
Abstract
Strongly associated with tobacco use, heavy alcohol consumption, and with high-risk human papillomavirus (HPV) infection, head and neck squamous cell carcinoma (HNSCC) is a frequently lethal, heterogeneous disease whose pathogenesis is a multistep and multifactorial process involving genetic and epigenetic events. The majority of HNSCC patients present with locoregional advanced stage disease and are treated with combined modality strategies that can markedly impair quality of life and elicit unpredictable results. A large fraction of those who undergo locoregional treatment and achieve a complete response later develop locoregional recurrences or second field tumors. Biomarkers that are thus able to stratify risk and enable clinicians to tailor treatment plans and to personalize post-therapeutic surveillance strategies are highly desirable. To date, only HPV status is considered a reliable independent predictor of treatment response and survival in patients with HNSCC arising from the oropharyngeal site. Recent studies suggest that telomere attrition, which may be an early event in human carcinogenesis, and telomerase activation, which is detected in up to 90 % of malignancies, could be potential markers of cancer risk and disease outcome. This review examines the current state of knowledge on and discusses the implications linked to telomere dysfunction and telomerase activation in the development and clinical outcome of HNSCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Head and neck cancer was expected to affect approximately 742,000 and 144,000 new patients, worldwide and in Europe, respectively, in 2015 [1]. The most common type of head and neck cancer is head and neck squamous cell carcinoma (HNSCC) which is a morbid, frequently lethal disease that develops in the epithelial cell lining of the upper aero-digestive tract (UADT, i.e., the oral cavity, pharynx, and larynx) [2]. While it has been strongly associated with tobacco use and heavy alcohol consumption, over the past two decades, high-risk alpha human papillomaviruses (HR α-HPV) have emerged as an important etiological factor for a subset of HNSCC arising from the oropharynx [3, 4]. The prevalence of HPV-driven HNSCC has been dramatically increasing in developed countries, predominantly affecting males at a younger age that those with tobacco- and alcohol-related carcinomas, and changes considerably across different geographical areas [5–7]. HPV-driven HNSCC and tobacco- and alcohol-related HNSCC are two biologically and clinically distinct entities [6, 8–14].
Primarily a locoregional disease at presentation [2], HNSCC frequently spreads to the neck lymph nodes, an event that usually occurs before it metastasizes to distant organs. Approximately one third of patients present with early stage squamous cell carcinoma (SCC) (small primary tumors with no evidence of lymph node metastases). This prognostically favored subgroup is generally treated with a single-modality approach consisting in surgery or radiotherapy alone, with cure rates reaching up to 90 % [15]. Most HNSCCs present, however, at an advanced stage, with the majority of patients facing combined modality therapy consisting in surgery followed by adjuvant radio-(chemo)therapy or upfront chemoradiotherapy with salvage surgery in non-responders [16, 17]. The extraordinary advancements in treatment strategies have not as yet produced the hoped for cure rates [18]; regrettably, the majority of HNSCC patients who show a complete response to locoregional treatment later develop locoregional recurrence, and approximately 35 and 30 %, respectively, develop distant metastases or second primary tumors of the UADT, lung, or esophagus [19, 20]. The median survival in patients with inoperable recurrent or metastatic disease is only 10 months in those receiving the best established chemotherapy regimen [21]. Intensive treatment strategies for locoregional advanced disease such as sequential or concurrent chemotherapy in addition to locoregional treatment, taxane-platinum combination chemotherapy regimens, and altered fractionation radiotherapy protocols have yielded encouraging results, although at the expense of substantial acute and late toxicity [22–24].
Traditional prognostic factors such as overall stage and neck metastases at presentation seem to have limited prediction accuracy and reproducibility [25]. In an era of biomarker-driven personalized cancer therapy, several investigations are currently examining new biological markers as prognostic and predictive factors in HNSCC [26]. But with the exception of positivity to HPV and its surrogate cyclin-dependent kinase inhibitor p16INK4a, no other prognostic biomarkers are presently being used in routine clinical practice in head and neck oncology [27].
HNSCC is a heterogeneous disease whose pathogenesis is a multistep, multifactorial process involving genetic and epigenetic aberrations [28]. Invasive HNSCC may be clinically preceded by visible pre-neoplastic lesions often appearing as white patches, particularly common in the oral cavity and larynx [29], exhibiting an annual transformation rate exceeding 3 % [30]. Termed leukoplakia, these white patches may harbor hyperplastic or dysplastic epithelial lesions and their molecular characterization may quantify the risk of transformation [31]. Genetic alterations in pre-neoplastic lesions of the UADT may reflect aberrations in cellular differentiation or cell cycle control. Suprabasal expression of low molecular weight keratins [29], loss of heterozygosity of the TP53 gene [32], microsatellite instability [33], and higher chromosomal aneuploidy rates [34] all increase the risk of malignant transformation. Unfortunately, several pre-neoplastic lesions in the UADT are not clinically detectable.
Slaughter et al. first formulated the theory of “field cancerization” in 1953 [35] to explain high recurrence rates following tumor resections or UADT metachronous second primary tumors in patients treated for HNSCC. According to this model, the emergence of malignant lesions is preceded by the development of precancerous fields characterized by genetic alterations linked to carcinogen exposure. Following crucial genetic hits, a cell within the field can become cancerous and eventually give rise to invasive SCC. The risk of a second tumor is, moreover, markedly higher in cases in which this more-prone-to-transformation mucosa partially persists after the primary tumor has been treated, and this mechanism has recently been described in molecular terms [36, 37]. Clusters of cells with cancer-associated genetic alterations such as TP53 mutations have been detected in biopsies of histologically normal mucosa of HNSCC patients, and, in particular, in those with multiple primary malignancies [38]. Proteomic analysis has recently detected abnormal proteomic profiling in tumor-adjacent and tumor-distant UADT mucosa samples without histological aberrations [39]. Identifying markers of field cancerization could, therefore, hold promise for improving risk assessment and personalized post-therapy surveillance in HNSCC patients.
Recent whole-exome sequencing studies have painted new pictures of the genetic landscape of HNSCC and have uncovered unexpected therapeutic targets [40]. HNSCC’s mutational landscape is dominated by tumor suppressor genes with activating oncogene mutations playing an additional relevant role [41–43]. TP53, CDKN2A, CCND1, PIK3CA, and NOTCH1 are the most commonly mutated genes in HNSCC. Telomerase reverse transcriptase (TERT) promoter mutations resulting in increased telomerase expression have also been detected in a significant proportion of HNSCC patients [44–46].
The tumor suppressor p53 protein and the retinoblastoma (RB) tumor suppressor protein pathways are the most frequently deregulated signaling pathways in HNSCC [47]. Since activated p53 triggers the expression of several genes involved in cell cycle arrest, DNA repair, or apoptosis, it plays a crucial role in tumor suppression [48]. RB inhibits E2F transcription factor by direct protein-protein interactions thus preventing transition to the S phase of the cell cycle and promoting cell cycle arrest in G1 [49].
Most HPV-negative tumors harbor inactivating mutations in the TP53 gene [50]. In HNSCC with wild-type TP53, the protein may be inactivated by binding to the HPV E6 oncoprotein or to the cellular over-expressed MDM2 oncogene. Overall, the p53 pathway is down-regulated in at least 80 % of HNSCC [2].
The p16INK4a-cyclin D1-RB axis is also frequently deregulated in HNSCC. HPV-negative HNSCC show inactivation mainly by deletion or promoter hypermethylation of the CDKN2A gene encoding p16INK4a [51] and frequently have CCND1 amplification [52], which encodes cyclin D1, with both leading to a decrease in the growth-suppressive hypo-phosphorylated RB form.
In HPV-driven HNSCC, the p53 and RB pathways are both inactivated at the protein level. The E6 protein promotes cell proliferation by stimulating ubiquitination and proteasome-dependent degradation of the p53 protein via the formation of a trimeric complex including E6, p53, and the cellular ubiquitin ligase E6AP. In addition to targeting p53, HR α-HPV E6 activates telomerase [53]. E7 viral oncoprotein targets the RB/E2F complex, resulting in the dissociation of RB-family proteins from E2F transcription factors and inducing S phase entry [54]. HPV-driven HNSCC shows, moreover, a higher incidence of activating mutations in the PIK3CA gene, fewer gross chromosomal aberrations, and approximately one half the mutation rate of its HPV-negative counterparts [43]. In addition, vascular endothelial growth factor (VEGF)-C and VEGF receptor 3 are involved in the molecular pathways that lead to newly formed intra- and peritumoral lymphatic vessels, thereby endorsing cancer cell diffusion to the regional lymph nodes and explaining the high propensity of HNSCC for neck node metastases [55].
Regardless of what the driving force in HNSCC carcinogenesis may be (HR α-HPV persistent infection or tobacco/alcohol-related alterations in the expression of oncogenes and tumor suppressor genes), an unlimited replicative potential continues to be the hallmark of cancer [56]. The telomere/telomerase interplay is a key element in determining genomic stability and cellular replicative potential [57], and both telomerase expression/activity and telomere dysfunction have been extensively investigated in human cancer [57–59] with most studies indicating that they are crucial, early events in tumorigenesis often detectable at the precursor lesion stage [60–65].
This review examines current knowledge on the implications of telomere dysfunction and telomerase up-regulation in the development and clinical outcome of HNSCC.
2 Telomeres and telomerase: structure and regulation
2.1 Telomeres
Specialized DNA structures located at the ends of chromosomes, telomeres are essential for stabilizing chromosomes by protecting them from end-to-end fusion and DNA degradation. In human cells, telomeres are composed of (TTAGGG)n tandem repeats with a 3′ single-stranded overhang [66]. Telomeres are associated with capping proteins of the shelterin complex. Shelterin proteins enable cells to distinguish their chromosome ends from DNA breaks and to repress DNA repair reactions as well as to regulate telomere-based telomere maintenance [67]. Shelterin consists of six interdependent core subunits: TRF1 and TRF2 (telomeric repeat-binding factors 1 and 2), TIN2 (TRF1-interacting nuclear factor 2), Rap1 (TRF2-interacting protein 1), POT1 (protection of telomeres 1), and TPP1 (POT1 and TIN2-interacting protein 1) [67]. TRF1 and TRF2 bind the double-stranded telomeric repeats, whereas POT1 is recruited to the single-stranded overhang through its specific single-stranded DNA-binding activity and its interaction with TPP1. TIN2 tethers TPP1/POT1 to TRF1 and TRF2 [68]. The single-stranded overhang forms the telomeric loop (T-loop) by invading the double-stranded TTAGG repeats. T-loop remodeling, which is promoted and maintained by the members of the shelterin complex, mainly TRF2, prevents the recognition of chromosome ends as sites of double-strand breaks, thus repressing DNA damage signaling pathways and classical non-homologous end joining at telomeres or homologous recombination [69]. In particular, TRF2 represses the ataxia telangiectasia mutated protein (ATM)-mediated DNA damage signal, while POT1 prevents the ataxia telangiectasia and Rad3-related (ATR)-mediated DNA damage response, and both prevent end-to-end telomere fusions [67, 70].
Since DNA polymerase is unable to completely replicate the 3′ end of chromosomes, the loss of telomeric repeats, which occurs at each round of DNA replication, progressively reduces telomeres to a critical length [71]. While random DNA damage activates a DNA damage response (DDR) until damage is removed, dysfunctional telomeres trigger persistent DDR activation [72]. Indeed, lacking key shelterin proteins, uncapped telomeres trigger a DDR resulting in cell cycle arrest, cellular senescence, and finally apoptosis, mediated by cell checkpoints, such as p53 and p16INK4a/RB signaling pathways [73].
TRF2 depletion predominantly leads to ATM activation, whereas deprotection of 3′ single-stranded overhang due to POT1 depletion activates ATR. ATM and ATR phosphorylate downstream targets [74], eventually leading to phosphorylation and p53 activation [75]. p53 induces transcription of the cyclin-dependent kinase inhibitor p21CIP1 to promote cell cycle arrest [76] and apoptosis by up-regulating several pro-apoptotic target genes [77].
When p53 and p16INK4a/RB signaling pathways are inactive, cells can bypass senescence and continue to proliferate, allowing further telomere erosion which leads to increased genomic instability, including chromosome breakage-fusion-bridge (BFB) cycles causing structural chromosomal rearrangements. This results principally in an additional proliferative constraint characterized by mitotic catastrophe with widespread cell death [78]. Telomerase or other mechanisms of telomere preservation can be stochastically re-activated in the rare cells that have entered the BFB process. In this scenario, telomeres are maintained and genetically unstable cells gain immortal growth capacity, a crucial event on the path towards malignancy [79].
Telomere/telomerase interplay is therefore critical in determining genomic instability and cellular transformation and, as such, each seems to be wearing the mask of Janus Bifrons, the two-faced Roman god. The consequence of telomere erosion depends, indeed, on the cellular milieu: in checkpoint-proficient cells, it leads to tumor suppression by senescence or apoptosis; while in checkpoint-compromised ones, it leads to tumor promotion by causing genetic instability (Fig. 1).

Short telomeres: senescence and cancer. Telomeres, which are essential to protect chromosomes from deterioration and from end-to-end fusion, are specialized DNA structures located at the ends of chromosomes composed of (TTAGGG)n tandem repeats that are associated with capping proteins. Human adult somatic cells have a limited capacity to divide (Hayflick limit) as DNA polymerase alone cannot replicate the 3′ end of the DNA strand resulting in a progressive loss of TTAGGG telomeric sequences. Critically short telomeres trigger a DNA damage response, resulting in cellular senescence, an efficient tumor suppressor mechanism, and apoptosis. Senescence involves intact DNA damage checkpoints, such as p53 and p16/RB signaling pathways. Checkpoint-compromised cells can escape cellular senescence and apoptosis. In this context, cells can experience an increased number of divisions and can ultimately enter to breakage-fusion-bridge events and dramatic genetic instability due to telomere erosion, which most commonly leads to cell death. Rarely, the cell reactivates telomerase expression to drive telomere maintenance and replicative immortality. TERT telomerase reverse transcriptase
2.2 Telomerase
More than 90 % of cancers acquire the capability to replicate indefinitely through telomerase expression [80], a ribonucleoprotein complex containing an internal RNA component (TR or TERC) and a catalytic protein, TERT, with telomere-specific reverse transcriptase activity [81]. Less than 10 % of human cancers, typically non-epithelial tumors, preserve their telomeres by a telomerase-independent alternative lengthening of telomeres (ALT) pathway [82].
TERT, which synthesizes de novo telomere sequences using TR as a template, is the rate-limiting component of the telomerase complex, and its expression is correlated with telomerase activity [81]. Expression of TERT, which is normally strongly repressed by multiple tumor suppressors and which plays a critical role in tumor formation and progression, is essential for unlimited cell growth and is under tight transcriptional control [83]. Regulation of telomerase operates at several levels: telomere-associated proteins, in particular TRF1 and POT1, are themselves negative regulators of telomerase as they repress telomerase access to telomeric ends [84]. Since longer telomeres contain larger amounts of TRF1, telomere length is usually maintained at stable average values in telomerase-expressing cells [85].
TERT gene transcription is probably the key determinant in telomerase activity regulation; more than 20 transcription factor-binding sites acting as activators or repressors have been identified within the TERT promoter. p53, which may be activated by telomere shortening, binds to transcription factor Sp1 and renders TERT core promoter inaccessible to TERT promoter activation in normal somatic cells [86]. Similarly, other cell cycle inhibitors such as p16INK4a [87] and p27KIP1 [88] may down-regulate TERT expression. Nuclear factor-kB (NF-kB), hypoxia-inducible factor (HIF)-1, and the ETS/MYC complex bind to the TERT promoter thus increasing TERT expression. NF-kB can also induce TERT translocation from the cytoplasm to the nucleus [89]. TERT promoter activity is also critically dependent on the chromatin environment and DNA methylation status [90]. SET and MYND domain-containing protein 3 (SMYD3), a dimethyltransferase and trimethyltransferase, are significantly up-regulated in several cancers. SMYD3 increases trimethylation of histone H3-K4 in the TERT promoter, thereby directly trans-activating the TERT gene [91]. TERT promoter methylation status has unveiled a complex methylation pattern with some studies reporting hypomethylation in the CpG island in the TERT promoter region while others have described increased DNA methylation in TERT-expressing cancer cells [80].
Although telomerase’s primary function is telomere elongation and/or maintenance, several different extra-telomeric functions have also been described. These non-canonical functions may affect some cellular processes including gene expression, signaling pathways for the regulation of cell survival, resistance to stress, and apoptosis [92]. Of interest, the re-activation of telomerase in cancer cells may affect the cancer’s invasiveness and metastasis through the interaction of TERT with the Wnt/β-catenin [93] and the NF-kB signaling pathways [94].
In a minority of cases, cancer cells can maintain their telomeres by ALT. ALT-mediated telomere elongation is achieved by telomere recombination between telomere-sister chromatid exchanges or adjacent chromosomal telomere [95]. The ALT phenotype is usually characterized by heterogeneous telomere lengths, the presence of extra-chromosomal telomeric DNA molecules amassed within ALT-associated promyelocytic leukemia (PML) bodies, and reduced compaction of telomeric chromatin [96].
3 Telomere dysfunction in HNSCC
In physiological conditions, the stratified squamous epithelium expresses low levels of telomerase activity [97]. Regulated telomerase activity has indeed been detectable in a subset of normal transit-amplifying stem cells residing in the basal layer providing them with an extended proliferative competence [98]. Consistently, non-cancerous epithelia show basal layers with longer telomeres with respect to parabasal and suprabasal ones [64].
Controlled telomerase expression is nevertheless inadequate to prevent telomere attrition during aging [99], and telomere shortening and DNA damage accumulate in human stem cells [100]. Furthermore, during suprabasal differentiation, telomerase expression is silenced via the recruitment of the RB-associated histone deacetylase repressor complex to the −98 E2F site of TERT promoter [101]. All epithelial cells may experience a progressive shortening of TTAGGG repeats that eventually results in replicative senescence, a state of growth arrest which is considered an effective tumor suppressor mechanism [76]. In human oral keratinocytes, senescence is associated with enhanced expression of discrete gene groups including G-protein-coupled receptors, matrix metalloproteinases (MMP), apolipoproteins, and mitochondrial proteins [102]. Senescent cells may acquire a senescent-associated secretory phenotype (SASP) characterized by increased secretion of pro-inflammatory cytokines such as interleukin (IL)-6, IL-1, and IL-8, matrix metalloproteinases, and reactive oxygen species [103]. SASP may create a pro-carcinogenic microenvironment in UADT, promoting tumor development, proliferation, and invasiveness [104].
As stated above, the abrogation of important cell cycle checkpoints, such as p53 and RB, may allow cells to bypass replicative senescence and to enter a state of crisis characterized by extremely short telomeres which may lead to BFB cycles and chromothripsis [105].
The presence of anaphase bridges in the majority of HNSCC is suggestive of their escape from replicative senescence [106]. Supporting the theory that genetic instability in head and neck carcinogenesis can be triggered by telomere dysfunction, telomere attrition has been associated with BFB events, accumulation of centrosomes, and multipolar cell divisions in cell lines from benign and malignant head and neck tumors [107, 108]. Also demonstrated in mice models, telomere attrition appears to be the main driver of genetic instability [109].
Findings indicating that telomere aberrations, mainly consisting in shortening, are consistently found in HNSCC precursors and in mucosa surrounding pre-neoplastic areas and invasive UADT carcinomas [63, 64, 110, 111] support the hypothesis that genomic instability driven by telomere dysfunction is an early event in the HNSCC oncogenetic process. Notably, telomere shortening has also been detected in nearby non-neoplastic esophageal epithelium from patients with invasive esophageal cancer [112] as well as in cancer-associated normal stromal cells and pre-neoplastic lesions, respectively, from the prostate [113] and the pancreas [114]. Telomere erosion is thus emerging as a very early, common genetic event in epithelial carcinogenesis [63]. Remarkably, recent whole-exome sequencing studies have disclosed that genomic instability is a dominant feature in HNSCC with about 80 % showing significant chromosomal instability and more than 40 % exhibiting whole genome duplication [115, 116].
Recently explained in molecular terms, field cancerization of UADT epithelium [117] offers a unique opportunity to study the multistep process of carcinogenesis during its earliest stages. Telomeres in the epithelium of the UADT from healthy subjects have been shown to shorten with age [118]. But the lengths of telomeres in histologically normal mucosa surrounding UADT cancer lesions do not appear to be correlated with patients’ ages, thus suggesting that the epithelium harbors some proliferative aberrations [63].
Studies examining telomere length in uninvolved, adjacent epithelium in carcinoma in situ of the oral cavity and in oral specimens from individuals without HNSCC have demonstrated that they are significantly shorter and the anaphase bridges are more abundant in the surrounding tissue of carcinomas in situ with respect to those in control specimens [64]. Telomere length analysis of orthokeratotic dysplasia, a precancerous condition frequently associated with SCC and of its surrounding background tissue, uncovered significantly shorter telomeres and a higher frequency of BFB with respect to those in controls [110].
A substantial number of HNSCC patients have been found to have extremely short telomeres in histologically normal mucosa surrounding SCC and, even more unexpectedly, short telomeres in background tissue were strongly and independently associated with mucosal failure [63]. It can be inferred from these findings that SCC arises in a telomere-shortened epithelial field characterized by genetic instability and a prone-to-transformation status. In this context, telomere shortening can be considered a biosensor of field cancerization that can identify patients at risk of local relapses or second field tumors. Supporting the hypothesis that telomere attrition precedes telomerase re-activation, TERT levels in mucosa surrounding HNSCC were not correlated with mucosal failure [63].
The amplification or re-activation of telomerase expression in stem or differentiated cells is considered a key event to escape crisis and to gain immortalization [97], and telomerase expression is necessary to convert and maintain an immortalized phenotype in human oral keratinocytes [119].
4 Telomerase in HNSCC
While telomerase activity is detectable in most tumors, it is usually absent in normal somatic cells [66]. Telomerase activity can be measured by means of polymerase chain reaction (PCR)-based telomere-repeat amplification protocol (TRAP), by scoring immunohistochemical TERT staining or by quantifying TERT mRNA using real-time PCR [81, 120, 121].
When real-time PCR or TRAP have been utilized, weak telomerase expression and activity have frequently been found in normal UADT epithelium [61, 63, 122–136]. High levels of telomerase expression have been detected in as much as 75–100 % of HNSCC [111, 126–128, 130–135, 137–148]. According to in vitro studies, telomerase is induced in a substantial proportion of HNSCCs in telomerase-deficient keratinocytes and not by telomerase overexpression in telomerase-positive cells. This can be inferred by the high anaphase bridge index, dicentric chromosomes, and multipolar spindles, which indicate that these cells have experienced a period of critically shortened TTAGGG repeats before telomerase re-activation [106, 107]. Telomerase re-activation or up-regulation can be achieved by gene amplification, promoter mutations, TERT mRNA alternative splicing, epigenetic changes, or through post-translational processing [97].
Liu et al. [149] reported that TR amplification was one of the most frequent amplifications observed in laryngeal SCC with the rate rising progressively with the increasing severity of the lesions: none were, indeed, found in normal epithelium and 100 % in invasive cancer. Telomerase activity, however, is strictly correlated with TERT levels, while higher TR levels do not affect its catalytic properties [81, 150]. Comprehensive genomic characterization of HNSCC has frequently uncovered amplification of chromosome 5p, which encompasses TERT gene, in both HPV-positive and HPV-negative carcinomas [42, 151].
TERT promoter is transcriptionally silenced upon cellular differentiation. In cancer cells, TERT expression is induced by several cellular transcription factors including NF-kB [152], β-catenin [153], and c-Myc [154]. Conversely, wild-type TP53 down-regulates TERT by forming a complex with the transcription activator Sp1 and, thus, inhibiting Sp1 binding to the TERT proximal promoter [86].
HR α-HPV E6 oncoprotein contributes to cell immortalization and transformation through both p53 degradation and telomerase activation. HPV-driven oropharyngeal SCCs express very high TERT levels [63]. The mechanisms underlying telomerase activation by E6 viral oncoprotein have not been completely elucidated. E6 can indirectly increase TERT expression by inducing p53 degradation. Furthermore, the increase in telomerase expression and activity in HPV-transformed cells could be the effect of E6-induced TERT transcription or post-transcriptional mechanisms. E6 from HR α-HPV can activate telomerase via degradation of NFX1-91, a transcription repressor of TERT [155]. In addition, HPV16 E6 physically and functionally interacts with telomerase complex and increases TERT catalytic activity [53]. Latent membrane protein 1 (LMP1) of Epstein–Barr virus may activate TERT via NF-kB and MAPK/ERK1/2 [156, 157]. In EBV-associated nasopharyngeal carcinoma, LMP1 enhances TERT expression and phosphorylation through the PI3K/AKT signaling pathway, and this makes the cells more resistant to irradiation [157].
Very recently, two prevalent and mutually exclusive mutations in the promoter of TERT (228C>T and 250C>T) emerged as the most frequently observed non-coding mutations in cancer and were associated with high levels of telomerase in multiple cancer types [158]. Notably, these mutations specifically promote TERT expression in telomerase-negative differentiated cell compartments, while their impact in telomerase-positive stem cell reservoirs appears to be neutral [150]. Thus, TERT promoter mutations uncouple cellular differentiation and telomerase silencing. These mutations result in generating de novo binding sites for transcription factors of the E26 transformation-specific family (ETS). In particular, the ETS GA-binding protein transcription factor α subunit selectively links the mutant form of the TERT promoter and increases TERT transcriptional activity [159]. More common in tumors derived from cells with low self-renewal rates, TERT promoter mutations have also been detected in a significant proportion of oral tongue [44, 45] and laryngeal SCCs [46]. SCCs of the base of the tongue, which are usually HPV-related, harbor, instead, wild-type TERT promoter [44]. HR α-HPV infection and TERT promoter mutations may be alternative mechanisms to up-regulate telomerase in HNSCC [44]. Remarkably, 228C > T and 250C > T mutations in the TERT promoter are more frequent in laryngeal tumors in smokers compared to that in non-smokers and are independently associated with poor overall survival (OS) [46].TERT promoter activity can be modified by a common polymorphism within the preexisting ETS2 binding site in the TERT promoter with patients with the rs2736098 variant and in particular those exposed to tobacco and alcohol having a lower risk of SCC of the oropharynx [160].
As histone deacetylase inhibitor FR901228 induces a significant increase in TERT expression in oral cancer cell lines, it is possible that the latter is epigenetically controlled in HNSCC through changes in DNA methylation or histone acetylation [161]. TERT regulation can also occur via post-translational protein modification [162]. Specific protein kinase C isoenzymes, namely α, β, δ, ɛ, and ζ (over-expressed in this malignancy), in HNSCC have been shown to regulate telomerase activity by phosphorylating TERT. This phosphorylation results in the interaction of telomerase and chaperone protein hsp90, an essential step for telomerase holoprotein integrity and enzymatic activity [163].
As telomerase expression is found in a large fraction of HNSCC, a subgroup of these tumors does not seem to require telomerase to maintain telomere, although the presence of Taq polymerase inhibitors and RNA degradation during specimen collection and processing cannot be excluded [133]. Human laryngeal cancer cell lines that survive after transfection with RNAi plasmid targeting TERT sequence show sustained proliferation and the presence of PML bodies suggesting that they are capable of employing an ALT pathway to maintain their telomeres [164]. This finding may pose a challenge if telomerase inhibitors are used to treat HNSCC.
As far as UADT pre-neoplastic lesions are concerned, telomerase expression is positively correlated with their severity in both oral and laryngeal SCC [122, 123, 134, 146, 147, 149, 165]. Interestingly, oral verrucous leukoplakia, an aggressive, premalignant lesion, exhibits telomerase activity levels approaching those found in invasive oral SCC [129]. Telomerase can be associated, alone or together with other markers, with a more aggressive phenotype and behavior in HNSCC. Several studies have, indeed, reported that TERT mRNA levels and telomerase activity are higher in poorly differentiated SCC [63, 126, 143, 146, 166–169], increase with tumor stage [63, 131, 142, 166], and are associated with lymph node involvement [63, 126, 131, 137, 139, 170] or extracapsular extension of lymph node metastases [125]. Numerous studies have analyzed telomerase’s ability to predict outcome (Table 1) [46, 63, 125, 127, 130, 136, 139, 140, 144, 171–177], and most have reported a correlation between increased telomerase expression and activity and a reduced response to treatment and a higher rate of regional and distant metastases ultimately resulting in poor clinical outcome [63, 125, 127, 136, 144, 171, 173–176]. A study examining a large sample of neoplastic tissues from 217 HNSCC patients reported that telomerase activity was an independent predictor of poor survival after adjustment was made for age, site, stage, histological grade, tumor depth, and extracapsular extension [125]. These results have been corroborated by studies focusing on other malignancies that have shown that telomerase activity correlates with progression and poor prognosis in lung [60], colorectal [62], breast [178], and prostate [179] cancers. These data are consistent with previous experimental observation and studies using in vivo mouse models demonstrating that a systemic injection of anti-telomerase ribozyme inhibiting telomerase activity significantly reduces metastatic progression in tumor-bearing mice [180, 181].
The associations between high telomerase expression and activity and more aggressive phenotypes and progression in HNSCC may be ascribable not only to its ability to maintain telomere lengths in rapidly proliferating cells but also to TERT’s extra-telomeric functions and their interactions with other cancer-related signaling cascades, such as Wnt/β-catenin and NF-kB pathways [92, 182]. TERT’s extra-telomeric functions are implicated in regulating several cancer hallmarks including cell proliferation, angiogenesis, invasion, and metastasis [57]. Sustaining the roles of telomerase non-canonical functions in tumor invasiveness and progression in HNSCC, the instauration of ALT mechanisms to elongate telomeres in laryngeal cancer cells following telomerase inhibition, while maintaining a transformed phenotype, has been associated with less aggressive tumor features [164].
The Wnt/β-catenin pathway is a crucial regulator of the self-renewal property of normal amplifying adult stem cells, and its deregulation plays a critical role in abnormal cell proliferation and oral oncogenesis [183, 184]. Activation of the canonical Wnt signaling pathway results in the stabilization and nuclear translocation of β-catenin. In the absence of Wnt stimulus, cytoplasmic β-catenin is constantly targeted to the proteasome by the multiprotein destruction complex, which includes protein kinases CK1γ and GSK3β [185]. When activated, the Wnt/β-catenin pathway results in increasingly higher levels of β-catenin that eventually move to the nucleus and interact with LEF/TCF transcription factors thus promoting the transcription of target genes such as MYC and cyclin D1 [186]. Epithelial-to-mesenchymal transition (EMT), an essential step in cancer invasiveness and metastatic dissemination which is characterized by loss of cell-cell adhesion and the acquisition of migratory properties, is regulated by several proteins including β-catenin and vimentin [187]. TERT can stimulate the EMT program and induce an undifferentiated stem-like phenotype, a process associated with β-catenin signaling activation [93]. TERT binds β-catenin thus preventing its degradation, enhancing its nuclear accumulation and transcriptional activity, and regulates vimentin transcription in cooperation with β-catenin [93]. It has recently been observed that the overexpression of TERT in oral SCC is sufficient to induce a mesenchymal phenotype, and this is strictly related to the activation of the Wnt/β-catenin pathway. Silencing TERT, instead, leads to the inhibition of Wnt/β-catenin signaling and the suppression of EMT in oral cancer [188]. Interestingly, β-catenin is a known activator of TERT expression [153].
The NF-kB pathway regulates the expression of several genes including those involved in cellular proliferation, differentiation, and apoptosis. NF-kB is constitutively expressed in HNSCC and plays a crucial role as the modulator of the gene expression program associated with maintaining the malignant phenotype, invasiveness, and metastasis in SCC [189, 190]. It has been established that NF-kB regulates TERT expression by binding to a site 350-bp upstream from the translational start site. TERT nevertheless directly interacts with the NF-kB p65 subunit and regulates the expression of NF-κB target genes, such as IL-6, tumor necrosis factor (TNF)-α, IL-8, MMP9, commonly over-expressed in HNSCC [191]. IL-6 and IL-8 both induce EMT and promote metastasis in HNSCC via activation, respectively, of JAK-STAT3-SNAIL and AKT signaling pathways [192, 193]. In addition to IL-6, TNF-α also converges upon STAT3 up-regulating it. Moreover, TNF-α increases cell motility, migration, and invasion of human hypopharyngeal cancer cells by inducing TWIST expression, a basic helix-loop-helix transcription factor and dominant regulator of the EMT program in many solid tumors [194]. Some have hypothesized that TERT provides oral cancer cells with invasion capability by modulating cathepsin D, MMP2, and MMP9 which, in turn, degrade the extracellular matrix and collagen IV, essential for basement membrane stability and integrity [195].
HPV-driven oropharyngeal SCCs show a higher propensity for lymph node metastasis with respect to their HPV-negative counterparts and are not uncommonly characterized by an atypical pattern of distant metastases [6]. Preliminary data have shown that these malignancies may express extremely elevated TERT levels [63]. It can be speculated that the consequent increase in telomerase’s extra-telomeric functions may be related to this clinical behavior. TERT may, thus, be strictly involved in regulating critical SCC-related pathways, such as Wnt/β-catenin and NF-kB signaling, in a feed-forward loop context that amplifies and sustains autonomous cancer cell proliferation and tumor progression (Fig. Fig. 2).

Epithelial carcinogenesis and telomere/telomerase interplay. The inactivation of the p53 and RB pathways are the main molecular determinants in head and neck carcinogenesis. In tobacco- and alcohol-related HNSCC, the abrogation of p53 and RB pathways may occur via mutation and genetic/epigenetic alterations. In HPV-driven carcinomas, p53 and RB pathways are inactivated at the protein level by E6 and E7 HR α-HPV oncoproteins, respectively. In this context, cells can bypass cellular senescence (a condition triggered by telomere shortening in which cells remain viable but are unable to divide) and experience an increased number of cell divisions of potentially premalignant clones characterized by extremely shortened telomeres and genetic instability. Different strategies may lead to re-activation of telomerase, a ribonucleoprotein complex containing an internal RNA component and a catalytic protein, TERT, with telomere-specific reverse transcriptase activity which synthesizes de novo telomere sequences. Cells can thus escape from apoptosis and maintain short but stable telomere lengths which are the key to cell immortality. Besides providing cells with unlimited proliferation potential, telomerase interacts with other cancer-related signaling cascades, such as Wnt/β-catenin and NF-kB pathways. In this scenario, telomerase plays additional non-canonical roles that may impact cancer progression by inducing crucial factors, such as MMP9, TNF-α, IL-6, and activating cellular programs leading to increased tumor cell motility/migration/invasion capability and epithelial-to-mesenchymal transition, in a context of feed-forward loops. HR α-HPV high-risk alpha human papillomaviruses, TERT telomerase reverse transcriptase, NF-kB nuclear factor-kB, EMT epithelial-to-mesenchymal transition, MMP matrix metalloproteinase, TNF tumor necrosis factor, RB protein retinoblastoma
5 Telomere length in peripheral blood mononuclear cells (PBMC) and risk of HNSCC
As telomere length shortens with age, the parameter can be used as a marker of biological aging. But the high rate of inter-individual variations in telomere length among age-matched peers [196] appears to suggest that it reflects an individual risk for age-related diseases and cancer [197]. As telomere lengths in PBMC are strongly correlated with those in cells of different tissues, they are considered a surrogate marker for other tissues [198]. In cancer epidemiological investigations, telomere lengths in PBMC are usually determined to estimate the correlation between biological aging and cancer risk.
The role of telomere length in PBMC and cancer risk is quite controversial depending on the tumor type and the familial or sporadic context. Nevertheless, as suggested by Zhang et al. [58], various neoplasms show intrinsic biological heterogeneity, and different biological pathways may be modulated in various ways by telomere status. Telomere shortening in PBMC could be considered a biosensor of endogenous and environmental damage: factors increasing oxidative stress, e.g., cigarette smoking, are associated with telomere shortening in PBMC, while telomere erosion is, in turn, a driver of genetic instability which may promote tumorigenesis [58, 199, 200].
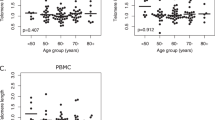
One large case–control study did not find a significant association between telomere length in PBMCs and HNSCC risk [201]. Conversely, two recently published case–control studies [202, 203] confirmed the results obtained a decade ago by Wu et al. [204] showing that telomeres were significantly shorter in PBMCs in HNSCC patients with respect to those in controls. When 266 patients with oral premalignant lesions or oral SCCs were compared with 394 age- and sex-matched controls, shortened telomeres were found to be an independent risk factor in PBMCs, although the risk was considerably higher in tobacco and alcohol consumers [202]. Telomere length also appeared to influence the malignant progression of pre-neoplastic lesions of the oral cavity. Short telomeres in PBMC may constitute an additional biomarker of oral habits and thus help to identify subjects at high risk of HNSCC. In another study [203] examining the association between telomere length in PBMCs and the risk of HR α-HPV-associated oropharyngeal SCC, the authors reported that short telomeres appeared to synergize with HPV type 16, increasing the risk of oropharyngeal SCC, particularly in the younger never smoker/drinker subgroup.
6 Telomerase in the peripheral blood compartment: can liquid biopsies be used as potential biomarkers for HNSCC?
Circulating tumor cells and tumor-driven nucleic acids, such as circulating cell-free DNA and RNA, could be useful in detecting real-time tumor dynamics and in monitoring drug sensitivity during treatment.
Telomerase expression and activity in the peripheral blood compartment of HNSCC patients has been estimated in PBMCs or quantified by measuring circulating cell-free TERT mRNA by some studies.
The rate of telomerase activity in PBMCs has been found to be significantly higher in HNSCC patients with respect to that in healthy controls and associated with advanced stage, lymph node metastases, and poor overall survival (OS) [205, 206]. Two mechanisms have been hypothesized to explain this finding [205]. First, PBMC could be activated by soluble factors, secreted either by the cancer or by the tumor microenvironment. Mean serum vascular endothelial growth factor levels have, indeed, been significantly linked with telomerase activity in PBMCs in HNSCC patients [207]. Alternatively, PBMC could be activated after tumor antigen processing and cross-presentation has taken place in draining lymph nodes.
Several authors have reported higher TERT mRNA plasma levels in cancer patients with respect to those in controls, and a correlation has been found between circulating TERT levels and more severe clinical-pathological features and disease outcomes [208–210]; thus, circulating TERT could be considered a noninvasive tool for detecting cancers and monitoring the course of treatment. To our knowledge, the only study that has investigated the significance of TERT mRNA plasma levels in HNSCC patients reported that the values were indeed significantly elevated before surgery and that they decreased significantly two days after surgery [211]. Additional studies are clearly warranted to verify the feasibility of using cell-free circulating plasma TERT mRNA to diagnose the carcinoma early and to monitor treatment response in these patients.
7 Conclusions
Although telomere dysfunction and telomerase activation appear to be dynamic processes during epithelial carcinogenesis, studies carried out until now have attempted to photograph them at a single point in time and have been unable to capture their mutable complex interaction. Our understanding of the complicated telomere/telomerase interplay in human cancer remains, in fact, for the time being incomplete. In the light of the comprehensive review of recently performed studies presented here, some conclusions can, however, be drawn about the clinicopathological and prognostic significance of telomere status and telomerase activity in HNSCC (Box 1).
Box 1. Key findings regarding telomeres and telomerase in HNSCC
• HNSCC precursors and normal mucosa surrounding pre-neoplastic areas and invasive carcinomas are characterized by shortened telomeres. |
• Short telomere lengths in mucosa surrounding HNSCC are strongly prognostic of mucosal failure. |
• Short telomere lengths in normal mucosa surrounding HNSCC can be considered a marker of “field cancerization.” |
• Telomerase activation plays a role in the majority of HNSCC cases. |
• The timing of telomerase expression and activation may differ depending on the genetic and epigenetic context. |
• Telomerase activity, which increases with tumor progression, is a prognostic marker of regional and distant failure. |
• The significance of telomere length and telomerase activity in peripheral blood cells and of circulating TERT mRNA levels in the plasma of patients with HNSCC are complex issues warranting further studies |
HNSCC head and neck squamous cell carcinoma, TERT telomerase reverse transcriptase
Analysis of SCC precursors and the normal mucosa surrounding pre-neoplastic and neoplastic areas has uncovered that extremely short telomeres are independently associated with mucosal failure and, thus, could represent potential biomarkers identifying patients at risk for relapses.
Several studies have shown that TERT mRNA levels and telomerase activity in HNSCC, which are associated with poor outcomes, gradually rise commensurately with the degree of epithelial aberrations and disease aggressiveness.
In conclusion, telomere/telomerase interplay and telomere shortening warrant further investigation in view of their ability to stratify HNSCC patients and the implications that they have on treatment and follow-up strategies in this particular patient population (Box 2).
Box 2. Questions that are still open with regard to telomeres and telomerase’s roles in the development of HNSCC
• Are the mechanisms, significance, and effects of telomerase re-activation different in HPV-negative with respect to HPV-driven HNSCC? |
• Are shortened telomeres in normal mucosa adjacent to HNSCC the consequence of greater cell proliferation or are they linked to an individual’s constitutive telomere length? |
• How does telomerase interact and cooperate with other cancer-related intracellular signaling pathways in head and neck tumorigenesis? |
• Are telomerase’s non-canonical functions critical for cancer invasion and metastasis HNSCC? |
• What is the significance of telomere length in peripheral blood mononuclear cells? |
• Is cell-free circulating plasma TERT mRNA a useful marker in diagnosing HNSCC and monitoring of treatment response? |
HNSCC head and neck squamous cell carcinoma, HPV human papillomavirus, TERT telomerase reverse transcriptase
References
Ferlay, J., Steliarova-Foucher, E., Lortet-Tieulent, J., Rosso, S., Coebergh, J. W., Comber, H., et al. (2013). Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. European Journal of Cancer, 49, 1374–403.
Leemans, C. R., Braakhuis, B. J., & Brakenhoff, R. H. (2011). The molecular biology of head and neck cancer. Nature Reviews Cancer, 11, 9–22.
Gillison, M. L., Koch, W. M., Capone, R. B., Spafford, M., Westra, W. H., Wu, L., et al. (2000). Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. Journal of the National Cancer Institute, 92, 709–20.
Snijders, P. J., Cromme, F. V., van den Brule, A. J., Schrijnemakers, H. F., Snow, G. B., Meijer, C. J., et al. (1992). Prevalence and expression of human papillomavirus in tonsillar carcinomas, indicating a possible viral etiology. International Journal of Cancer, 51, 845–50.
Chaturvedi, A. K., Anderson, W. F., Lortet-Tieulent, J., Curado, M. P., Ferlay, J., Franceschi, S., et al. (2013). Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 31, 4550–9. doi:10.1200/JCO.2013.50.3870.
Boscolo-Rizzo, P., Del Mistro, A., Bussu, F., Lupato, V., Baboci, L., Almadori, G., et al. (2013). New insights into human papillomavirus-associated head and neck squamous cell carcinoma. Acta Otorhinolaryngol Ital Organo Uff Della Soc Ital Otorinolaringol E Chir Cerv-Facc, 33, 77–87.
Baboci, L., Holzinger, D., Boscolo-Rizzo, P., Tirelli, G., Spinato, R., Lupato, V., et al. (2016). Low prevalence of HPV-driven head and neck squamous cell carcinoma in North-East Italy. Papillomavirus Research, 2, 133–40. doi:10.1016/j.pvr.2016.07.002.
Andl, T., Kahn, T., Pfuhl, A., Nicola, T., Erber, R., Conradt, C., et al. (1998). Etiological involvement of oncogenic human papillomavirus in tonsillar squamous cell carcinomas lacking retinoblastoma cell cycle control. Cancer Research, 58, 5–13.
Braakhuis, B. J. M., Snijders, P. J. F., Keune, W.-J. H., Meijer, C. J. L. M., Ruijter-Schippers, H. J., Leemans, C. R., et al. (2004). Genetic patterns in head and neck cancers that contain or lack transcriptionally active human papillomavirus. Journal of the National Cancer Institute, 96, 998–1006.
Jung, A. C., Briolat, J., Millon, R., de Reyniès, A., Rickman, D., Thomas, E., et al. (2010). Biological and clinical relevance of transcriptionally active human papillomavirus (HPV) infection in oropharynx squamous cell carcinoma. International Journal of Cancer, 126, 1882–94. doi:10.1002/ijc.24911.
Lindquist, D., Romanitan, M., Hammarstedt, L., Näsman, A., Dahlstrand, H., Lindholm, J., et al. (2007). Human papillomavirus is a favourable prognostic factor in tonsillar cancer and its oncogenic role is supported by the expression of E6 and E7. Molecular Oncology, 1, 350–5. doi:10.1016/j.molonc.2007.08.005.
van Houten, V. M., Snijders, P. J., van den Brekel, M. W., Kummer, J. A., Meijer, C. J., van Leeuwen, B., et al. (2001). Biological evidence that human papillomaviruses are etiologically involved in a subgroup of head and neck squamous cell carcinomas. International Journal of Cancer, 93, 232–5. doi:10.1002/ijc.1313.
Wiest, T., Schwarz, E., Enders, C., Flechtenmacher, C., & Bosch, F. X. (2002). Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene, 21, 1510–7. doi:10.1038/sj.onc.1205214.
Hernandez, B. Y., Rahman, M., Lynch, C. F., Cozen, W., Unger, E. R., Steinau, M., et al. (2016). p16(INK4A) expression in invasive laryngeal cancer. Papillomavirus Research, 2, 52–5. doi:10.1016/j.pvr.2016.03.001.
Suh, Y., Amelio, I., Guerrero Urbano, T., & Tavassoli, M. (2014). Clinical update on cancer: molecular oncology of head and neck cancer. Cell Death & Disease, 5, e1018. doi:10.1038/cddis.2013.548.
Boscolo-Rizzo, P., Gava, A., Marchiori, C., Baggio, V., & Da Mosto, M. C. (2011). Functional organ preservation in patients with locoregionally advanced head and neck squamous cell carcinoma treated by platinum-based multidrug induction chemotherapy and concurrent chemoradiotherapy. Annals of Oncology: Official Journal of the European Society of Medical Oncology ESMO, 22, 1894–901. doi:10.1093/annonc/mdq681.
Boscolo-Rizzo, P., Gava, A., Baggio, V., Marchiori, C., Stellin, M., Fuson, R., et al. (2011). Matched survival analysis in patients with locoregionally advanced resectable oropharyngeal carcinoma: platinum-based induction and concurrent chemoradiotherapy versus primary surgical resection. International Journal of Radiation Oncology, Biology, Physics, 80, 154–60. doi:10.1016/j.ijrobp.2010.01.032.
Pulte, D., & Brenner, H. (2010). Changes in survival in head and neck cancers in the late 20th and early 21st century: a period analysis. The Oncologist, 15, 994–1001. doi:10.1634/theoncologist.2009-0289.
Vermorken, J. B., & Specenier, P. (2010). Optimal treatment for recurrent/metastatic head and neck cancer. Annals of Oncology: Official Journal of the European Society of Medical Oncology ESMO, 21 Suppl 7, vii252–61. doi:10.1093/annonc/mdq453.
Griffioen, G. H. M. J., Louie, A. V., de Bree, R., Smit, E. F., Paul, M. A., Slotman, B. J., et al. (2015). Second primary lung cancers following a diagnosis of primary head and neck cancer. Lung Cancer Amsterdam Netherlands, 88, 94–9. doi:10.1016/j.lungcan.2015.01.011.
Vermorken, J. B., Mesia, R., Rivera, F., Remenar, E., Kawecki, A., Rottey, S., et al. (2008). Platinum-based chemotherapy plus cetuximab in head and neck cancer. The New England Journal of Medicine, 359, 1116–27. doi:10.1056/NEJMoa0802656.
Vermorken, J. B., Remenar, E., van Herpen, C., Gorlia, T., Mesia, R., Degardin, M., et al. (2007). Cisplatin, fluorouracil, and docetaxel in unresectable head and neck cancer. The New England Journal of Medicine, 357, 1695–704.
Bourhis, J., Overgaard, J., Audry, H., Ang, K. K., Saunders, M., Bernier, J., et al. (2006). Hyperfractionated or accelerated radiotherapy in head and neck cancer: a meta-analysis. Lancet, 368, 843–54.
Pignon, J. P., le Maitre, A., Maillard, E., Bourhis, J., & Group, M.-N. C. (2009). Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): an update on 93 randomised trials and 17,346 patients. Radiotherapy & Oncology, Journal of the European Society for Therapeutic Radiology and Oncology, 92, 4–14.
Ang, K. K., Harris, J., Wheeler, R., Weber, R., Rosenthal, D. I., Nguyen-Tan, P. F., et al. (2010). Human papillomavirus and survival of patients with oropharyngeal cancer. The New England Journal of Medicine, 363, 24–35.
Kang, H., Kiess, A., & Chung, C. H. (2015). Emerging biomarkers in head and neck cancer in the era of genomics. Nature Reviews. Clinical Oncology, 12, 11–26. doi:10.1038/nrclinonc.2014.192.
O’Rorke, M. A., Ellison, M. V., Murray, L. J., Moran, M., James, J., & Anderson, L. A. (2012). Human papillomavirus related head and neck cancer survival: a systematic review and meta-analysis. Oral Oncology, 48, 1191–201. doi:10.1016/j.oraloncology.2012.06.019.
Chung, C. H., Parker, J. S., Karaca, G., Wu, J., Funkhouser, W. K., Moore, D., et al. (2004). Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell, 5, 489–500.
Sakr, W. A., Gale, N., Gnepp, D. R., & Crissman, J. D. (2009). Chapter 1—squamous intraepithelial neoplasia of the upper aerodigestive tract. Diagn. Surg. Pathol. Head Neck (Secondth ed., pp. 1–44). Philadelphia: W.B. Saunders.
Liu, W., Wang, Y.-F., Zhou, H.-W., Shi, P., Zhou, Z.-T., & Tang, G.-Y. (2010). Malignant transformation of oral leukoplakia: a retrospective cohort study of 218 Chinese patients. BMC Cancer, 10, 685. doi:10.1186/1471-2407-10-685.
Torres-Rendon, A., Stewart, R., Craig, G. T., Wells, M., & Speight, P. M. (2009). DNA ploidy analysis by image cytometry helps to identify oral epithelial dysplasias with a high risk of malignant progression. Oral Oncology, 45, 468–73. doi:10.1016/j.oraloncology.2008.07.006.
Zhang, L., & Rosin, M. P. (2001). Loss of heterozygosity: a potential tool in management of oral premalignant lesions? Journal of Oral Pathology & Medicine; Official Publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology, 30, 513–20.
Ha, P. K., Pilkington, T. A., Westra, W. H., Sciubba, J., Sidransky, D., & Califano, J. A. (2002). Progression of microsatellite instability from premalignant lesions to tumors of the head and neck. International Journal of Cancer, 102, 615–7. doi:10.1002/ijc.10748.
Partridge, M., Emilion, G., Pateromichelakis, S., A’Hern, R., Phillips, E., & Langdon, J. (1998). Allelic imbalance at chromosomal loci implicated in the pathogenesis of oral precancer, cumulative loss and its relationship with progression to cancer. Oral Oncology, 34, 77–83.
Slaughter, D. P., Southwick, H. W., & Smejkal, W. (1953). Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer, 6, 963–8.
Braakhuis, B. J. M., Bloemena, E., Leemans, C. R., & Brakenhoff, R. H. (2010). Molecular analysis of surgical margins in head and neck cancer: more than a marginal issue. Oral Oncology, 46, 485–91. doi:10.1016/j.oraloncology.2010.01.019.
Dakubo, G. D., Jakupciak, J. P., Birch-Machin, M. A., & Parr, R. L. (2007). Clinical implications and utility of field cancerization. Cancer Cell International, 7, 2. doi:10.1186/1475-2867-7-2.
van Houten, V. M. M., Tabor, M. P., van den Brekel, M. W. M., Kummer, J. A., Denkers, F., Dijkstra, J., et al. (2002). Mutated p53 as a molecular marker for the diagnosis of head and neck cancer. The Journal of Pathology, 198, 476–86. doi:10.1002/path.1242.
Roesch-Ely, M., Nees, M., Karsai, S., Ruess, A., Bogumil, R., Warnken, U., et al. (2007). Proteomic analysis reveals successive aberrations in protein expression from healthy mucosa to invasive head and neck cancer. Oncogene, 26, 54–64. doi:10.1038/sj.onc.1209770.
Ausoni, S., Boscolo-Rizzo, P., Singh, B., Da Mosto, M. C., Spinato, G., Tirelli, G., et al. (2016). Targeting cellular and molecular drivers of head and neck squamous cell carcinoma: current options and emerging perspectives. Cancer Metastasis Reviews. doi:10.1007/s10555-016-9625-1.
Stransky, N., Egloff, A. M., Tward, A. D., Kostic, A. D., Cibulskis, K., Sivachenko, A., et al. (2011). The mutational landscape of head and neck squamous cell carcinoma. Science, 333, 1157–60.
Network, C. G. A. (2015). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature, 517, 576–82.
Agrawal, N., Frederick, M. J., Pickering, C. R., Bettegowda, C., Chang, K., Li, R. J., et al. (2011). Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science, 333, 1154–7.
Killela, P. J., Reitman, Z. J., Jiao, Y., Bettegowda, C., Agrawal, N., Diaz, L. A., et al. (2013). TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proceedings of the National Academy of Sciences, 110, 6021–6. doi:10.1073/pnas.1303607110.
Vinothkumar, V., Arunkumar, G., Revathidevi, S., Arun, K., Manikandan, M., Rao, A. K. D. M., et al. (2015). TERT promoter hot spot mutations are frequent in Indian cervical and oral squamous cell carcinomas. Tumour Biology : The Journal of the International Society for Oncodevelopmental Biology and Medicine. doi:10.1007/s13277-015-4694-2.
Qu, Y., Dang, S., Wu, K., Shao, Y., Yang, Q., Ji, M., et al. (2014). TERT promoter mutations predict worse survival in laryngeal cancer patients. International Journal of Cancer, 135, 1008–10.
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502, 333–9.
Liu, J., Zhang, C., & Feng, Z. (2014). Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochimica et Biophysica Sinica, 46, 170–9. doi:10.1093/abbs/gmt144.
Nevins, J. R. (2001). The Rb/E2F pathway and cancer. Human Molecular Genetics, 10, 699–703. doi:10.1093/hmg/10.7.699.
Wichmann, G., Rosolowski, M., Krohn, K., Kreuz, M., Boehm, A., Reiche, A., et al. (2015). The role of HPV RNA transcription, immune response-related gene expression and disruptive TP53 mutations in diagnostic and prognostic profiling of head and neck cancer. International Journal of Cancer, 137, 2846–57. doi:10.1002/ijc.29649.
Shi H, Chen X, Lu C, Gu C, Jiang H, Meng R, et al. Association between P16INK4a promoter methylation and HNSCC: a meta-analysis of 21 published studies. PLoS ONE 2015;10. doi:10.1371/journal.pone.0122302.
Hanken, H., Gröbe, A., Cachovan, G., Smeets, R., Simon, R., Sauter, G., et al. (2014). CCND1 amplification and cyclin D1 immunohistochemical expression in head and neck squamous cell carcinomas. Clinical Oral Investigations, 18, 269–76. doi:10.1007/s00784-013-0967-6.
Liu, X., Dakic, A., Zhang, Y., Dai, Y., Chen, R., & Schlegel, R. (2009). HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proceedings of the National Academy of Sciences, 106, 18780–5. doi:10.1073/pnas.0906357106.
Ghittoni, R., Accardi, R., Hasan, U., Gheit, T., Sylla, B., & Tommasino, M. (2010). The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes, 40, 1–13. doi:10.1007/s11262-009-0412-8.
Karatzanis, A. D., Koudounarakis, E., Papadakis, I., & Velegrakis, G. (2012). Molecular pathways of lymphangiogenesis and lymph node metastasis in head and neck cancer. European Archives of Oto-Rhino-Laryngol: Official Journal of European Federation of Oto-Rhino-Laryngol Societies EUFOS: Affiliated with the German Society for Oto-Rhino-Laryngology - Head and Neck Surgery, 269, 731–7. doi:10.1007/s00405-011-1809-2.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144, 646–74. doi:10.1016/j.cell.2011.02.013.
Low, K. C., & Tergaonkar, V. (2013). Telomerase: central regulator of all of the hallmarks of cancer. Trends in Biochemical Sciences, 38, 426–34. doi:10.1016/j.tibs.2013.07.001.
Zhang, C., Chen, X., Li, L., Zhou, Y., Wang, C., & Hou, S. (2015). The association between telomere length and cancer prognosis: evidence from a meta-analysis. PLoS One, 10, e0133174. doi:10.1371/journal.pone.0133174.
Li, Y., & Tergaonkar, V. (2014). Noncanonical functions of telomerase: implications in telomerase-targeted cancer therapies. Cancer Research, 74, 1639–44. doi:10.1158/0008-5472.CAN-13-3568.
Fernandez-Marcelo, T., Gomez, A., Pascua, I., de Juan, C., Head, J., Hernando, F., et al. (2015). Telomere length and telomerase activity in non-small cell lung cancer prognosis: clinical usefulness of a specific telomere status. Journal of Experimental & Clinical Cancer Research , 34, 78. doi:10.1186/s13046-015-0195-9.
Downey, M. G., Going, J. J., Stuart, R. C., & Keith, W. N. (2001). Expression of telomerase RNA in oesophageal and oral cancer. Journal of Oral Pathology & Medicine, 30, 577–81.
Bertorelle, R., Briarava, M., Rampazzo, E., Biasini, L., Agostini, M., Maretto, I., et al. (2013). Telomerase is an independent prognostic marker of overall survival in patients with colorectal cancer. British Journal of Cancer, 108, 278–84. doi:10.1038/bjc.2012.602.
Boscolo-Rizzo, P., Rampazzo, E., Perissinotto, E., Piano, M. A., Giunco, S., Baboci, L., et al. (2015). Telomere shortening in mucosa surrounding the tumor: biosensor of field cancerization and prognostic marker of mucosal failure in head and neck squamous cell carcinoma. Oral Oncology, 51, 500–7. doi:10.1016/j.oraloncology.2015.02.100.
Aida, J., Izumo, T., Shimomura, N., Nakamura, K., Ishikawa, N., Matsuura, M., et al. (2010). Telomere lengths in the oral epithelia with and without carcinoma. European Journal of Cancer, 46, 430–8.
Rampazzo, E., Bertorelle, R., Serra, L., Terrin, L., Candiotto, C., Pucciarelli, S., et al. (2010). Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. British Journal of Cancer, 102, 1300–5. doi:10.1038/sj.bjc.6605644.
Blackburn, E. H., Epel, E. S., & Lin, J. (2015). Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science, 350, 1193–8. doi:10.1126/science.aab3389.
Palm, W., & de Lange, T. (2008). How shelterin protects mammalian telomeres. Annual Review of Genetics, 42, 301–34. doi:10.1146/annurev.genet.41.110306.130350.
Schmutz, I., & de Lange, T. (2016). Shelterin. Current Biology CB, 26, R397–9. doi:10.1016/j.cub.2016.01.056.
Doksani, Y., Wu, J. Y., de Lange, T., & Zhuang, X. (2013). Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell, 155, 345–56. doi:10.1016/j.cell.2013.09.048.
Xu, L., Li, S., & Stohr, B. A. (2013). The role of telomere biology in cancer. Annual Review of Pathology, 8, 49–78. doi:10.1146/annurev-pathol-020712-164030.
Harley, C. B., Futcher, A. B., & Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature, 345, 458–60. doi:10.1038/345458a0.
Fumagalli, M., Rossiello, F., Clerici, M., Barozzi, S., Cittaro, D., Kaplunov, J. M., et al. (2012). Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nature Cell Biology, 14, 355–65. doi:10.1038/ncb2466.
Deng, Y., Chan, S., & Chang, S. (2008). Telomere dysfunction and tumor suppression—the senescence connection. Nature Reviews Cancer, 8, 450–8. doi:10.1038/nrc2393.
Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nature Reviews Cancer, 3, 155–68. doi:10.1038/nrc1011.
Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C. W., Chessa, L., et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–7.
Herbig, U., Jobling, W. A., Chen, B. P. C., Chen, D. J., & Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Molecular Cell, 14, 501–13. doi:10.1016/S1097-2765(04)00256-4.
Artandi, S. E., & Attardi, L. D. (2005). Pathways connecting telomeres and p53 in senescence, apoptosis, and cancer. Biochemical and Biophysical Research Communications, 331, 881–90. doi:10.1016/j.bbrc.2005.03.211.
Murnane, J. P. (2012). Telomere dysfunction and chromosome instability. Mutation Research, 730, 28–36. doi:10.1016/j.mrfmmm.2011.04.008.
Davoli, T., & de Lange, T. (2012). Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell, 21, 765–76. doi:10.1016/j.ccr.2012.03.044.
Akincilar, S. C., Unal, B., & Tergaonkar, V. (2016). Reactivation of telomerase in cancer. Cellular and Molecular Life Sciences CMLS, 73, 1659–70. doi:10.1007/s00018-016-2146-9.
Nakamura, T. M., Morin, G. B., Chapman, K. B., Weinrich, S. L., Andrews, W. H., Lingner, J., et al. (1997). Telomerase catalytic subunit homologs from fission yeast and human. Science, 277, 955–9.
Shay, J. W., Reddel, R. R., & Wright, W. E. (2012). Cancer: cancer and telomeres—an alternative to telomerase. Science, 336, 1388–90. doi:10.1126/science.1222394.
Shay, J. W., & Wright, W. E. (2011). Role of telomeres and telomerase in cancer. Seminars in Cancer Biology, 21, 349–53. doi:10.1016/j.semcancer.2011.10.001.
Smogorzewska, A., van Steensel, B., Bianchi, A., Oelmann, S., Schaefer, M. R., Schnapp, G., et al. (2000). Control of human telomere length by TRF1 and TRF2. Molecular and Cellular Biology, 20, 1659–68.
Smogorzewska, A., & de Lange, T. (2004). Regulation of telomerase by telomeric proteins. Annual Review of Biochemistry, 73, 177–208. doi:10.1146/annurev.biochem.73.071403.160049.
Xu, D., Wang, Q., Gruber, A., Björkholm, M., Chen, Z., Zaid, A., et al. (2000). Downregulation of telomerase reverse transcriptase mRNA expression by wild type p53 in human tumor cells. Oncogene, 19, 5123–33. doi:10.1038/sj.onc.1203890.
Bazarov, A. V., Van Sluis, M., Hines, W. C., Bassett, E., Beliveau, A., Campeau, E., et al. (2010). p16INK4a-mediated suppression of telomerase in normal and malignant human breast cells. Aging Cell, 9, 736–46. doi:10.1111/j.1474-9726.2010.00599.x.
Kanzawa, T., Komata, T., Kyo, S., Germano, I. M., Kondo, Y., & Kondo, S. (2003). Down-regulation of telomerase activity in malignant glioma cells by p27KIP1. International Journal of Oncology, 23, 1703–8.
Akiyama, M., Hideshima, T., Hayashi, T., Tai, Y.-T., Mitsiades, C. S., Mitsiades, N., et al. (2003). Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Research, 63, 18–21.
Wang, S., & Zhu, J. (2003). Evidence for a relief of repression mechanism for activation of the human telomerase reverse transcriptase promoter. The Journal of Biological Chemistry, 278, 18842–50. doi:10.1074/jbc.M209544200.
Liu, C., Fang, X., Ge, Z., Jalink, M., Kyo, S., Björkholm, M., et al. (2007). The telomerase reverse transcriptase (hTERT) gene is a direct target of the histone methyltransferase SMYD3. Cancer Research, 67, 2626–31. doi:10.1158/0008-5472.CAN-06-4126.
Saretzki, G. (2014). Extra-telomeric functions of human telomerase: cancer, mitochondria and oxidative stress. Current Pharmaceutical Design, 20, 6386–403.
Liu, Z., Li, Q., Li, K., Chen, L., Li, W., Hou, M., et al. (2013). Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene, 32, 4203–13. doi:10.1038/onc.2012.441.
Ding, D., Xi, P., Zhou, J., Wang, M., & Cong, Y.-S. (2013). Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-kB-dependent transcription. The FASEB Journal: Official Publication of the Federal American Society of Experimental Biology, 27, 4375–83. doi:10.1096/fj.13-230904.
Cesare, A. J., & Reddel, R. R. (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nature Reviews Genetics, 11, 319–30. doi:10.1038/nrg2763.
Episkopou, H., Draskovic, I., Van Beneden, A., Tilman, G., Mattiussi, M., Gobin, M., et al. (2014). Alternative lengthening of telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Research, 42, 4391–405. doi:10.1093/nar/gku114.
Shay, J. W. (2016). Role of telomeres and telomerase in aging and cancer. Cancer Discovery. doi:10.1158/2159-8290.CD-16-0062.
Härle-Bachor, C., & Boukamp, P. (1996). Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proceedings of the National Academy of Sciences of the United States of America, 93, 6476–81.
Choudhury, A. R., Ju, Z., Djojosubroto, M. W., Schienke, A., Lechel, A., Schaetzlein, S., et al. (2007). Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nature Genetics, 39, 99–105. doi:10.1038/ng1937.
Günes, C., & Rudolph, K. L. (2013). The role of telomeres in stem cells and cancer. Cell, 152, 390–3. doi:10.1016/j.cell.2013.01.010.
Crowe, D. L., Nguyen, D. C., & Ohannessian, A. (2005). Mechanism of telomerase repression during terminal differentiation of normal epithelial cells and squamous carcinoma lines. International Journal of Oncology, 27, 847–54.
Kang, M. K., Kameta, A., Shin, K.-H., Baluda, M. A., Kim, H.-R., & Park, N.-H. (2003). Senescence-associated genes in normal human oral keratinocytes. Experimental Cell Research, 287, 272–81. doi:10.1016/S0014-4827(03)00061-2.
Davalos, A. R., Coppe, J.-P., Campisi, J., & Desprez, P.-Y. (2010). Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Reviews, 29, 273–83. doi:10.1007/s10555-010-9220-9.
Coppe, J.-P., Boysen, M., Sun, C. H., Wong, B. J. F., Kang, M. K., Park, N.-H., et al. (2008). A role for fibroblasts in mediating the effects of tobacco-induced epithelial cell growth and invasion. Molecular Cancer Research MCR, 6, 1085–98. doi:10.1158/1541-7786.MCR-08-0062.
Ernst, A., Jones, D. T. W., Maass, K. K., Rode, A., Deeg, K. I., Jebaraj, B. M. C., et al. (2016). Telomere dysfunction and chromothripsis. International Journal of Cancer, 138, 2905–14. doi:10.1002/ijc.30033.
Gordon, K. E., Ireland, H., Roberts, M., Steeghs, K., McCaul, J. A., MacDonald, D. G., et al. (2003). High levels of telomere dysfunction bestow a selective disadvantage during the progression of human oral squamous cell carcinoma. Cancer Research, 63, 458–67.
Gisselsson, D., Jonson, T., Yu, C., Martins, C., Mandahl, N., Wiegant, J., et al. (2002). Centrosomal abnormalities, multipolar mitoses, and chromosomal instability in head and neck tumours with dysfunctional telomeres. British Journal of Cancer, 87, 202–7. doi:10.1038/sj.bjc.6600438.
McCaul, J. A., Gordon, K. E., Minty, F., Fleming, J., & Parkinson, E. K. (2008). Telomere dysfunction is related to the intrinsic radio-resistance of human oral cancer cells. Oral Oncology, 44, 261–9. doi:10.1016/j.oraloncology.2007.02.010.
Artandi, S. E., Chang, S., Lee, S. L., Alson, S., Gottlieb, G. J., Chin, L., et al. (2000). Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature, 406, 641–5. doi:10.1038/35020592.
Aida, J., Kobayashi, T., Saku, T., Yamaguchi, M., Shimomura, N., Nakamura, K., et al. (2012). Short telomeres in an oral precancerous lesion: Q-FISH analysis of leukoplakia. Journal of Oral Pathology & Medicine, 41, 372–8.
Meeker, A. K., Hicks, J. L., Iacobuzio-Donahue, C. A., Montgomery, E. A., Westra, W. H., Chan, T. Y., et al. (2004). Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clinical Cancer Research, 10, 3317–26.
Kammori, M., Poon, S. S. S., Nakamura, K.-I., Izumiyama, N., Ishikawa, N., Kobayashi, M., et al. (2007). Squamous cell carcinomas of the esophagus arise from a telomere-shortened epithelial field. International Journal of Molecular Medicine, 20, 793–9.
Heaphy, C. M., Gaonkar, G., Peskoe, S. B., Joshu, C. E., De Marzo, A. M., Lucia, M. S., et al. (2015). Prostate stromal cell telomere shortening is associated with risk of prostate cancer in the placebo arm of the Prostate Cancer Prevention Trial. The Prostate, 75, 1160–6. doi:10.1002/pros.22997.
Hong, S.-M., Heaphy, C. M., Shi, C., Eo, S.-H., Cho, H., Meeker, A. K., et al. (2011). Telomeres are shortened in acinar-to-ductal metaplasia lesions associated with pancreatic intraepithelial neoplasia but not in isolated acinar-to-ductal metaplasias. Modern Pathology: Official Journal of the United States and Canadian Academy of Pathology Inc, 24, 256–66. doi:10.1038/modpathol.2010.181.
Pickering, C. R., Zhang, J., Yoo, S. Y., Bengtsson, L., Moorthy, S., Neskey, D. M., et al. (2013). Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discovery, 3, 770–81.
Zack, T. I., Schumacher, S. E., Carter, S. L., Cherniack, A. D., Saksena, G., Tabak, B., et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nature Genetics, 45, 1134–40. doi:10.1038/ng.2760.
Braakhuis, B. J. M., Tabor, M. P., Kummer, J. A., Leemans, C. R., & Brakenhoff, R. H. (2003). A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Research, 63, 1727–30.
Nakamura, K.-I., Izumiyama-Shimomura, N., Sawabe, M., Arai, T., Aoyagi, Y., Fujiwara, M., et al. (2002). Comparative analysis of telomere lengths and erosion with age in human epidermis and lingual epithelium. The Journal of Investigative Dermatology, 119, 1014–9. doi:10.1046/j.1523-1747.2002.19523.x.
Kang, M. K., Guo, W., & Park, N. H. (1998). Replicative senescence of normal human oral keratinocytes is associated with the loss of telomerase activity without shortening of telomeres. Cell Growth & Differentiation: The Molecular Biology Journal of the American Association for Cancer Research, 9, 85–95.
Fujimoto, R., Kamata, N., Yokoyama, K., Ueda, N., Satomura, K., Hayashi, E., et al. (2001). Expression of telomerase components in oral keratinocytes and squamous cell carcinomas. Oral Oncology, 37, 132–40.
Kilian, A., Bowtell, D. D. L., Abud, H. E., Hime, G. R., Venter, D. J., Keese, P. K., et al. (1997). Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Human Molecular Genetics, 6, 2011–9. doi:10.1093/hmg/6.12.2011.
Luzar, B., Poljak, M., Marin, I. J., & Gale, N. (2003). Telomerase reactivation is an early event in laryngeal carcinogenesis. Modern Pathology, 16, 841–8.
Liao, J., Mitsuyasu, T., Yamane, K., & Ohishi, M. (2000). Telomerase activity in oral and maxillofacial tumors. Oral Oncology, 36, 347–52.
Liao, C. T., Chen, I. H., Chang, J. T., Wang, H. M., Hsieh, L. L., & Cheng, A. J. (2003). Lack of correlation of betel nut chewing, tobacco smoking, and alcohol consumption with telomerase activity and the severity of oral cancer. Chang Gung Medical Journal, 26, 637–45.
Liao, C. T., Chang, J. T.-C., Wang, H. M., Chen, I. H., Lin, C. Y., Chen, T. M., et al. (2004). Telomerase as an independent prognostic factor in head and neck squamous cell carcinoma. Head & Neck, 26, 504–12.
Kunicka, Z., Mucha, I., & Fajkus, J. (2008). Telomerase activity in head and neck cancer. Anticancer Research, 28, 3125–9.
Chen, M., Yang, B., & Zhang, X. (2008). Effects of telomerase activity on carcinogenesis and survival in laryngeal carcinoma. The Journal of International Medical Research, 36, 336–42.
Curran, A. J., Gullane, P. J., Irish, J., Macmillan, C., Freeman, J., & Kamel-Reid, S. (2000). Telomerase activity is upregulated in laryngeal squamous cell carcinoma. Laryngoscope, 110, 391–6.
Chang, L. Y., Lin, S. C., Chang, C. S., Wong, Y. K., Hu, Y. C., & Chang, K. W. (1999). Telomerase activity and in situ telomerase RNA expression in oral carcinogenesis. Journal of Oral Pathology & Medicine, 28, 389–96.
Fabricius, E. M., Gurr, U., & Wildner, G. P. (2002). Telomerase activity levels in the surgical margin and tumour distant tissue of the squamous cell carcinoma of the head-and-neck. Analytical Cellular Pathology, 24, 25–39.
Hohaus, S., Cavallo, S., Bellacosa, A., Genuardi, M., Galli, J., Cadoni, G., et al. (1996). Telomerase activity in human laryngeal squamous cell carcinomas. Clinical Cancer Research, 2, 1895–900.
Yajima, Y., Noma, H., Furuya, Y., Nomura, T., Yamauchi, T., Kasahara, K., et al. (2004). Quantification of telomerase activity of regions unstained with iodine solution that surround oral squamous cell carcinoma. Oral Oncology, 40, 314–20.
Patel, M. M., Patel, D. D., Parekh, L. J., Raval, G. N., Rawal, R. M., Bhatavdekar, J. M., et al. (1999). Evaluation of telomerase activation in head and neck cancer. Oral Oncology, 35, 510–5.
Mutirangura, A., Supiyaphun, P., Trirekapan, S., Sriuranpong, V., Sakuntabhai, A., Yenrudi, S., et al. (1996). Telomerase activity in oral leukoplakia and head and neck squamous cell carcinoma. Cancer Research, 56, 3530–3.
Califano, J., Ahrendt, S. A., Meininger, G., Westra, W. H., Koch, W. M., & Sidransky, D. (1996). Detection of telomerase activity in oral rinses from head and neck squamous cell carcinoma patients. Cancer Research, 56, 5720–2.
Fabricius, E. M., Kruse-Boitschenko, U., Khoury, R., Wildner, G. P., Raguse, J. D., & Klein, M. (2009). Immunohistochemical determination of the appropriate anti-hTERT antibodies for in situ detection of telomerase activity in frozen sections of head and neck squamous cell carcinomas and tumor margin tissues. International Journal of Oncology, 34, 1257–79.
Sumida, T., Sogawa, K., Hamakawa, H., Sugita, A., Tanioka, H., & Ueda, N. (1998). Detection of telomerase activity in oral lesions. Journal of Oral Pathology & Medicine, 27, 111–5.
Mao, L., El-Naggar, A. K., Fan, Y. H., Lee, J. S., Lippman, S. M., Kayser, S., et al. (1996). Telomerase activity in head and neck squamous cell carcinoma and adjacent tissues. Cancer Research, 56, 5600–4.
Koscielny, S., Eggeling, F., Dahse, R., & Fiedler, W. (2004). The influence of reactivation of the telomerase in tumour tissue on the prognosis of squamous cell carcinomas in the head and neck. Journal of Oral Pathology & Medicine, 33, 538–42.
Lee, B. K., Diebel, E., Neukam, F. W., Wiltfang, J., & Ries, J. (2001). Diagnostic and prognostic relevance of expression of human telomerase subunits in oral cancer. International Journal of Oncology, 19, 1063–8.
Fiedler, M., Ressler, S., Campo-Fernandez, B., Laich, A., Jansen, L., Widschwendter, A., et al. (2005). Expression of the high-risk human papillomavirus type 18 and 45 E7 oncoproteins in cervical carcinoma biopsies. The Journal of General Virology, 86, 3235–41.
Curran, A. J., Denis, K. S., Irish, J., Gullane, P. J., MacMillan, C., & Kamel-Reid, S. (1998). Telomerase activity in oral squamous cell carcinoma. Archives of Otolaryngology – Head & Neck Surgery, 124, 784–8.
Kannan, S., Tahara, H., Yokozaki, H., Mathew, B., Nalinakumari, K. R., Nair, M. K., et al. (1997). Telomerase activity in premalignant and malignant lesions of human oral mucosa. Cancer Epidemiology, Biomarkers & Prevention, 6, 413–20.
Ogawa, Y., Nishioka, A., Hamada, N., Terashima, M., Inomata, T., Yoshida, S., et al. (1998). Changes in telomerase activity of advanced cancers of oral cavity and oropharynx during radiation therapy: correlation with clinical outcome. International Journal of Molecular Medicine, 2, 301–7.
Miyoshi, Y., Tsukinoki, K., Imaizumi, T., Yamada, Y., Ishizaki, T., Watanabe, Y., et al. (1999). Telomerase activity in oral cancer. Oral Oncology, 35, 283–9.
Fujita, H., Nagata, M., Hoshina, H., Nagashima, K., Seki, Y., Tanaka, K., et al. (2004). Clinical significance and usefulness of quantification of telomerase activity in oral malignant and nonmalignant lesions. International Journal of Oral and Maxillofacial Surgery, 33, 693–9.
Soria, J. C., Morat, L., Commo, F., Dabit, D., Perie, S., Sabatier, L., et al. (2001). Telomerase activation cooperates with inactivation of p16 in early head and neck tumorigenesis. British Journal of Cancer, 84, 504–11.
Sainger, R. N., Shah, F. D., Telang, S. D., Shah, P. M., & Patel, P. S. (2009). Telomere attrition and telomerase activity are associated with GSTM1 polymorphism in oral cancer. Cancer Biomarkers, 5, 189–95.
Liu, Y., Dong, X. L., Tian, C., & Liu, H. G. (2012). Human telomerase RNA component (hTERC) gene amplification detected by FISH in precancerous lesions and carcinoma of the larynx. Diagnostic Pathology, 7, 34.
Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. eLife 2015;4. doi:10.7554/eLife.07918.
Hayes, D. N., Van Waes, C., & Seiwert, T. Y. (2015). Genetic landscape of human papillomavirus-associated head and neck cancer and comparison to tobacco-related tumors. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 33, 3227–34. doi:10.1200/JCO.2015.62.1086.
Yin, L., Hubbard, A. K., & Giardina, C. (2000). NF-kappa B regulates transcription of the mouse telomerase catalytic subunit. The Journal of Biological Chemistry, 275, 36671–5. doi:10.1074/jbc.M007378200.
Zhang, Y., Toh, L., Lau, P., & Wang, X. (2012). Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. The Journal of Biological Chemistry, 287, 32494–511. doi:10.1074/jbc.M112.368282.
Greenberg, R. A., O’Hagan, R. C., Deng, H., Xiao, Q., Hann, S. R., Adams, R. R., et al. (1999). Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene, 18, 1219–26. doi:10.1038/sj.onc.1202669.
Gewin, L., Myers, H., Kiyono, T., & Galloway, D. A. (2004). Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes & Development, 18, 2269–82. doi:10.1101/gad.1214704.
Terrin, L., Dal Col, J., Rampazzo, E., Zancai, P., Pedrotti, M., Ammirabile, G., et al. (2008). Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. Journal of Virology, 82, 10175–87. doi:10.1128/JVI.00321-08.
Yang, L., Xu, Z., Liu, L., Luo, X., Lu, J., Sun, L., et al. (2014). Targeting EBV-LMP1 DNAzyme enhances radiosensitivity of nasopharyngeal carcinoma cells by inhibiting telomerase activity. Cancer Biology & Therapy, 15, 61–8. doi:10.4161/cbt.26606.
Weinhold, N., Jacobsen, A., Schultz, N., Sander, C., & Lee, W. (2014). Genome-wide analysis of noncoding regulatory mutations in cancer. Nature Genetics, 46, 1160–5. doi:10.1038/ng.3101.
Bell, R. J. A., Rube, H. T., Kreig, A., Mancini, A., Fouse, S. D., Nagarajan, R. P., et al. (2015). The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science, 348, 1036–9. doi:10.1126/science.aab0015.
Liu, Z., Li, G., Wei, S., Niu, J., Wang, L.-E., Sturgis, E. M., et al. (2010). Genetic variations in TERT-CLPTM1L genes and risk of squamous cell carcinoma of the head and neck. Carcinogenesis, 31, 1977–81. doi:10.1093/carcin/bgq179.
Murakami, J., Asaumi, J., Kawai, N., Tsujigiwa, H., Yanagi, Y., Nagatsuka, H., et al. (2005). Effects of histone deacetylase inhibitor FR901228 on the expression level of telomerase reverse transcriptase in oral cancer. Cancer Chemotherapy and Pharmacology, 56, 22–8. doi:10.1007/s00280-004-0976-x.
Blackburn EH, Collins K. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol. 2011;3. doi:10.1101/cshperspect.a003558.
Chang, J. T., Lu, Y.-C., Chen, Y.-J., Tseng, C.-P., Chen, Y.-L., Fang, C.-W., et al. (2006). hTERT phosphorylation by PKC is essential for telomerase holoprotein integrity and enzyme activity in head neck cancer cells. British Journal of Cancer, 94, 870–8. doi:10.1038/sj.bjc.6603008.
Chen, W., Xiao, B.-K., Liu, J.-P., Chen, S.-M., & Tao, Z.-Z. (2010). Alternative lengthening of telomeres in hTERT-inhibited laryngeal cancer cells. Cancer Science, 101, 1769–76. doi:10.1111/j.1349-7006.2010.01611.x.
Luzar, B., Poljak, M., Marin, I. J., Eberlinc, A., Klopcic, U., & Gale, N. (2004). Human telomerase catalytic subunit gene re-expression is an early event in oral carcinogenesis. Histopathology, 45, 13–9.
Chrysovergis, A., Gorgoulis, V. G., Giotakis, I., Tsiambas, E., Karameris, A., Kittas, C., et al. (2011). Simultaneous over activation of EGFR, telomerase (h TERT), and cyclin D1 correlates with advanced disease in larynx squamous cell carcinoma: a tissue microarray analysis. Medical Oncology, 28, 871–7.
Sharma, H., Sen, S., Mathur, M., Bahadur, S., & Singh, N. (2004). Combined evaluation of expression of telomerase, survivin, and anti-apoptotic Bcl-2 family members in relation to loss of differentiation and apoptosis in human head and neck cancers. Head & Neck, 26, 733–40.
Kumar, S. K., Zain, R. B., Ismail, S. M., & Cheong, S. C. (2005). Human telomerase reverse transcriptase expression in oral carcinogenesis—a preliminary report. Journal of Experimental & Clinical Cancer Research, 24, 639–46.
Luzar, B., Poljak, M., Marin, I. J., Fischinger, J., & Gale, N. (2001). Quantitative measurement of telomerase catalytic subunit (hTERT) mRNA in laryngeal squamous cell carcinomas. Anticancer Research, 21, 4011–5.
Thurnher, D., Knerer, B., Formanek, M., & Kornfehl, J. (1998). Non-radioactive semiquantitative testing for the expression levels of telomerase activity in head and neck squamous cell carcinomas may be indicative for biological tumour behaviour. Acta Oto-Laryngologica, 118, 423–7.
Chen, H. H., Yu, C. H., Wang, J. T., Liu, B. Y., Wang, Y. P., Sun, A., et al. (2007). Expression of human telomerase reverse transcriptase (hTERT) protein is significantly associated with the progression, recurrence and prognosis of oral squamous cell carcinoma in Taiwan. Oral Oncology, 43, 122–9.
Freier, K., Pungs, S., Flechtenmacher, C., Bosch, F. X., Lichter, P., Joos, S., et al. (2007). Frequent high telomerase reverse transcriptase expression in primary oral squamous cell carcinoma. Journal of Oral Pathology & Medicine, 36, 267–72.
Samny, T. E., Halaby, H. E., Fiky, L. E., Nassar, M., Makhzangy, A. E., & Eissa, S. (2005). Prognostic value of telomerase and DNA ploidy in laryngeal carcinoma. European Archives of Oto-Rhino-Laryngology, 262, 799–803.
Eissa, S., Kenawy, G., Moteleb, F. A., El-Makhzangyc, A. N., & Nassar, M. (2005). Real-time PCR hTERT mRNA pattern in tumor core, edge, resection margin, and lymph nodes in laryngeal tumors: relation to proliferative index and impact on prognosis. Clinical Biochemistry, 38, 873–8.
Pannone, G., Maria, S. D., Zamparese, R., Metafora, S., Serpico, R., Morelli, F., et al. (2007). Prognostic value of human telomerase reverse transcriptase gene expression in oral carcinogenesis. International Journal of Oncology, 30, 1349–57.
Patel, M. M., Parekh, L. J., Jha, F. P., Sainger, R. N., Patel, J. B., Patel, D. D., et al. (2002). Clinical usefulness of telomerase activation and telomere length in head and neck cancer. Head & Neck, 24, 1060–7.
Luzar, B., Poljak, M., Fischinger, J., Klopcic, U., & Gale, N. (2005). Telomerase catalytic subunit gene expression does not influence survival of patients with squamous cell carcinoma of the larynx and hypopharynx: a case–control study. The Journal of Laryngology and Otology, 119, 917–21.
Poremba, C., Heine, B., Diallo, R., Heinecke, A., Wai, D., Schaefer, K.-L., et al. (2002). Telomerase as a prognostic marker in breast cancer: high-throughput tissue microarray analysis of hTERT and hTR. The Journal of Pathology, 198, 181–9. doi:10.1002/path.1191.
Bantis, A., Patsouris, E., Gonidi, M., Kavantzas, N., Tsipis, A., Athanassiadou, A.-M., et al. (2009). Telomerase RNA expression and DNA ploidy as prognostic markers of prostate carcinomas. Tumori, 95, 744–52.
Nosrati, M., Li, S., Bagheri, S., Ginzinger, D., Blackburn, E. H., Debs, R. J., et al. (2004). Antitumor activity of systemically delivered ribozymes targeting murine telomerase RNA. Clinical Cancer Research, 10, 4983–90. doi:10.1158/1078-0432.CCR-04-0134.
Bagheri, S., Nosrati, M., Li, S., Fong, S., Torabian, S., Rangel, J., et al. (2006). Genes and pathways downstream of telomerase in melanoma metastasis. Proceedings of the National Academy of Sciences of the United States of America, 103, 11306–11. doi:10.1073/pnas.0510085103.
Artandi, S. E., Alson, S., Tietze, M. K., Sharpless, N. E., Ye, S., Greenberg, R. A., et al. (2002). Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proceedings of the National Academy of Sciences of the United States of America, 99, 8191–6. doi:10.1073/pnas.112515399.
Yang, J., Wei, D., Wang, W., Shen, B., Xu, S., & Cao, Y. (2015). TRAF4 enhances oral squamous cell carcinoma cell growth, invasion and migration by Wnt-β-catenin signaling pathway. International Journal of Clinical and Experimental Pathology, 8, 11837–46.
Iwai, S., Yonekawa, A., Harada, C., Hamada, M., Katagiri, W., Nakazawa, M., et al. (2010). Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. International Journal of Oncology, 37, 1095–103.
Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013; 5. doi:10.1101/cshperspect.a007898.
Smalley, M. J., & Dale, T. C. (1999). Wnt signalling in mammalian development and cancer. Cancer Metastasis Reviews, 18, 215–30.
Smith, A., Teknos, T. N., & Pan, Q. (2013). Epithelial to mesenchymal transition in head and neck squamous cell carcinoma. Oral Oncology, 49, 287–92. doi:10.1016/j.oraloncology.2012.10.009.
Zhao, T., Hu, F., Qiao, B., Chen, Z., & Tao, Q. (2015). Telomerase reverse transcriptase potentially promotes the progression of oral squamous cell carcinoma through induction of epithelial-mesenchymal transition. International Journal of Oncology, 46, 2205–15. doi:10.3892/ijo.2015.2927.
Loercher, A., Lee, T. L., Ricker, J. L., Howard, A., Geoghegen, J., Chen, Z., et al. (2004). Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Research, 64, 6511–23. doi:10.1158/0008-5472.CAN-04-0852.
Allen, C. T., Ricker, J. L., Chen, Z., & Van Waes, C. (2007). Role of activated nuclear factor-kB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head & Neck, 29, 959–71. doi:10.1002/hed.20615.
Ghosh, A., Saginc, G., Leow, S. C., Khattar, E., Shin, E. M., Yan, T. D., et al. (2012). Telomerase directly regulates NF-kB-dependent transcription. Nature Cell Biology, 14, 1270–81. doi:10.1038/ncb2621.
Zhang, R.-L., Peng, L.-X., Yang, J.-P., Zheng, L.-S., Xie, P., Wang, M.-Y., et al. (2016). IL-8 suppresses E-cadherin expression in nasopharyngeal carcinoma cells by enhancing E-cadherin promoter DNA methylation. International Journal of Oncology, 48, 207–14. doi:10.3892/ijo.2015.3226.
Yadav, A., Kumar, B., Datta, J., Teknos, T. N., & Kumar, P. (2011). IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Molecular Cancer Research MCR, 9, 1658–67. doi:10.1158/1541-7786.MCR-11-0271.
Yu, L., Mu, Y., Sa, N., Wang, H., & Xu, W. (2014). Tumor necrosis factor α induces epithelial-mesenchymal transition and promotes metastasis via NF-kB signaling pathway-mediated TWIST expression in hypopharyngeal cancer. Oncology Reports, 31, 321–7. doi:10.3892/or.2013.2841.
Park, Y.-J., Kim, E. K., Bae, J. Y., Moon, S., & Kim, J. (2016). Human telomerase reverse transcriptase (hTERT) promotes cancer invasion by modulating cathepsin D via early growth response (EGR)-1. Cancer Letters, 370, 222–31. doi:10.1016/j.canlet.2015.10.021.
Aubert, G., & Lansdorp, P. M. (2008). Telomeres and aging. Physiological Reviews, 88, 557–79. doi:10.1152/physrev.00026.2007.
Gu, J. (2015). Leukocyte telomere length and cancer risk: a dynamic problem. EBioMedicine, 2, 493–4. doi:10.1016/j.ebiom.2015.05.006.
Daniali, L., Benetos, A., Susser, E., Kark, J. D., Labat, C., Kimura, M., et al. (2013). Telomeres shorten at equivalent rates in somatic tissues of adults. Nature Communications, 4, 1597. doi:10.1038/ncomms2602.
Hou, L., Zhang, X., Gawron, A. J., & Liu, J. (2012). Surrogate tissue telomere length and cancer risk: shorter or longer? Cancer Letters, 319, 130–5. doi:10.1016/j.canlet.2012.01.028.
Hou, L., Savage, S. A., Blaser, M. J., Perez-Perez, G., Hoxha, M., Dioni, L., et al. (2009). Telomere length in peripheral leukocyte DNA and gastric cancer risk. Cancer Epidemiology, Biomarkers & Prevention: A Publication of the American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology, 18, 3103–9. doi:10.1158/1055-9965.EPI-09-0347.
Liu, Z., Ma, H., Wei, S., Li, G., Sturgis, E. M., & Wei, Q. (2011). Telomere length and TERT functional polymorphisms are not associated with risk of squamous cell carcinoma of the head and neck. Cancer Epidemiology, Biomarkers & Prevention: A Publication of the American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology, 20, 2642–5. doi:10.1158/1055-9965.EPI-11-0890.
Bau, D.-T., Lippman, S. M., Xu, E., Gong, Y., Lee, J. J., Wu, X., et al. (2013). Short telomere lengths in peripheral blood leukocytes are associated with an increased risk of oral premalignant lesion and oral squamous cell carcinoma. Cancer, 119, 4277–83. doi:10.1002/cncr.28367.
Zhang, Y., Sturgis, E. M., Dahlstrom, K. R., Wen, J., Liu, H., Wei, Q., et al. (2013). Telomere length in peripheral blood lymphocytes contributes to the development of HPV-associated oropharyngeal carcinoma. Cancer Research, 73, 5996–6003. doi:10.1158/0008-5472.CAN-13-0881.
Wu, X., Amos, C. I., Zhu, Y., Zhao, H., Grossman, B. H., Shay, J. W., et al. (2003). Telomere dysfunction: a potential cancer predisposition factor. Journal of the National Cancer Institute, 95, 1211–8.
Lee, B.-J., Wang, S.-G., Choi, J.-S., Lee, J.-C., Goh, E.-K., & Kim, M.-G. (2006). The prognostic value of telomerase expression in peripheral blood mononuclear cells of head and neck cancer patients. American Journal of Clinical Oncology, 29, 163–7. doi:10.1097/01.coc.0000207372.64733.b0.
Hong, D.-Y., Lee, B.-J., Lee, J.-C., Choi, J.-S., Wang, S.-G., & Ro, J.-H. (2009). Expression of VEGF, HGF, IL-6, IL-8, MMP-9, telomerase in peripheral blood of patients with head and neck squamous cell carcinoma. Clinical and Experimental Otorhinolaryngology, 2, 186–92. doi:10.3342/ceo.2009.2.4.186.
Hong DY, Lee BJ, Lee JC, Choi JS, Wang SG, Ro JH. Expression of VEGF, HGF, IL-6, IL-8, MMP-9, telomerase in peripheral blood of patients with head and neck squamous cell carcinoma 2009; 2:186–92.
Terrin, L., Rampazzo, E., Pucciarelli, S., Agostini, M., Bertorelle, R., Esposito, G., et al. (2008). Relationship between tumor and plasma levels of hTERT mRNA in patients with colorectal cancer: implications for monitoring of neoplastic disease. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 14, 7444–51. doi:10.1158/1078-0432.CCR-08-0478.
March-Villalba, J. A., Martínez-Jabaloyas, J. M., Herrero, M. J., Santamaria, J., Aliño, S. F., & Dasí, F. (2012). Cell-free circulating plasma hTERT mRNA is a useful marker for prostate cancer diagnosis and is associated with poor prognosis tumor characteristics. PLoS One, 7, e43470. doi:10.1371/journal.pone.0043470.
Kang, Y., Zhang, J., Sun, P., & Shang, J. (2012). Circulating cell-free human telomerase reverse transcriptase mRNA in plasma and its potential diagnostic and prognostic value for gastric cancer. International Journal of Clinical Oncology, 18, 478–86. doi:10.1007/s10147-012-0405-9.
Li, Y. R., Wu, J. M., Wang, L., Huang, X., Shi, J., & Hu, L. H. (2005). Human telomerase reverse transcriptase expression and its clinical significance in laryngeal squamous cell carcinoma. Acta Oto-Laryngologica, 125, 409–14.
Acknowledgments
The authors would like to express their sincere gratitude to Prof. Carlo Marchiori for his charismatic guidance, support, and encouragement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None to declare
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Boscolo-Rizzo, P., Da Mosto, M.C., Rampazzo, E. et al. Telomeres and telomerase in head and neck squamous cell carcinoma: from pathogenesis to clinical implications. Cancer Metastasis Rev 35, 457–474 (2016). https://doi.org/10.1007/s10555-016-9633-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-016-9633-1