithelium becomes dysregulated by HPV, which leads to dysplasia and carcinoma in situ. The key event of carcinogenesis that leads to cell immortalization is telomerase activation by viral proteins E6 and E7. These proteins also inhibit apoptosis, which induces genome instability and immune modulation. Telomerase is an enzymatic complex responsible for telomere extension in proliferating cells. It becomes inactive in cells that are unable to divide. Telomere extension is crucial for cancer cells because this allows uncontrolled cell division and tumorigenesis [14].
In this review article, we describe the role of telomerase and telomeres in the molecular mechanism of carcinogenesis induced by HR HPV. First, we describe the virus structure and its life cycle in cervical epithelium. Then we highlight the molecular mechanisms of oncogenesis, with a particular emphasis on telomerase activation induced by virus proteins. Finally, we discuss the potential application of hTERT analysis and telomere length as indicators of early tumorigenesis.
2 Virus Structure
HPVs belong to small viruses with a spherical shape and diameter of 50–60 nm. The capsid of the virus is composed of L1 and L2 proteins (Fig. 1). The viral genome consists of a double-stranded DNA of 8000 base pairs, circularly coiled and associated with histone-like proteins. Functionally, the HPV genome can be divided into three regions. The first non-coding long control region (LCR) is responsible for the regulation of E6 and E7 gene transcription. The second region contains six open reading frames and codes for non-structural proteins (E1, E2, E4, E5, E6, and E7) involved in viral DNA replication and oncogenesis. The third region represents 10% of the entire genome, codes for structural L1 and L2 proteins, and varies between different HPV types [15, 16].

modified from Fernandes et al. [15])
Structure of human papillomavirus (
3 Life Cycle of High-Risk (HR) Human Papillomavirus (HPV) in Cervical Epithelium
HPV uses a unique strategy for propagation. Initially, HPV gains entry to the cells of the basal epithelium layer via interaction of L1 with heparin sulphate proteoglycans [17] and perhaps also laminin [18]. Conformational changes in viral capsid based on furin cleavage of L2 allows virus binding to the keratinocyte receptor, which is important for internalization. Virus endocytosis is most likely mediated by caveolae- and clatrin-independent pathways [19]. One of the novel pathways involving tetraspanin-enriched microdomains has been proposed by Spoden et al. [20, 21]. Viral proteins E1 and E2 are responsible for the maintenance of a low number of genome copies, since genomes are transferred to the nucleus and are established as episomes after internalization. Early viral genes are expressed (E1, E2, E6, and E7) and viral genomes are replicated at the same time as the cell’s DNA. Active cell division generates daughter cells, which migrate from the basal layer and undergo differentiation. In differentiated cells, production of E4 protein induces amplification of viral genome replication. Products of late genes L1 and L2 are needed to form new virions, which reach the cornified layer of the epithelium and are released here (Fig. 2) [22, 23]. The role of individual proteins produced during the HPV cell cycle is described in Table 1.

modified from Tomaić [24]). HPV human papillomavirus
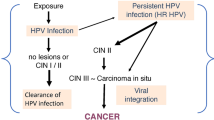
Normal and infected epithelium. Human papillomavirus penetrates the mucosal epithelium via microinjuries. The infection begins with the basal cell layer and successively progresses to involve cells from higher layers. Expression of the human papillomavirus genes (E6 and E7) promotes uncontrolled cell proliferation. If the immune system does not eliminate viruses, a long period of infection may cause cancer development (
HR HPV infection is associated with cervical intraepithelial neoplasia (CIN) and can cause cervical cancer as a result of the development of lesions after persistent infection. Cervical cancer is located preferentially in the cervical transformation zone (TZ) between the squamous epithelium of the ectocervix and the columnar epithelium of the endocervix. Because basal cells of the TZ are able to differentiate they are susceptible to HPV infection [22].
According to the Bethesda Classification of cervical dysplasia, the term CIN has now been changed to squamous intraepithelial lesion (SIL). In cervical cytology, three types of lesions are distinguished: low-grade or high-grade SIL (LSIL and HSIL, respectively) and atypical squamous cells (ASC). LSIL corresponds to CIN1 and HSIL corresponds to CIN2 and CIN3 [25].
In lesions such as mild dysplasia (CIN1) and moderate dysplasia (CIN2), low levels of E6 and E7 expression are observed in cells in the lower layers. Cells are stimulated to divide. Cells in mid layers are usually in the S or the G2 phase of the cell cycle and they produce E4 protein. In turn, cells in the upper layer leave the cell cycle and produce L1 and L2 proteins. In those cells, amplified viral genomes are packed into capsids. Lesions such as CIN3 (severe dysplasia, carcinoma in situ) and invasive cancer lesions reveal high levels of E6 and E7 expression [26].
4 Molecular Mechanism of HR HPV Cancer Development
The most important role in the process of carcinogenesis is attributed to the HPV E6 and E7 proteins. These proteins interact with selected human suppressors of neoplastic transformation, thereby affecting the course of the cell cycle and apoptosis [27, 28].
The P53 and RB genes are the most important human genes encoding proteins regulating the cell cycle and apoptosis. The p53 protein plays an important role in maintaining genomic stability in conditions harmful to the cell. It controls genetic material replication, participates in DNA repair and induces programmed cell death in the case of irreversible cell damage [29]. The content of p53 in normal cells is not high, but it increases rapidly when DNA in the cells is damaged. In addition, p53 can arrest the cell cycle at the G1/S phase and can maintain the integrity of the cell genome by enhancing the expression of the DNA repair protein. If DNA is severely damaged, p53 induces apoptosis, thereby avoiding the transfer of damaged genes to the next generation of cells. Continuous production and degradation of p53 in normal cells maintains its content at a certain level [30].
It has been demonstrated that the HPV E6 protein can interact with p53 and cause its degradation [28, 31]. In the first step, the E6 protein binds to the cellular E6-AP ubiquitin ligase, to which p53 binds, which in turn leads to the degradation of this complex in the proteasome. The consequence of excessive degradation of p53 is that the cells cannot normally repair DNA damages. As a result, the cell genome cannot maintain its integrity as gene mutations accumulate, and this finally leads to malignant transformation of the cells [32, 33]. p53 also has the ability to interact with the p21 and p27 proteins, important cyclin-dependent kinase (Cdk) inhibitors that inhibit the cellular transition from the G1 to S phase. The inactivation of p53 results in the inability to arrest the cell cycle and inhibit the apoptosis of damaged cells [34, 35]. Additionally, E6 protein up-regulates the expression of the Bcl-2 (B cell lymphoma 2) apoptosis-inhibiting protein and down-regulates the expression of the Bax (Bcl-2-like protein 4) apoptosis-promoting protein, leading to the inhibition of apoptosis [36, 37].
In turn, the HPV E7 protein negatively affects cellular pRb (retinoblastoma protein). pRb forms a complex with E2F (transcription factor family) in a non-phosphorylated form and stops the cell cycle in G1 in an intact cell (Fig. 3). The phosphorylated form of Rb cannot bind E2F, which causes the activation of genes involved in the cell transition from the G1 to the S phase of the cell cycle. The E7 protein interacts with the pRb, resulting in the release of the E2F factor, and this leads to the constitutive expression of genes responsible for the production of proteins necessary for cellular DNA replication [38, 39]. It has been shown that viral E7 protein can also bind to cellular p21 and p27 proteins. This process triggers the activation of Cdk4/6 and Cdk2 kinases dependent on D and E cyclins, the consequence of which is transition from G1 to the S phase and apoptosis inhibition [35, 39].

modified from Jayshree et al. [39]). Cdk4/6 and Cdk2 cyclin-dependent kinases, CycD cyclin D, E2F transcription factor, E6 and E7 human papillomavirus proteins, E6AP ubiquitin ligase, pRB retinoblastoma protein
Human papillomaviruses block apoptosis (
E7 protein can also promote cell dysplasia by stimulating the expression of human Pygopus (hPygo)2 gene, which codes for a protein component of the Wnt/β-catenin transcription complex. This protein is important for Wnt-dependent transcriptional activation during embryonic development. It is also over-expressed in various cancer cell lines. In HPV-positive cervical cancer cells, the level of hPygo2 messenger RNA (mRNA) and protein are higher than in uninfected cells. It has been experimentally confirmed that reduction of E7 expression by RNA interference promotes pRb binding to the hPygo2 promoter. In turn activation of hPygo2 is dependent on E74-like factor-1 (Elf-1). Thus, E7-mediated attenuation of pRb induces hPygo2 expression via Elf-1 in cervical cancer [40].
5 Telomerase Activation: An Important Target for HPV
The activation of telomerase, a holoenzyme responsible for the elongation of chromosome ends called telomeres, is one of the known mechanisms of HPV action. Telomeres protect chromosomes from degradation and fusion. They consist of multiple repetitions of the 5’-TTAGGG-3’ sequence. In human cells, the length of telomeres varies from 5000 to 15,000 nucleotides [41]. Telomeres are shortened with each cell division by 100–200 nucleotides. Reaching the critical telomere length is a signal for the cell to enter programmed cell death. Telomeres are extended by telomerase in actively dividing cells, e.g., stem cells; however, this enzyme remains inactive in fully differentiated cells. Telomerase is an enzyme composed of the TERC (telomerase RNA component) subunit, which is a template for telomeric sequence amplification, and the hTERT (human telomerase catalytic) subunit with the reverse transcriptase function. The telomerase complex also includes the dyskerin complex proteins (dyskerin, NOP10, NHP2, and GAR1). Moreover, the Shelterin protein complex, consisting of TRF1, TRF2, TIN2, POT1, RAP1, and TPP1 proteins, has telomerase-protective functions [42].
In vitro studies on keratinocytes have shown that neoplastic transformation of cells infected with HR HPV is mainly due to the viral E6 protein that induces telomerase activation [43]. The activity of this enzyme is not detected in cells in the absence of the E6 protein. The E6 protein acts on several levels: it directly interacts with hTERT and telomeric DNA [44]; and it mediates the regulation of the hTERT promoter and participates in the epigenetic and post-transcriptional regulation of hTERT [45]. The E6 protein interacts with E6AP ubiquitin ligase in the degradation of p53, but not telomerase degradation. Experimentally, it has been shown that reduction of E6 and E6AP levels by microRNA leads to the reduction of hTERT transcription and lower activity of telomerase in cells [46]. The E6/E6AP proteins bind to the hTERT promoter and activate it by interacting with the c-Myc protein (expression regulator) [47]. At the same time, the hTERT repressor complex (composed of the upstream transcription factor [USF] 1 and USF2 transcription factors) is replaced by c-Myc protein in the presence of E6 protein and, as a result, the hTERT gene transcription is increased [48] (Fig. 4). In turn, the second of the hTERT repressors—NFX1-91—is degraded in the cellular E6AP ubiquitin ligase-dependent manner [49]. Histone acetyltransferase (HAT) activity is increased and histone deacetylase (HDAC) activity is decreased as a result of NFX1-91 degradation, which binds SIN3 (transcription regulator family member A [mSin3A]), a transcriptional co-repressor responsible for HDACs recrutation. As a result, histone protein acetylation increases [50]. It has been shown that inhibiting endogenous NFX1-123 reduces the E6 protein capacity to activate telomerase [51]. In vitro studies have also indicated that DNA methylation patterns in HPV-positive cells are changing during HPV infection. In cells with HR E6 and E7 hypermethylation of specific hTERT promoter regions and hypomethylation of other regions were observed [52,53,54,55].

(modified from Howie et al. [31]). E6AP ubiquitin ligase
Telomerase activation by human papillomavirus protein E6. E6/E6AP affects hTERT promoter repressors—USF1/2—which bind to cis elements of promotor (X1 boxes, E boxes, and GC-rich sequences) and NFX1-91, and recruits histone deacetylase (HDAC) through mSin3A. cMYC/Max heterodimer, Sp1, and histone acetyltransferases (HAT) bind to the hTERT promoter and activate hTERT expression. hTERT activation is also increased by NFX1-123 with cytoplasmic poly(A) binding proteins (PABPCs) which cooperate with E6/E6AP
The results of these studies also indicate post-transcriptional regulation of telomerase activity by E6, mediated by cellular protein NFX1-123 (a longer splice variant of the NFX1 gene) that stabilizes binding to hTERT mRNA. Interaction of NFX1-123 via poly(A) binding protein interacting motif (PAM2) with cytoplasmic poly(A) binding proteins (PABPCs) (which bind to the poly(A) tail of mRNA) leads to an increase of protein expression through mRNA stabilization. PAM2 is critical for increasing hTERT expression in E6 expressing keratinocytes [45, 56].
Thus, the interaction of viral E6 protein with E6AP plays a key role in the induction of telomerase and its hTERT subunit.
Research also indicates another possible mechanism of action of the E6 protein. Chen et al. [57] have demonstrated that HPV16 E6 interacts physically with the KDM5C demethylase of H3K4 histone, which causes its degradation in the proteosome. A lower level of KDM5C expression was observed in the HPV16-infected cancer cell lines than in the HPV16-negative cell lines. Restoration of the original level of KDM5C resulted in significant inhibition of the neoplastic capacity of the CaSki line (HPV16-positive cell line derived from epidermoid carcinoma of the cervix metastatic to the small bowel mesentery).
The role in maintaining telomere length is also attributed to the E7 protein, which can regulate telomere elongation by means of the alternative lengthening of telomeres (ALT) pathway. The exact mechanism of this phenomenon remains unknown; however, Zhang et al. [58] postulated that the E7 protein could affect the degradation of p130 belonging to the pRb family, the role of which is ALT inhibition.
6 Potential Application of hTERC Analysis and Telomere Length as an Indicator of Early Tumorigenesis
The activation of telomerase, which, by extending the telomeres, leads to cell immortalization, is one of the first and also a key event indicating the initation of cell transformation. The length of telomeres in cancer cells results from a particular balance between the proliferative shortening of telomeres and their elongation by telomerase activity. Research on certain cancers, including colon cancer and cervical cancer, has provided information on tumor cell-specific dynamics of telomere length changes and confirmed that there was a correlation between telomere length and cancer stage. The shortest telomeres are found in cancer cells in the first stage of development [59,60,61]. In more advanced cancers, telomeres are longer, which indicates that stabilization of their length is achieved relatively late during tumorigenesis, after a period of active proliferation and telomere shortening [59]. Research on a group of over 100 healthy women, with ASCUS (ASC of undetermined significance), LSIL, HSIL, and invasive cervical cancer showed that telomerase activity increased with lesion development [62,63,64,65,66,67], and that a higher level of hTERT expression was characteristic of more advanced CIN3 lesions [68]. A high level of hTERT expression with a low level of TGFBR2 (transforming growth factor-β receptor type II) expression correlated with poor prognosis in patients with cervical cancer [69]. Studies conducted by Zappacosta et al. [70] have indicated the possible application of the hTERC analysis in the diagnostics of CIN2 lesions. Studies on a group of 54 women with ASCUS+ lesions showed that 96.3% had oncogenic HPV-DNA, and the expression of the hTERC gene was confirmed by fluorescence in situ hybridization (FISH) in more than 70% of women with CIN2+ lesions. In turn, Zheng et al. [71] examined 373 patients classified into CIN1, CIN2, and CIN3 groups depending on lesion advancement. Patients with invasive cervical cancer formed a separate group. The expression of the hTERC gene was confirmed in 41.86% of patients with CIN2 lesions, 78.29% of patients with CIN3 lesions, and 89.47% of patients with invasive carcinoma. The sensitivity of the hTERC FISH method, as a prognostic test in predicting lesion progression, was estimated at 88.89%. Similar conclusions were drawn from results obtained by subsequent research teams [71,72,73,74]. Ravaioli et al. [75] showed that use of the FISH method for detection of hTERC expression was a more reliable method, allowing for prediction of the progression of CIN2 to CIN3 lesions, as compared with the immunohistochemistry (IHC) method. The progression from CIN2 to CIN3 lesions is more frequent in patients with CIN lesions, who are hTERT-positive. Although the sensitivity of both methods is similar and amounts to about 86%, the FISH method is characterized by specificity two times higher than that of IHC. A new potential method of testing telomerase activity—asymmetric polymerase chain reaction (PCR) (A-PCR)—is characterized by insensitivity to PCR inhibitors and may find application in monitoring precancerous changes and the effect of therapy on cancer cells in the near future [76]. In turn, measurement of hTERT in serum may be useful in diagnosing and assessing the clinical stage of cervical cancer. Porika et al. [77] demonstrated that hTERT serum levels correlate with telomerase activity in 80.2% of squamous cell carcinoma and 73.8% of adenocarcinoma patients. This indicates a correlation with clinical stage, tumor size, and lymph node metastasis.
It seems that telomere length measurement can also be an indicator of early changes at the cellular level, before they can be detected by cytological methods. So far, the usefulness of the telomere length assessment has been demonstrated in patients with Barrett’s esophagus, whose telomere length could be a biomarker indicating the risk of developing cancer since shortened telomeres were observed before the tumor lesion [78]. The simultaneous occurrence of short telomeres and elevated levels of hTERT correlated with tumor aggressiveness and shorter survival in patients with neck and head cancer. A higher level of hTERT expression was observed among patients with an additionally confirmed HPV infection than in patients without HPV infection. At the same time, this group was characterized by a longer survival. The authors concluded that the high level of hTERT expression might have had other prognostic significance in patients with HPV infection-induced tumors [79, 80]. In turn, long telomeres correlated with poor prognosis in patients with esophageal squamous cell carcinoma and HPV infection. The length of telomere decreased in the following order: cancer, tissues adjacent to the tumor, and healthy tissue of the esophagus [81]. A study on the relationship between the length of leukocyte telomeres in patients with HPV16 infection and the risk of oral squamous cell carcinoma confirmed an increased risk, especially in young, non-smoking, and non-alcoholic patients [82, 83]. A study on a group of 16 patients showed that CIN lesions were associated with shortened telomeres, suggesting that telomere shortening precedes carcinogenesis [60]. Studies with mouse models have also indicated that CIN2 lesions have significantly shorter telomeres than corresponding normal squamous epithelia [84]. Additionally, Meeker et al. [85] reported that telomere shortening was a prevalent alteration in many epithelial lesions, including, among others, those of the prostate, pancreas, breast, and uterine cervix. The authors confirmed that shortening of the telomeres was a predominant abnormality in 88.6% of epithelial cancer precursor lesions and they concluded that telomere length abnormalities appear to be one of the earliest and most prevalent genetic alterations acquired in the multistep process of malignant transformation.
7 Summary
Carcinogenesis induced by HR HPV is a multistep process initiated by viral proteins, which can lead to apoptosis inhibition, genome instability, and uncontrolled cell division. Telomere shortening appears to be one of the earliest genetic alterations acquired in malignant transformation. Telomerase activation and telomere extension are crucial steps in the immortalization of cells. Effective detection of molecular events in the early carcinogenesis stages is important for prevention of cervical cancer development. Hence, the aim of many reports is to search for new biomarkers that would be useful in predicting the risk of progression and could be a potential tool for triage in cervical cancer screening. The level of hTERT expression correlates with dysplasia and telomerase activity, and increases with lesion development [62,63,64,65,66,67]. Likewise, telomere length measurement may be useful as a monitoring tool for early precancerous lesions. The problematic aspect of telomere measurement may be which selection method to use for quantifying telomere length. The most popular methods, such as terminal restriction fragment (TRF), quantitative PCR (qPCR), and a few subtypes of FISH, are most useful in measuring relative telomere length instead of absolute telomere length [86]. Furthermore, the length of telomeres seems to be an individual feature that can be different between tissues [87]. The correlation observed between telomere length and higher risk of some cancers [88, 89] seems to be promising for the future, but there is a need to refine methods to measure telomere lengths so they can be useful as a diagnostic tool.
References
Merkhofer C, Maslow J. Human papilloma virus (HPV) infection and non-cervical oncogenic disease states. Virol Mycol. 2015;4:144. https://doi.org/10.4172/2161-0517.1000144.
Boshart M, Gissmann L, Ikenberg H, Kleinheinzl A, Scheurlen W, Zur Hausen H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984;3:1151–7.
Gissmann L, Boshart M, Durst M, Ikenberg H, Wagner D, Zur Hausen H. Presence of human papillomavirus in genital tumors. J Invest Dermatol. 1984;83:265–85.
Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, et al. International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–27.
Myers ER, McCrory DC, Nanda K, Bastian L, Matchar DB. Mathematical model for the natural history of human papillomavirus infection and cervical carcinogenesis. Am J Epidemiol. 2000;151:1158–71.
Bosch FX, Lorincz A, Muñoz N, Meijer CJ, Shah KV. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 2002;55:244–65.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:359–86.
Tulay P, Serakinci N. The role of human papillomaviruses in cancer progression. J Cancer Metastasis Treat. 2016;2:201–13.
Abramowitz L, Jacquard AC, Jaroud F, Haesebaert J, Siproudhis L, Pradat P, et al. Human papillomavirus genotype distribution in anal cancer in France: the EDiTH V study. Int J Cancer. 2011;129:433–9.
De Vuyst H, Clifford GM, Nascimento MC, Madeleine MM, Franceschi S. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: a meta-analysis. Int J Cancer. 2009;124:1626–36.
Miralles-Guri C, Bruni L, Cubilla AL, Castellsagué X, Bosch FX, de Sanjosé S. Human papilloma virus prevalence and type distribution in penile carcinoma. J Clin Pathol. 2009;62:870–8.
St Guily JL, Jacquard AC, Prétet JL, Haesebaert J, Beby-Defaux A, Clavel C, et al. Human papillomavirus genotype distribution in oropharynx and oral cavity cancer in France–the EDiTH VI study. J Clin Virol. 2011;51:100–4.
Scarbrough Lefebvre CD, Van Kriekinge G, Gonçalves MA, de Sanjose S. Appraisal of the burden of genital warts from a healthcare and individual patient perspective. Public Health. 2011;125:464–75.
Ibeanu OA. Molecular pathogenesis of cervical cancer. Cancer Biol Ther. 2011;11:295–306.
Fernandes JV, de Medeiros Fernandes TAA. Human papillomavirus: biology and pathogenesis. In: Vanden Broeck D (Ed). Human papillomavirus and related diseases: from bench to bedside—a clinical perspective. InTechOpen. 2012. https://doi.org/10.5772/27154. https://www.intechopen.com/books/human-papillomavirus-and-related-diseases-from-bench-to-bedside-a-clinical-perspective/human-papillomavirus-biology-and-pathogenesis. Accessed 11 May 2018.
Morshed K, Polz-Gruszka D, Szymański M, Polz-Dacewicz M. Human papillomavirus (HPV)—structure, epidemiology and pathogenesis. Otolaryngol Pol. 2014;68:213–9.
Giroglou T, Florin L, Schäfer F, Streeck RE, Sapp M. Human papillomavirus infection requires cell surface heparan sulfate. J Virol. 2001;75:1565–70.
Culp TD, Budgeon LR, Marinkovich MP, Meneguzzi G, Christensen ND. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J Virol. 2006;80:8940–50.
Schelhaas M, Shah B, Holzer M, Blattmann P, Kühling L, Day PM, et al. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012;8:e1002657.
Spoden G, Kühling L, Cordes N, Frenzel B, Sapp M, Boller K, et al. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J Virol. 2013;87:7765–73.
Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, et al. Clathrin- and caveolin-independent entry of human papillomavirus type 16–involvement of tetraspanin-enriched microdomains (TEMs). PLoS One. 2008;3:e3313.
Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, et al. The biology and life-cycle of human papillomaviruses. Vaccine. 2012;30(suppl 5):F55–70.
Pinidis P, Tsikouras P, Iatrakis G, Zervoudis S, Koukouli Z, Bothou A, et al. Human papilloma virus’ life cycle and carcinogenesis. Maedica (Buchar). 2016;11:48–54.
Tomaić V. Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers (Basel). 2016;8:95.
Wright TC, Kurman RJ, Ferenczy A. Precancerous lesions of the cervix. In: Kurman RJ, editor. Blaustein’s pathology of the female genital tract. 5th ed. New York: Springer; 2002. p. 253–324.
Von Knebel Doeberitz M. New markers for cervical dysplasia to visualise the genomic chaos created by aberrant oncogenic papillomavirus infections. Eur J Cancer. 2002;38:2229–42.
Ghittoni R, Accardi R, Chiocca S, Tommasino M. Role of human papillomaviruses in carcinogenesis. Ecancermedicalscience. 2015;29(9):526.
Tommasino M. The human papillomavirus family and its role in carcinogenesis. Semin Cancer Biol. 2014;26:13–21.
McLaughlin-Drubin ME, Münger K. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143:195–208.
Fu Z, Tian H, Wang F, Zhao J. Carcinogenic mechanisms of oncoproteins in high-risk human papillomavirus. Int J Clin Exp Med. 2016;9:20439–47.
Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384:324–34.
Shai A, Pitot HC, Lambert PF. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010;70:5064–73.
Faridi R, Zahra A, Khan K, Idrees M. Oncogenic potential of Human Papillomavirus (HPV) and its relation with cervical cancer. Virol J. 2011;8:269.
Finzer P, Aguilar-Lemarroy A, Rosl F. The role of human papillomavirus oncoproteins E6 and E7 in apoptosis. Cancer Lett. 2002;188:15–24.
Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–60.
Cai Q, Lv L, Shao Q, Li X, Dian A. Human papillomavirus early proteins and apoptosis. Arch Gynecol Obstet. 2013;287:541–8.
McCloskey R, Menges C, Friedman A, Patel D, McCance DJ. Human papillomavirus type 16 E6/E7 upregulation of nucleophosmin is important for proliferation and inhibition of differentiation. J Virol. 2010;84:5131–9.
Adams PD. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim Biothys Acta. 2001;1471:M123–33.
Jayshree RS, Sreenivas A, Tessy M, Krishna S. Cell intrinsic & extrinsic factors in cervical carcinogenesis. Indian J Med Res. 2009;130:286–95.
Tzenov YR, Andrews PG, Voisey K, Popadiuk P, Xiong J, Popadiuk C, et al. Human papillomavirus (HPV) E7-mediated attenuation of retinoblastoma (Rb) induces hPygopus2 expression via Elf-1 in cervical cancer. Mol Cancer Res. 2013;11:19–30.
Schmidt JC, Cech TC. Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev. 2015;29:1095–105.
Wysoczańska B. Maintaining telomere length. Adv Hyg Med Exp. 2013;67:1319–30.
Miller J, Dakic A, Chen R, Palechor-Ceron N, Dai Y, Kallakury B, et al. HPV16 E7 protein and hTERT proteins defective for telomere maintenance cooperate to immortalize human keratinocytes. PLoS Pathog. 2013;9:e1003284.
Liu X, Dakic A, Zhang Y, Dai Y, Chen R, Schlegel R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc Natl Acad Sci USA. 2009;106:18780–5.
Katzenellenbogen R. Telomerase Induction in HPV infection and oncogenesis. Viruses. 2017;9:pii: E180.
Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82.
Veldman T, Liu X, Yuan H, Schlegel R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci USA. 2003;100:8211–6.
McMurray HR, McCance DJ. Human papillomavirus type 16, E6 activates TERT gene transcription through induction of c-Myc and release of USF mediated repression. J Virol. 2003;77:9852–61.
Gewin L, Myers H, Kiyono T, Galloway DA. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16, E6/E6-AP complex. Genes Dev. 2004;18:2269–82.
Xu M, Luo W, Elzi DJ, Grandori C, Galloway DA. NFX1 interacts with mSin3a/histone deacetylase to repress hTERT transcription in keratinocytes. Mol Cell Biol. 2008;28:4819–28.
Katzenellenbogen RA, Vliet-Gregg P, Xu M, Galloway DA. Nfx1-123 increases hTERT expression and telomerase activity posttranscriptionally in human papillomavirus type 16 E6 keratinocytes. J Virol. 2009;83:6446–56.
Schutze DM, Kooter JM, Wilting SM, Meijer CJ, Quint W, Snijders PJ, et al. Longitudinal assessment of DNA methylation changes during HPVE6E7-induced immortalization of primary keratinocytes. Epigenetics. 2015;10:73–81.
DeWild J, Kooter JM, Overmeer RM, Claassen-Kramer D, Meijer CJ, Snijders PJ, et al. hTERT promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer. 2010;10:271.
Jiang J, Zhao LJ, Zhao C, Zhang G, Zhao Y, Li JR, et al. Hypomethylated CpG around the transcription start site enables TERT expression and HPV16 E6 regulates TERT methylation in cervical cancer cells. Gynecol Oncol. 2012;124:534–41.
Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007;67:194–201.
Katzenellenbogen RA, Egelkrout EM, Vliet-Gregg P, Gewin LC, Gafken PR, Galloway DA. Nfx1-123 and poly(A) binding proteins synergistically augment activation of telomerase in human papillomavirus type 16E6 expressing cells. J Virol. 2007;81:3786–96.
Chen X, Loo JX, Shi X, Xiong W, Guo Y, Ke H, et al. E6 protein expressed by high-risk HPV activates super-enhancers of the EGFR and c-MET oncogenes by destabilizing the histone demethylase KDM5C. Cancer Res. 2018;78:1418–30.
Zhang W, Tian Y, Chen JJ, Zhao W, Yu X. A postulated role of p130 in telomere maintenance by human papillomavirus oncoprotein E7. Med Hypotheses. 2012;79:178–80.
Gertler R, Rosenberg R, Stricker D, Friederichs J, Hoos A, Werner M, et al. Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J Clin Oncol. 2004;22:1807–14.
Zhang A, Wang J, Zheng B, Fang X, Angström T, Liu C, et al. Telomere attrition predominantly occurs in precursor lesions during in vivo carcinogenic process of the uterine cervix. Oncogene. 2004;23:7441–7.
Barczak W, Suchorska WM, Sobecka A, Bednarowicz K, Machczynski P, Golusinski P, et al. hTERT C250T promoter mutation and telomere length as a molecular markers of cancer progression in patients with head and neck cancer. Mol Med Rep. 2017;16:441–6.
Reddy VG, Khanna N, Jain SK, Das BC, Singh N. Telomerase—a molecular marker for cervical cancer screening. Int J Gynecol Cancer. 2001;11:100–6.
Sen S, Reddy VG, Guleria R, Jain SK, Kapila K, Singh N. Telomerase—a potential molecular marker of lung and cervical cancer. Clin Chem Lab Med. 2002;40:994–1001.
Sharma A, Rajappa M, Saxena A, Sharma M. Telomerase activity as a tumor marker in Indian women with cervical intraepithelial neoplasia and cervical cancer. Mol Diagn Ther. 2007;11:193–201.
Barbosa LC, da Silva ID, Corrêa JC, Ribalta JC. Survivin and telomerase expression in the uterine cervix of women with human papillomavirus-induced lesions. Int J Gynecol Cancer. 2011;21:15–21.
Castro-Duque AF, Loango-Chamorro N, Ruiz-Hoyos BM, Landázuri P. Telomerase activity associated with progression of cervical lesions in a group of Colombian patients. Rev Bras Ginecol Obstet. 2015;37:559–64.
Molano M, Martín DC, Moreno-Acosta P, Hernández G, Cornall A, Buitrago O, et al. Telomerase activity in cervical scrapes of women with high-grade cervical disease: a nested case-control study. Oncol Lett. 2018;15:354–60.
Branca M, Giorgi C, Ciotti M, Santini D, Di Bonito L, Costa S, et al. Upregulation of telomerase (hTERT) is related to the grade of cervical intraepithelial neoplasia, but is not an independent predictor of high-risk human papillomavirus, virus persistence, or disease outcome in cervical cancer. Diagn Cytopathol. 2006;34:739–48.
Yang H, Zhang H, Zhong Y, Wang Q, Yang L, Kang H, et al. Concomitant underexpression of TGFBR2 and overexpression of hTERT are associated with poor prognosis in cervical cancer. Sci Rep. 2017;7:41670.
Zappacosta R. Ianieri MM,·Buca D,·Repetti E,·Ricciardulli A,·Liberati M. Clinical role of the detection of human telomerase RNA component gene amplification by fluorescence in situ hybridization on liquid-based cervical samples: comparison with human papillomavirus-DNA testing and histopathology. Acta Cytol. 2015;59:345–54.
Zheng X, Liang P, Zheng Y, Yi P, Liu Q, Han J, et al. Clinical significance of hTERC gene detection in exfoliated cervical epithelial cells for cervical lesions. Int J Gynecol Cancer. 2013;23:785–90.
Li Y, Zeng WJ, Ye F, Wang XY, Lü WG, Ma D, et al. Application of hTERC in thin prep samples with mild cytologic abnormality and HR-HPV positive. Gynecol Oncol. 2011;120:73–83.
Liu H, Liu S, Wang H, Xie X, Chen X, Zhang X, Zhang Y. Genomic amplification of the human telomerase gene (hTERC) associated with human papillomavirus is related to the progression of uterine cervical dysplasia to invasive cancer. Diagn Pathol. 2012;7:147.
Zhao XY, Cui Y, Jiang SF, Liu KJ, Han HQ, Liu XS, et al. Human telomerase gene and high-risk human papillomavirus infection are related to cervical intraepithelial neoplasia. Asian Pac J Cancer Prev. 2015;16:693–7.
Ravaioli S, Tumedei MM, Amadori A, Puccetti M, Chiadini E, Bravaccini S. Role of telomerase in cervical lesions as prognostic marker: a comparison between immunohistochemistry and fluorescence in situ hybridization. J Low Genit Tract Dis. 2017;21:42–6.
Yaku H, Yoshida Y, Okazawa H, Kiyono Y, Fujita Y, Miyoshi D. Highly sensitive telomerase assay insusceptible to telomerase and polymerase chain reaction inhibitors for cervical cancer screening using scraped cells. Anal Chem. 2017;89:6948–53.
Porika M, Tippani R, Mohammad A, Bollam SR, Panuganti SD, Abbagani S. Evaluation of serum human telomerase reverse transcriptase as a novel marker for cervical cancer. Int J Biol Markers. 2011;26:22–6.
Gertler R, Doll D, Maak M, Feith M, Rosenberg R. Telomere length and telomerase subunits as diagnostic and prognostic biomarkers in Barrett carcinoma. Cancer. 2008;112:2173–80.
Boscolo-Rizzo P, Da Mosto MC, Rampazzo E, Giunco S, Del Mistro A, Menegaldo A, et al. Telomeres and telomerase in head and neck squamous cell carcinoma: from pathogenesis to clinical implications. Cancer Metastasis Rev. 2016;35:457–74.
Boscolo-Rizzo P, Rampazzo E, Perissinotto E, Piano MA, Giunco S, Baboci L, et al. Telomere shortening in mucosa surrounding the tumor: biosensor of field cancerization and prognostic marker of mucosal failure in head and neck squamous cell carcinoma. Oral Oncol. 2015;51:500–7.
Zhang DH, Chen JY, Hong CQ, Yi DQ, Wang F, Cui W. High-risk human papillomavirus infection associated with telomere elongation in patients with esophageal squamous cell carcinoma with poor prognosis. Cancer. 2014;120:2673–83.
Zhang Y, Sturgis EM, Dahlstrom KR, Wen J, Liu H, Wei Q, et al. Telomere length in peripheral blood lymphocytes contributes to the development of HPV-associated oropharyngeal carcinoma. Cancer Res. 2013;73:5996–6003.
Yu Q, Yang J, Liu B, Li W, Hu G, Qiu H, et al. Combined effects of leukocyte telomere length, p53 polymorphism and human papillomavirus infection on esophageal squamous cell carcinoma in a Han Chinese population. Cancer Epidemiol. 2014;38:569–75.
Maida Y, Kyo S, Forsyth NR, Takakura M, Sakaguchi J, Mizumoto Y, et al. Distinct telomere length regulation in premalignant cervical and endometrial lesions: implications for the roles of telomeres in uterine carcinogenesis. J Pathol. 2006;210:214–23.
Meeker AK, Hicks JL, Iacobuzio-Donahue CA, Montgomery EA, Westra WH, Chan TY, et al. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin Cancer Res. 2004;10:3317–26.
Montpetit AJ, Alhareeri AA, Montpetit M, Starkweather AR, Elmore LW, Filler K, et al. Telomere length: a review of methods for measurement. Nurs Res. 2014;63:289–99.
Dlouha D, Maluskova J, Kralova Lesna I, Lanska V, Hubacek JA. Comparison of the relative telomere length measured in leukocytes and eleven different human tissues. Physiol Res. 2014;63:S343–50.
Hou L, Savage SA, Blaser MJ, Perez-Perez G, Hoxha M, Dioni L, et al. Telomere length in peripheral leukocyte DNA and gastrin cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18:3103–9.
Prescott J, Wentzensen I, Savage S, De Vivo I. Epidemiologic evidence for a role of dysfunction in cancer etiology. Mutat Res. 2012;730:75–84.
Acknowledgements
The authors wish to thank Natasha Ng for her suggestions for improving the language in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Anna Pańczyszyn, Ewa Boniewska-Bernacka, and Grzegorz Głąb declare no conflicts of interest.
Funding
The article was funded from the internal grant of Department of Biotechnology and Molecular Biology, University of Opole, Opole, Poland.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pańczyszyn, A., Boniewska-Bernacka, E. & Głąb, G. Telomeres and Telomerase During Human Papillomavirus-Induced Carcinogenesis. Mol Diagn Ther 22, 421–430 (2018). https://doi.org/10.1007/s40291-018-0336-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-018-0336-x