
Abstract
The age-related loss of skeletal muscle mass and physical function leads to a loss of independence and an increased reliance on health-care. Mitochondria are crucial in the aetiology of sarcopenia and have been identified as key targets for interventions that can attenuate declines in physical capacity. Exercise training is a primary intervention that reduces many of the deleterious effects of ageing in skeletal muscle quality and function. However, habitual levels of physical activity decline with age, making it necessary to implement adjunct treatments to maintain skeletal muscle mitochondrial health and physical function. This review provides an overview of the effects of ageing and exercise training on human skeletal muscle mitochondria and considers several supplements that have plausible mechanistic underpinning to improve physical function in ageing through their interactions with mitochondria. Several supplements, including MitoQ, urolithin A, omega-3 polyunsaturated fatty acids (n3-PUFAs), and a combination of glycine and N-acetylcysteine (GlyNAC) can improve physical function in older individuals through a variety of inter-dependent mechanisms including increases in mitochondrial biogenesis and energetics, decreases in mitochondrial reactive oxygen species emission and oxidative damage, and improvements in mitochondrial quality control. While there is evidence that some nicotinamide adenine dinucleotide precursors can improve physical function in older individuals, such an outcome seems unrelated to and independent of changes in skeletal muscle mitochondrial function. Future research should investigate the safety and efficacy of compounds that can improve skeletal muscle health in preclinical models through mechanisms involving mitochondria, such as mitochondrial-derived peptides and mitochondrial uncouplers, with a view to extending the human health-span.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Skeletal muscle mitochondria are key targets for interventions that can attenuate age-related declines in muscle mass and physical function. |
Exercise training counteracts many of the deleterious effects of ageing on skeletal muscle mitochondria. However, complementary interventions are needed as many older adults fail to meet physical activity guidelines. |
Several supplements, including MitoQ, urolithin A, n-3 PUFAs and GlyNAC improve physical function in older individuals through mechanisms involving mitochondria, including increases in mitochondrial biogenesis and bioenergetics, decreases in mitochondrial ROS emission and oxidative damage, and improvements in mitochondrial quality control. |
1 Introduction
The number of adults aged 65 years or older globally is predicted to increase more than twofold by 2050, reaching an estimated 1.6 billion, or one in every six persons alive [1]. While global life expectancy has increased during the past three decades, health-span (the length of time a person remains healthy) has lagged, with older adults (greater than 65 years) now likely to live their last decade in poor health [2]. At the population level, ageing will have a significant economic impact as a reduced labour force and associated reduction in tax revenue, coupled with a greater demand for government programs that support older individuals, will place unsustainable fiscal pressure on government budgets. In Australia (population 25 million), this is projected to equate to an annual cost of $36 billion by 2028–29 [3]. Consequently, there is an urgent need to focus research resources on cost-effective strategies that contribute to the maintenance of functional longevity.
One of the most notable morphological changes observed with ageing is a progressive loss of skeletal muscle mass and quality, which is accompanied by a deterioration in physical function [4]. Age-related declines in muscle mass and function (i.e., sarcopenia) are related to reductions in the ability to undertake tasks of daily living [5], an increased risk of falls and fractures [6], and numerous metabolic impairments [7], ultimately leading to a cascade of events that precipitate a loss of physical independence and an increased reliance on care. Age-associated changes in skeletal muscle mitochondrial content, bioenergetics, reactive oxygen species (ROS) emission, and quality control have been identified and studied as putative candidates underpinning the loss of muscle mass and physical function that occurs with ageing [8, 9]. As such, several of these factors may be key targets for interventions that can attenuate declines in muscle structure and function.
To date, pharmacological interventions to combat sarcopenia have failed to produce clinically relevant outcomes regarding improvements in strength and physical performance. As such, exercise training is still arguably the most potent, cost-effective, and practical intervention for the preservation of muscle mass and function for most of the population throughout adult life. However, levels of habitual physical activity progressively decline with increasing age, and the majority of older adults fail to meet the physical activity guidelines outlined by the World Health Organisation [10]. As a result, alternative interventions that improve mitochondrial function and promote functional longevity independently or in combination with exercise are required. Other lifestyle interventions that have been shown to impact mitochondria include sleep [11], periods of energy restriction [12], and nutritional interventions such as supplements. This review summarises the effects of ageing and exercise training on human skeletal muscle mitochondria and discusses novel complementary supplement interventions that have plausible mechanistic underpinning to improve physical function in ageing through their interactions with mitochondria.
2 Mitochondria and Ageing Skeletal Muscle: Use It or Lose It
Ageing results in a decline in cardiovascular fitness (measured as an individual’s maximal aerobic capacity (V̇O2 max) beginning in the third decade of life [13]. While age-related declines in maximal aerobic capacity are largely related to reductions in maximal cardiac output [14], they may also be linked to age-related loss of muscle mass and declines in mitochondrial content and/or function. Beyond adenosine triphosphate (ATP) production, mitochondria play important roles in multiple cellular processes such as calcium uptake and extrusion, iron-sulphur cluster synthesis, and mitochondrial DNA (mtDNA) maintenance and expression [15]. Mitochondria also act as systemic signalling hubs that transduce information within and between cells [17]. For a comprehensive summary of known mitochondrial functions, readers are referred to several recent reviews [15,16,17].
Experimental evidence from mice and human studies suggest that mitochondria are crucial in the aetiology of sarcopenia [18]. Specifically, dysregulation in mitochondrial turnover via the mitochondrial-specific form of autophagy, mitophagy, is linked to elevated mitochondrial ROS emission and the activation of proteolytic pathways responsible for muscle protein breakdown and ‘anabolic resistance’ [19]. Muscle of older individuals also exhibits substantial loss of neuromuscular junction integrity and loss of innervation resulting in increased mitochondrial ROS emission in the denervated fibres and neighbouring innervated fibers in the same muscle [20, 21]. Age-related declines in mitochondrial bioenergetics (e.g., mitochondrial capacity and efficiency) have been proposed as a factor underpinning low physical function [22, 23], fatiguability [24], and loss of exercise efficiency (defined as the ratio of mechanical work rate over energy expenditure) with ageing [25]. Furthermore, there is some evidence that age-related changes in mitochondrial positioning within the muscle away from the calcium release units on the sarcoplasmic reticulum may underlie fatigue due to disruptions in calcium handling [26]. Despite many years of research investigating potential associations between mitochondria and ageing, there exists considerable debate as to the direct role of mitochondrial ‘dysfunction’ in age-related declines in muscle mass and function. This, in part, is because it is difficult to isolate the direct effects of ageing per se on mitochondria from the effects of low levels of physical activity, since these events are inter-related (i.e., age-related changes in mitochondrial content can contribute to a decline in physical activity and vice versa). Indeed, the age and level of habitual physical activity of a cohort under investigation can be a major confounder when interpreting the impact of a given intervention on mitochondrial content and/or function. Deeper investigation of the mitochondrial changes that accompany ageing is therefore crucial to understand how mitochondria might be targeted to promote the maintenance of physical function. Given that skeletal muscle contains highly specialised and metabolically distinct mitochondrial subpopulations [27, 28], future research investigating the effects of ageing and exercise on mitochondria should also take into account the independent pathways of these distinctly different populations.
2.1 Mitochondrial Content and Plasticity
One of the most inconsistent findings in the literature concerns the impact of ageing on skeletal muscle mitochondrial content, with some studies reporting no change in mitochondrial content in individuals aged over 65 years compared to their young counterparts [29,30,31] and others observing a decrease [32,33,34]. Such differences may, in part, be related to the various measures used to determine mitochondrial content, including maximal enzyme activities, mitochondrial protein levels, and mitochondrial density/volume. Mitochondrial content is typically up- or down-regulated based on the metabolic activity of the tissue/organ under investigation. In the context of exercise training, mitochondrial adaptations are largely dependent on the frequency, intensity and duration of an individual’s habitual activity levels [35, 36]. As such, the divergent levels of physical activity and exercise patterns that often accompany ageing are likely to contribute to the inconsistent results between investigations. Studies involving activity-matched old and young participants show little or no decline in several indices of mitochondrial content in ageing muscle [29]. Furthermore, comparisons between elderly subjects differing in training status demonstrate that lifelong training maintains the expression and activity of proteins associated with oxidative metabolism like those observed in muscle of young individuals [37, 38]. Whether differences in physical activity status alone can explain the discordant results remains to be determined.
Irrespective of age, mitochondria have been shown to respond similarly to an acute exercise challenge and exercise training in both young and old individuals, with similar exercise-induced mitochondrial signalling responses observed in skeletal muscle of older compared with younger males, independent of prior training status [39]. This is consistent with data indicating normal mitochondrial remodelling responses to long-term exercise training in the elderly [40, 41]. Therefore, it appears that ageing is not accompanied by diminished mitochondrial plasticity in response to both acute and chronic exercise stimuli [42].
2.2 Mitochondrial Bioenergetics
Considerable discussion surrounds the concept of decreased mitochondrial capacity with ageing. Several cross-sectional studies have demonstrated decreased mitochondrial capacity with advancing age [34, 40, 41, 43,44,45], although this finding is not universal [29, 46,47,48,49]. Such variability may be observed because many studies fail to consider important covariates, such as physical activity levels, cardiorespiratory fitness [43], and adiposity [44], which are likely to confound the relationship between mitochondrial capacity and age. Several studies have also used isolated mitochondria preparations [34, 45, 50], which tend to amplify age-related deficits in mitochondrial function compared to measurements conducted on permeabilized myofibers where the mitochondrial reticulum is maintained [51]. Contemporary research has shown that maximal mitochondrial respiration is not influenced by chronological age per se but is closely related to cardiorespiratory fitness and body composition [52], suggesting the aetiology of lower mitochondrial bioenergetics in ageing muscle may be primarily due to low levels of physical activity. Indeed, exercise training preserves mitochondrial capacity in older individuals when measured in both isolated mitochondria and permeabilized myofibers [40, 41], with several studies reporting no differences in mitochondrial capacity when young and old participants are matched for physical activity levels, V̇O2 max and body mass index (BMI) [29, 48, 53].
While maximal mitochondrial respiration does not appear to be largely influenced by ageing, a recent study reported that ageing is associated with a decline in skeletal muscle mitochondrial respiration in the presence of non-saturating adenosine diphosphate (ADP) concentrations that more closely reflect the in vivo metabolic milieu [46]. Furthermore, the ability of ADP to stimulate respiration (i.e., apparent sensitivity or Km) was also decreased in skeletal muscle of older individuals [46]. This is consistent with previous work in rodents, and suggests the underlying cause of mitochondrial defects in ageing muscle may be due to dysregulation of the adenosine nucleotide translocator (ANT), which transports ADP into the mitochondrial matrix [54]. Crucially, in that study, resistance exercise training reversed the age-related derangements in mitochondrial ADP sensitivity, further highlighting the capacity for exercise training to improve or potentially preserve some aspects of mitochondrial bioenergetics that occur with ageing [46].
2.3 Mitochondrial Reactive Oxygen Species Emission
Mitochondrial ROS play important roles in cellular signalling and in the promotion of several beneficial cellular adaptations to stress [55]. However, oxidative stress has been implicated in age-related muscle atrophy [19], suggesting there is a ‘threshold’ beyond which further elevations in ROS levels become deleterious. The notion that increased mitochondrial ROS production contributes to ageing in humans is a current topic of lively scientific debate [56,57,58]. Studies using isolated mitochondria report increased maximal mitochondrial H2O2 release with ageing [59]. However, isolating mitochondria potentiates ROS emission [60] and exaggerates the impact of ageing [51]. In humans, the maximal capacity for mitochondria to produce ROS is not increased with ageing when assessed using methods that preserve mitochondrial structure [29, 46]. In contrast, muscle immobilisation induces an increase in mitochondrial ROS in otherwise healthy individuals [61, 62], suggesting regular physical activity may be an effective way of attenuating increases in mitochondrial ROS in ageing skeletal muscle.
The transport of ADP into mitochondria and the subsequent binding of ADP to ATP synthase decreases membrane potential and the overall rate of mitochondrial superoxide production while simultaneously stimulating aerobic metabolism. While maximal rates of mitochondrial H2O2 emission are unaltered with ageing, the ability of ADP to reduce mitochondrial ROS emission is impaired in older individuals, leading to elevated mitochondrial ROS emission at submaximal ATP concentrations [46]. Resistance-based exercise training reverses age-related declines in mitochondrial ADP sensitivity while concurrently increasing the capacity for mitochondrial H2O2 emission, possibly through the induction of mitochondrial biogenesis [46]. Consequently, age-related increases in mitochondrial H2O2 emission in the presence of non-saturating ADP concentrations are not impacted by resistance exercise training [46]. Short-term resistance exercise training has also been shown to increase mitochondrial ROS emission during recovery from muscle immobilisation [62], suggesting that resistance training does not restore perturbations of mitochondrial redox homeostasis. Future work should investigate the ability of aerobic-based exercise training to improve ADP sensitivity and restore redox balance in aged muscle as this type of training may reverse immobilisation-induced increases in mitochondrial ROS emission [61].
2.4 Mitochondrial Quality Control
The mitochondrial reticulum is a dynamic structure undergoing constant remodelling. Mitochondrial fusion allows mitochondrial components to be exchanged and diluted while mitochondrial fission segregates damaged mitochondria for removal by mitophagy. These processes are essential for the maintenance of a healthy, functioning mitochondrial network. Several studies have investigated whether mitochondrial quality control is influenced by ageing, with inconclusive results. While some investigations report no age-related differences in the skeletal muscle expression of the major regulators of mitochondrial fusion (optic atrophy-1 (OPA1), mitofusin 1 (MFN1) and mitofusin 2 (MFN2)) and fission (mitochondrial fission 1 protein (FIS1), dynamin-related protein 1 (DRP1)) [22, 52, 63, 64], others find upregulation of mitochondrial dynamics proteins MFN2 and mitochondrial dynamics protein 49 (MiD49) in aged skeletal muscle, with the increased abundance of MFN2 exclusive to type II (‘fast twitch’, glycolytic) muscle fibres [30]. In that study [30], high-intensity interval training reversed the increase in MFN2 expression in type II fibres. Upregulation of MFN2 and MiD49 with ageing may be a protective mechanism against mitochondrial dysfunction, particularly in type II muscle fibre. In this context, exercise, particularly resistance-based training, may have a unique protective effect in negating the need for increased turnover of mitochondria. Therefore, age-related changes in mitochondrial quality control are likely related to low levels of habitual physical activity.
The effect of ageing on mitophagy is poorly understood. Decreased expression of mitophagy genes BNIP, DRP1 and Parkin has been observed in skeletal muscle of physically inactive elderly women [65]. In contrast, no differences in skeletal muscle Parkin protein expression between young and old physically active individuals have been reported [29]. To date, studies investigating the effect of ageing on mitophagy have focused on static measures of mRNA and protein in whole-cell lysates, which may be inadequate to capture and accurately quantify this dynamic process or elucidate what is happening at the mitochondrial level [29]. As a result, the impact of ageing on mitophagy is unclear.
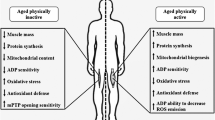
In summary, exercise training can attenuate, and in some cases reverse, many of the deleterious effects of ageing on mitochondria, including declines in mitochondrial content and bioenergetics, increased ROS emission, and perturbations of mitochondrial quality control (Fig. 1). Consequently, exercise training is an essential primary intervention to reduce many of the age-related declines in skeletal muscle mass and function that take place with an inactive lifestyle. However, levels of habitual physical activity steadily decline with age, and many older individuals fail to meet the physical activity guidelines outlined by the World Health Organisation [10]. Therefore, complementary interventions that promote the maintenance of skeletal muscle mitochondrial health and overall physical function with ageing are needed. While no current pharmacotherapies are effective in the treatment of sarcopenia, there have been intense research efforts focussed on the potential for supplements to improve mitochondrial function and promote muscle health and physical function in older individuals.

Exercise training attenuates many of the deleterious effects of ageing on mitochondria. Exercise training restores mitochondrial content, energetics, ROS emission, and quality control in aged skeletal muscle leading to improved muscle mass and physical function. ROS reactive oxygen species
3 Targeting the Mitochondria Through Supplements
Supplements can impact mitochondria through several mechanisms, including increases in mitochondrial biogenesis and bioenergetics, decreases in mitochondrial ROS emission and oxidative damage, and improvements in mitochondrial quality control (Table 1). The following section provides a contemporary evaluation of the evidence for the potential of several novel supplements to improve physical function in older individuals via their interactions with mitochondria. A thorough literature search of online databases was conducted to identify supplements that can improve physical function in older individuals. Supplements were included if there was evidence from human studies that this improvement may be mediated by mechanisms involving mitochondria. All authors agreed on the final list of supplements to be included in the review.
3.1 MitoQ
Despite being a major source of cellular ROS, mitochondria are particularly sensitive to ROS-induced oxidative damage, and ageing is associated with increased skeletal muscle mitochondrial ROS emission in the presence of non-saturating ADP concentrations [46]. ROS production by mitochondria can lead to oxidative damage to mitochondrial proteins, membranes and DNA, impairing the ability of mitochondria to synthesise ATP and carry out their normal metabolic functions [66]. Furthermore, elevated ROS emission activates proteolytic pathways in skeletal muscle and oxidative stress has been implicated in the pathophysiology of age-related muscle atrophy [19]. Consequently, there has been growing interest in the potential for mitochondria-targeted antioxidants to attenuate age-related mitochondrial oxidative stress and maintain skeletal muscle health with ageing [67].
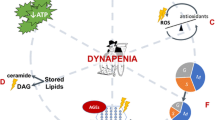
Coenzyme Q10 (CoQ10) is an essential component of the mitochondrial electron transport chain with antioxidant properties. Studies investigating the efficacy of CoQ10 to improve physical function in older individuals have shown equivocal results [68], which may, in part, be related to its poor bioavailability [69]. Conversely, mitochondria-targeted coenzyme Q10 (Mitoquinone, MitoQ) consists of a ubiquinone moiety attached to a lipophilic cation, which enables it to pass through the plasma membrane and accumulate in mitochondria via the plasma and mitochondrial membrane potential [70]. Within mitochondria, MitoQ localises to the inner mitochondrial membrane where it acts as an antioxidant, primarily by preventing lipid peroxidation (Fig. 2A). Mitochondrial membranes are rich in phospholipids that are susceptible to ROS-induced peroxidation, and lipid peroxidation byproducts can also damage mtDNA. This ROS-induced damage to mitochondrial lipids and DNA may compromise the mitochondrial membrane structure, ultimately generating a chain reaction and further amplifying mitochondrial ROS. As such, the antioxidant properties of MitoQ may act to indirectly reduce mitochondrial ROS production by protecting mitochondrial components from oxidative damage. MitoQ may also affect mitochondrial superoxide production by interacting with membrane potential (MitoQ uptake into mitochondria decreases proton motive force) and the coenzyme Q pool redox state [71]. MitoQ has been safely administered as a single oral 160 mg dose [72] or as a daily dose up to 80 mg [73]. However, supplementation with MitoQ at doses over 40 mg is sometimes associated with mild gastrointestinal side effects in some individuals. As a result, most studies provide MitoQ at a dose of 20 mg/day. MitoQ supplementation has been shown to decrease mitochondrial ROS emissions, increase protein expression of the antioxidant enzyme catalase [74], and attenuate exercise-induced DNA damage in skeletal muscle [75], suggesting MitoQ is available to skeletal muscle. However, there are currently no published data on the extent to which supplementation increases MitoQ levels in human skeletal muscle.

Supplements that promote physical function in ageing through their interactions with mitochondria. (A) MitoQ localises to the inner mitochondrial membrane and prevents the formation of lipid peroxidation products. MitoQ may also prevent mitochondrial superoxide production by interacting with proton motive forces and the coenzyme Q pool redox state. (B) Urolithin A activates mitochondrial biogenesis and PINK1-Parkin-mediated mitophagy. Parkin is recruited to PINK1 and ubiquitinates mitochondrial proteins. PINK1 then phosphorylates ubiquitin chains, leading to the accumulation of phospho-ubiquitinated mitochondrial proteins that serve as docking sites for adapter proteins that bind LC3. LC3 allows the formation of a phagosome membrane around mitochondria, which then fuses with and is broken down by lysosomes. (C) Incorporation of n-3 PUFAs EPA and DHA into mitochondrial membranes maintains mitochondrial energetics and decreases ROS emission. (D) GlyNAC provides precursors for the restoration of cellular GSH levels. GSH enters the mitochondrial matrix via different carriers, particularly the OGC and the DIC, where it aids in the detoxification of ROS. GlyNAC also increases the expression of regulators of mitochondrial biogenesis and mitophagy. PINK1 PTEN induced kinase 1, L• lipid radical, LOO• lipid peroxy radical, H2O2 hydrogen peroxide, O2•− superoxide, MnSOD manganese-dependent superoxide dismutase, •OH hydroxyl radical, GPx4 glutathione peroxide 4, GSH reduced glutathione, GSSG oxidised glutathione, ATP adenosine triphosphate, ADP adenosine diphosphate, ANT adenine nucleotide translocase, VDAC voltage-dependent anion channel, OGC 2-oxoglutarate carrier, DIC dicarboxylate carrier, ETC electron transport chain, DHA docosahexaenoic acid, EPA eicosapentaenoic acid, GlyNAC glycine and N-acetylcystine, GCL glutamate cysteine ligase, Glu-Cys γ-glutamylcysteine, GSy glutathione synthetase
Human studies that have investigated the effects of MitoQ supplementation on skeletal muscle mass and function in an ageing context are limited. Six weeks of daily 20 mg MitoQ supplementation improved leg-extension power in healthy middle-aged and older individuals [76]. However, the effects of MitoQ supplementation on skeletal muscle mitochondrial respiration and ROS emission were not determined in that study [76]. Six weeks of MitoQ supplementation (20 mg/day) decreased maximal mitochondrial H2O2 emission rates and increased catalase expression in skeletal muscle of middle-aged men [74]. Given that high ROS levels impair muscle contractile function [77], the improvements in physical function observed in older individuals following MitoQ supplementation may be related to a decrease in ROS levels in skeletal muscle. Despite observing decreases in skeletal muscle ROS emission, mitochondrial content and respiration were unaffected following 6 weeks of 20 mg/day MitoQ supplementation in middle-aged men [74]. The effects of MitoQ supplementation on mitochondrial function may be greater in individuals who have higher mitochondrial ROS emission and lower levels of mitochondrial function. Therefore, studies involving longer supplementation periods are needed to determine the long-term effects of MitoQ supplementation on mitochondrial and physical function in older adults.
As outlined in earlier sections of this review, exercise is crucial for maintaining muscle mass and function at all ages but becomes increasingly important with advancing age. Research into the mechanisms underlying the beneficial effects of exercise has identified ROS as key signalling molecules that regulate exercise adaptations in skeletal muscle. However, several ROS-stimulated exercise responses, including acute stress responses [78] and anabolic responses [79], are attenuated in aged skeletal muscle regardless of training status, which may blunt several of the health-promoting effects of exercise in older individuals. Age-related increases in mitochondrial ROS emission have been linked to the failure of ROS-stimulated exercise responses in skeletal muscle of older individuals [80]. It has been proposed that increased mitochondrial ROS emission in aged muscle stimulates retrograde mitonuclear communication to increase the expression of a range of cytoprotective proteins that protect against oxidative damage [80]. This adaptive response may modify the local cytosol redox environment to induce a more reductive state in key cysteines of specific signalling proteins. In support of this hypothesis, expression of both cytosolic and mitochondrial forms of the antioxidant enzyme thioredoxin is elevated in muscles from old mice compared to younger mice [81]. Furthermore, redox proteomic analysis of human skeletal muscle has shown that ageing is associated with decreased levels of cysteine oxidation in several proteins [82]. Shifting to a more reduced cytosolic environment may prevent the transient oxidation of redox-sensitive signalling proteins during exercise and attenuate health-promoting exercise adaptations. Given that age-related increases in mitochondrial ROS emissions have been linked to the failure of redox responses to exercise in older individuals, interventions that decrease mitochondrial ROS emission in older individuals may restore these responses. MitoQ supplementation has been shown to increase training-induced improvements in peak cycling power output in middle-aged men, which may be related to the augmentation of the skeletal muscle transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and vascular endothelial growth factor (VEGF) mRNA following acute exercise [83]. Further studies are required to determine whether MitoQ supplementation can restore redox responses to exercise in aged skeletal muscle.
3.2 Urolithin A
Urolithin A is a natural compound produced in the large intestine by gut bacteria from ingested ellagitannins and ellagic acid, which are polyphenols present in a variety of plant products and foods, including pomegranates, strawberries, raspberries and walnuts [84, 85]. However, the ingestion of ellagitannin-rich foods is not always sufficient to elevate circulating levels of urolithin A, as specific gut microbiota are needed to convert ellagitannin into urolithin A. Urolithin A was only detectable in the plasma of 12% of healthy participants screened, while supplementation with urolithin A bypasses the need for microbiome-mediated conversion of urolithin A precursors and results in significantly elevated urolithin A levels in healthy adults [86]. Specifically, pharmacokinetic data indicate urolithin A is bioavailable at doses ranging from 250 to 2,000 mg, has a relatively long half-life (t1/2 = 17–22 h), and can be detected in skeletal muscle following oral supplementation [87].
Recent studies in humans provide evidence that urolithin A supplementation has positive effects on physical function in older individuals. Four months of daily supplementation with 1000 mg urolithin A improved muscular endurance during repeated contractions in older adults who presented with average physical performance measures (defined as 6-min walk distance ≤ 550 m) and low-average maximal ATP synthesis rates (≤ 1.0 mM/s measured using magnetic resonance spectroscopy) [88]. Clinically relevant improvements in whole-body measures of physical performance (6-min walk test distance and V̇O2peak) have also been observed following urolithin A supplementation [89]. Improved lower-body muscle strength was observed following 4 months of daily supplementation with 500 mg urolithin A supplementation in overweight middle-aged individuals [89]. Improvements in muscle strength following urolithin A supplementation were associated with an enrichment of gene sets related to muscle contraction and ribosome in skeletal muscle biopsies [89], suggesting urolithin A may activate an ‘anabolic’ response in skeletal muscle. Ageing is associated with anabolic resistance, a blunted stimulation of muscle protein synthesis in response to common anabolic stimuli such as exercise and dietary protein [79]. Given that age-related anabolic resistance contributes to the progressive decline in muscle mass and strength, further research is required to establish whether activation of anabolic responses in skeletal muscle by urolithin A has beneficial effects on muscle mass and strength in older individuals.
Improvements in physical performance following urolithin A supplementation are consistently associated with improvements in markers of skeletal muscle mitochondrial health (Fig. 2B). Urolithin A supplementation decreases plasma acylcarnitines [87,88,89], suggesting a decrease in incomplete fatty acid (FA) oxidation. Consistent with this finding, urolithin A increases the expression of several mitochondrial genes and tricarboxylic acid (TCA) cycle and oxidative phosphorylation protein levels in skeletal muscle [87, 89]. Collectively, these data suggest that urolithin A stimulates mitochondrial biogenesis and improves mitochondrial FA metabolism in skeletal muscle. This premise is supported by proteomic analysis of human skeletal muscle revealing that urolithin A supplementation increases markers of PTEN-induced kinase 1 (PINK1)/Parkin-mediated mitophagy [89].
In addition to its benefits on mitochondrial health, urolithin A also decreases plasma biomarkers of inflammation, including C-reactive protein (CRP) and inflammatory cytokines [88, 89]. Therefore, urolithin A may offer a potential dual benefit to muscle health during ageing by improving both mitochondrial function and attenuating age-related chronic inflammation. Urolithin A induction of mitophagy could mediate its anti-inflammatory effect, as removing dysfunctional mitochondria reduces the production of ROS and the release of mtDNA and cardiolipins, both known triggers of inflammatory responses [90]. Alternatively, urolithin A-mediated reduction of inflammatory markers may contribute to blunting their negative regulation of mitochondrial biogenesis effectors, such as the PGC-1α and sirtuin 1 (SIRT1), thereby allowing the generation of new mitochondria [90]. PGC-1α upregulates mitochondrial and metabolic gene expression, synchronizes several exercise-associated aspects of muscle plasticity and function, while concomitantly suppressing a broad inflammatory response [91]. Therefore, upregulation of PGC-1α in skeletal muscle following both exercise and supplementation with urolithin A may confer several local and whole-body effects, conferring protection against age-related declines in skeletal muscle mass and physical function [92] (Fig. 3).

Both exercise and supplements such as urolithin A or GlyNAC activate cell signalling networks in skeletal muscle that target the peroxisome proliferator-activated receptor-γ coactivator 1α isoform 1 (PGC-1α), which may confer protective effects on skeletal muscle during ageing. Specifically, classical signalling kinases such as the 5’AMP activated protein kinase (AMPK) [147] or p38 MAPK [148] can phosphorylate PGC-1α, stabilizing the protein and promoting cellular accumulation. Within the nucleus, PGC-1α can bind to a host of transcription factors, including nuclear estrogen related receptors (ERRs) [149], respiratory factors (NRF) 1 and 2 [150], as well as peroxisome proliferator-activated receptor receptors (PPARs) [151]. This promotes the development of a more oxidative phenotype in muscle through mitochondrial biogenesis and increased expression of proteins associated with lipid uptake, transport, and metabolism [151, 152]. Furthermore, PGC-1α can exert an anti-inflammatory role in muscle by regulating the expression of proinflammatory cytokines such as interleukin-6 and tumor necrosis factor-alpha (TNF-α) [153, 154] and decreasing ROS production in part via the induction of genes that activate mitochondrial uncoupling proteins [155, 156]. GlyNAC glycine and N-acetylcysteine, PGC-1α peroxisome proliferator-activated receptor-γ coactivator 1α isoform, 1ESRRA estrogen-related receptor α, NFE2L1 nuclear factor erythroid-derived 2 like-1, NFE2L2 nuclear factor erythroid‐derived 2‐like 2, PPARA,peroxisome proliferator activated receptor alpha, ROS reactive oxygen species
3.3 Omega-3 Polyunsaturated Fatty Acids
The omega-3 polyunsaturated FA (n-3 PUFAs) eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are critical for the structure and function of biological membranes. While n-3 PUFAs can be obtained following the consumption of fatty fish such as salmon, herring, mackerel, tuna and sardines, supplementation with EPA and DHA results in incorporation of these n-3 PUFAs into phospholipid membranes within skeletal muscle [93,94,95]. Population recommendations for EPA and DHA intake are typically 250–500 mg/day for EPA and DHA combined. However, studies investigating the effects of n-3 PUFA supplementation on physical function typically use higher doses (intakes of up to 5 g/day), which are difficult to obtain from food sources. While this dose is considered safe for human supplementation, the minimal n-3 PUFA regimen required to improve physical function is currently unknown.
A growing body of evidence suggests that n-3 PUFA supplementation positively impacts skeletal muscle anabolism and strength gains [96]. Eight weeks of n-3 PUFA supplementation (1.9 g/day EPA and 1.5 g/day DHA) increases rates of muscle protein synthesis in young, middle-aged and older adults in response to a hyperaminoacidemic-hyperinsulinemic clamp [97, 98]. Furthermore, n-3 PUFA supplementation (2.1 g/day EPA and 0.6 g/day DHA) amplified the improvements in muscle strength and quality observed in older women, but not men, following 18 weeks of resistance exercise training [99]. n-3 PUFA supplementation has also been shown to improve skeletal muscle mass and strength in older men and women [100, 101] and attenuate immobilisation-induced declines in skeletal muscle mass in healthy young women [102]. Collectively, these observations indicate that n-3 PUFAs may protect against age-related declines in muscle mass and function and muscle atrophy during acute periods of disuse (e.g., before and after elective surgery). However, while some studies report improvements in muscle anabolism and physical function following n-3 PUFA supplementation, this is not a universal finding. Six months of n-3 PUFA supplementation (1.6 g/day EPA and 2.3 g/day DHA) failed to improve lean mass, muscle strength or whole-body physical performance in 107 older males and females [103]. Similarly, chronic (3 years) n-3 PUFA supplementation (0.3 g/day EPA and 0.7 g/day DHA) failed to improve whole-body physical performance when taken alone or in combination with strength training in a cohort of 2,000 healthy men and women aged 70 years and over [104]. These conflicting results may, in part, be explained by differences in the dose administered, or by differences in baseline physical function and nutrition status of the participants as individuals with reduced physical function and/or lower protein intakes may experience greater benefits following n-3 PUFA supplementation.
The mechanisms underlying improved muscle anabolism following n-3 PUFA supplementation are unknown but increasing evidence suggests that these effects may be mediated by mitochondria (Fig. 2C). Incorporation of n-3 PUFAs into phospholipid membranes impacts the organisation and function of membrane proteins, which is important in mitochondria where electron transfer is tightly coupled between the complexes of the electron transport chain embedded in the inner mitochondrial membrane. Sixteen weeks of n-3 PUFA supplementation (2.7 g/day EPA and 1.2 g/day DHA) decreased mitochondrial H2O2 emissions in skeletal muscle of older individuals [105], suggesting n-3 PUFA supplementation may be an effective strategy to attenuate age-related increases in skeletal muscle mitochondrial ROS production and oxidative stress. Improvements in ADP sensitivity have also been observed in skeletal muscle of young individuals following n-3 PUFA supplementation [93, 106]. This is important from an ageing perspective as mitochondrial ADP sensitivity is decreased in skeletal muscle of older individuals, resulting in increased mitochondrial ROS emission [46]. Age-related anabolic resistance and loss of muscle mass and strength have also been linked to elevated skeletal muscle mitochondrial ROS levels [80, 107]. Therefore, increasing mitochondrial ADP sensitivity and reducing mitochondrial ROS levels may be one mechanism through which n-3 PUFA supplementation improves muscle mass and strength in older individuals.
Supplementation with n-3 PUFAs (3 g/day EPA and 2 g/day DHA) attenuates reductions in mitochondrial protein content and respiration independent of changes in mitochondrial H2O2 emissions after 2 weeks of single-leg immobilisation in young women [106]. These mitochondrial responses were associated with an attenuated loss of MRI-measured skeletal muscle volume and leg lean mass [102]. Whether the effects of n-3 PUFA supplementation on mitochondrial protein content and respiration are a cause or a consequence of changes in muscle mass is currently unknown. However, the loss of muscle mass during periods of disuse is preceded by reductions in mitochondrial content [108]. Given that cytosolic protein synthesis is an energetically costly process, and mitochondria are the primary source of cellular energy, retrograde signalling mechanisms may exist to maintain the balance between cytosolic and mitochondrial protein synthesis/bioenergetics [109]. Therefore, maintenance of mitochondrial bioenergetics during disuse may sustain mitochondrial and cytosolic protein transcription and translation [96]. Such a hypothesis has not been tested in humans and requires further investigation.
3.4 Glycine and N-Acetylcysteine (GlyNAC)
Glutathione (GSH) is the most abundant endogenous intracellular antioxidant. GSH donates reducing equivalents to neutralize H2O2 to water in a reaction where GSH itself becomes oxidized to GSSG. Therefore, GSH exists and cycles between a reduced (GSH) and oxidised (GSSG) state to maintain an optimal cellular redox state. GSH is a tripeptide composed of three amino acids: glycine, cysteine and glutamic acid. In older adults, impaired GSH synthesis caused by decreased availability of glycine and cysteine (but not glutamic acid) contributes to the development of GSH deficiency, leading to increased levels of oxidative stress [110]. GSH deficiency cannot be corrected through oral GSH supplementation as GSH is digested in the gut. However, daily oral supplementation with GlyNAC, a combination of GSH precursor amino acids glycine and cysteine (provided as N-acetylcysteine, NAC), can correct glycine and cysteine deficiencies, restore impaired GSH synthesis, and increase intracellular GSH concentrations in older individuals [110,111,112]. There is now widespread acceptance that ROS play an important role in cell signalling and that antioxidant supplementation is a double-edged sword: too much can be as detrimental as too little. Therefore, GlyNAC supplementation is an attractive intervention as it provides precursor amino acids to allow cells to autoregulate their own GSH synthesis based on cellular need.
GlyNAC supplementation (12–24 weeks, 100 mg/kg/day glycine and cysteine) improved gait speed, muscle strength and exercise capacity in older individuals [112, 113] and patients with HIV who develop the onset of geriatric conditions at an early age [114]. GlyNAC supplementation has also been shown to attenuate age-related increases in muscle protein breakdown [113], which has important implications for the maintenance of muscle mass during ageing. Multiple factors could be responsible for the observed improvements in physical function following GlyNAC supplementation as GlyNAC promotes positive effects on mitochondria, oxidative stress and inflammation, all of which interact and contribute to the decline in skeletal muscle mass and function occurring with ageing (Fig. 2D). Improvements in physical function following GlyNAC supplementation are consistently associated with increases in fat oxidation measured using calorimetry [112,113,114,115] and increases in protein expression of regulators of mitochondrial biogenesis (PGC-1α) and energy metabolism including mitochondrial FA transport (CPT1b), β-oxidation (HADHA), electron complexes (I, II, III, IV), ATP synthesis (ATP synthase F1 subunit alpha; ATP5A), and mitophagy (PINK1) [112, 114]. While evidence from human studies suggests that GlyNAC improves physical function in ageing via interactions with mitochondria, these results should be interpreted with caution as the majority of data derive from pilot trials with small sample sizes and no placebo group [113,114,115].
3.5 NAD+ Precursors
Nicotinamide adenine dinucleotide (NAD+) is a crucial universal cofactor. In its reduced form, NADH fuels oxidative phosphorylation by supplying the mitochondrial electron transport chain with electrons from glycolysis and the TCA cycle. NAD+ also has an important role as a substrate in multiple enzymatic processes. NAD+ is replenished through three pathways: the Preiss-Handler pathway, the kynurenine pathway and the salvage pathway [116]. The Preiss-Handler pathway converts nicotinic acid (NA) through nicotinic acid mononucleotide (NAMN) and nicotinic acid adenine dinucleotide (NAAD) to NAD+. The kynurenine pathway begins with tryptophan and converges with the Preiss-Handler pathway at the level of NAMN. Nicotinic acid riboside (NAR) also converges with the Preiss-Handler pathway at the level of NAMN. In the salvage pathway, NAM is converted to NAD+ through nicotinamide mononucleotide (NMN). Ageing is associated with a systemic decrease in NAD+ levels [116], which may contribute to declines in mitochondrial and physical function. As a result, there has been growing interest in the potential for supplementation with NAD+ precursors, including nicotinamide riboside (NR), NMN and NA to increase NAD+ levels and attenuate age-related pathology.
NR is the most well studied of all the NAD+ precursors [117], with its uptake mediated by the nucleoside transporter protein family [118]. Once inside the cell, NR can be phosphorylated by the NR kinases (NRK1 and NRK2) [119] or be deribosylated by the purine nucleoside phosphorylase to become NAM [120]. In principle, both conversions allow the production of NAD+ via entry into the salvage pathway. However, animal studies provide evidence that few tissues are exposed to large amounts of NR since the majority of orally administered NR is converted to NAM in the small intestine and then to NA by the gut microbiota [121,122,123]. The small amount of orally administered NR that enters the blood is rapidly converted to NAM in whole blood, meaning NR is not easily detectable in blood following oral administration [124]. Acute supplementation with 250 mg and chronic supplementation with between 100 and 2,000 mg NR per day for between 7 days and 5 months increased NAD+ and a range of its related metabolites in whole blood and peripheral blood mononuclear cells (PBMCs) [125,126,127,128,129,130]. However, to date, there is no evidence that oral NR increases muscle NAD+ levels, which likely explains why the majority of studies investigating the effects of NR supplementation on mitochondrial content and respiration have failed to show positive results [131,132,133,134]. Despite this, acute NR supplementation has been shown to increase peak isometric torque and attenuate fatigue in older individuals [130]. Given that high ROS levels impact skeletal muscle contractile function [77, 135], the improvements in physical function observed in that study may be related to improvements in redox homeostasis as NR supplementation decreased urine F2-isoprostanes and increased erythrocyte glutathione levels in the older group [130]. Further research is needed to elucidate the mechanisms through which NR improves physical function in older individuals as these effects appear to be unrelated to the capacity for oral NR supplementation to impact skeletal muscle NAD+ levels.
Oral supplementation with the NAD+ precursor NMN increases NAD+ and its metabolites in blood [126, 127, 136]. However, there is currently no evidence that NMN supplementation increases NAD+ levels in human skeletal muscle. Despite this, several studies have investigated the effect of NMN on physical function. Twelve weeks of daily 250 mg NMN supplementation improved walking speed and grip strength in older men [136]. In contrast, shorter periods of NMN supplementation (4–10 weeks) failed to impact muscle strength, fatigue and V̇O2peak in individuals with prediabetes or middle-aged and older adults with overweight/obesity. These observations may be explained by the failure of NMN to impact muscle mitochondrial oxidative capacity [126, 127]. Taken together, the results from studies investigating the effect of oral NMN on physical function are conflicting, and further research is required to elucidate potential mechanisms by which NMN acts on skeletal muscle to exert any beneficial effect, if present. This is particularly important given that oral NMN supplementation has not been shown to impact muscle NAD+ levels.
NA can be incorporated into the NAD+ pool in various tissues via the Preiss-Handler pathway, but synthesis of NAD+ from NA in muscle appears to be low or non-existent [124, 137]. However, 10 months of NA treatment (escalating dose starting at 250 mg/day and reaching 1 g/day by 3 months) in patients with mitochondrial myopathy and NAD+ deficiency restored muscle NAD+ levels, increased mitochondrial respiratory enzyme activity and mitochondrial mass, and improved muscle strength and walking speed [138]. Results from that investigation [138] suggest that human trials involving NAD+ precursors may be of benefit to individuals with NAD+ deficiency, but further studies are needed in a variety of clinical populations to determine the effects of NA supplementation on muscle NAD+ levels and mitochondrial and physical function.
4 Application to Master Athletes
While most older adults reduce their levels of habitual physical activity with advancing age, master athletes are a unique subpopulation in that they continue to maintain high levels of physical activity late in life, including structured and performance-driven exercise training. Master athletes exhibit remarkable physical function relative to their sedentary counterparts [139]. Furthermore, some master athletes even display muscular strength, performance, myofiber properties and function comparable with young, healthy individuals [140]. However, despite completing high levels of physical training that partially protect against the decline in musculoskeletal and cardiorespiratory health that occurs with ageing, master athletes still experience lower levels of performance compared to their young, trained counterparts [139]. Over 60% of master athletes report using nutritional supplements [141, 142], suggesting this population is aware of the potential benefits of such interventions to mitigate age-related declines in health and performance. Given that master athletes exhibit age-related declines in performance [139], attenuated adaptive responses [78, 143], and impaired recovery [144], the supplements discussed in this review could have ergogenic effects in this population through several mechanisms, including improvements in mitochondrial function, improved adaptive responses [83, 105], and decreased muscle damage/inflammation [89, 112, 145]. While studies in master athletes are limited, 6 weeks of MitoQ supplementation has been shown to attenuate exercise-induced oxidative stress and improve cycling time trial performance in master cyclists (average age 44 years) [146]. Further studies are required to determine whether the supplements discussed in this review have performance benefits for master athletes.
5 Conclusion
This review has highlighted the critical role of both aerobic- and strength-based exercise training in attenuating many of the deleterious effects of ageing on skeletal muscle mitochondria, including decreases in mitochondrial content and bioenergetics, increased mitochondrial ROS emission, and perturbations of mitochondrial quality control. While exercise training is essential for the attenuation of age-related declines in skeletal muscle mass and function, many older adults have low levels of habitual physical activity. Therefore, there is a need to identify complementary interventions that can improve physical function in ageing either independently or in synergy with exercise. There is both mechanistic and functional evidence to demonstrate that several supplements can improve physical function in older individuals through direct or indirect effects on skeletal muscle mitochondria. In this context, MitoQ, urolithin A, n-3 PUFAs and GlyNAC improve physical function in older individuals through several mechanisms including increases in mitochondrial biogenesis and bioenergetics, decreases in mitochondrial ROS emission and oxidative damage, and improvements in mitochondrial quality control. While there is some evidence that some NAD+ precursors can improve physical function in older individuals, such an outcome seems unrelated to and independent of changes in skeletal muscle mitochondrial function. While this review has summarised the evidence for supplements that improve mitochondrial function and skeletal muscle health in elderly humans, there are several compounds that have shown similar effects in preclinical models (e.g., mitochondrial-derived peptides and mitochondrial uncouplers). Future studies should investigate the safety and efficacy of these compounds with a view to extending the human health-span.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
United Nations, D.o.E.a.S.A., Population Division, World Population Ageing 2019: Highlights. 2019. (ST/ESA/SER.A/430).
World health statistics 2024: monitoring health for the SDGs, Sustainable Development Goals. Geneva: World Health Organization; 2024. Licence: CC BY-NC-SA 3.0 IGO.
Office, P.B., Australia’s ageing population: Understanding the fiscal impacts over the next decade. 2019.
Roubenoff R. Sarcopenia and its implications for the elderly. Eur J Clin Nutr. 2000;54(Suppl 3):S40–7.
Janssen I, Heymsfield SB, Ross R. Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J Am Geriatr Soc. 2002;50(5):889–96.
Sherrington C, et al. Exercise for preventing falls in older people living in the community. Cochrane Database Syst Rev. 2019;1(1):Cd012424.
Crescenzo R, et al. Skeletal muscle mitochondrial energetic efficiency and aging. Int J Mol Sci. 2015;16(5):10674–85.
Carter HN, Chen CC, Hood DA. Mitochondria, muscle health, and exercise with advancing age. Physiology (Bethesda). 2015;30(3):208–23.
Dai DF, et al. Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan. 2014;3:6.
Organisation WH. Global status report on physical activity 2022. 2022.
Saner NJ, et al. Exercise mitigates sleep-loss-induced changes in glucose tolerance, mitochondrial function, sarcoplasmic protein synthesis, and diurnal rhythms. Mol Metab. 2021;43: 101110.
Civitarese AE, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4(3): e76.
Wilson TM, Tanaka H. Meta-analysis of the age-associated decline in maximal aerobic capacity in men: relation to training status. Am J Physiol Heart Circ Physiol. 2000;278(3):H829–34.
Tanaka H, Seals DR. Endurance exercise performance in Masters athletes: age-associated changes and underlying physiological mechanisms. J Physiol. 2008;586(1):55–63.
Monzel AS, Enríquez JA, Picard M. Multifaceted mitochondria: moving mitochondrial science beyond function and dysfunction. Nat Metab. 2023;5(4):546–62.
Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61(5):654–66.
Hood DA, et al. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu Rev Physiol. 2019;81:19–41.
Marzetti E, et al. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol. 2013;45(10):2288–301.
Gomez-Cabrera MC, et al. Redox modulation of muscle mass and function. Redox Biol. 2020;35: 101531.
Muller FL, et al. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293(3):R1159–68.
Pollock N, et al. Denervated muscle fibers induce mitochondrial peroxide generation in neighboring innervated fibers: Role in muscle aging. Free Radic Biol Med. 2017;112:84–92.
Joseph AM, et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell. 2012;11(5):801–9.
Coen PM, et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci. 2013;68(4):447–55.
Santanasto AJ, et al. Skeletal muscle mitochondrial function and fatigability in older adults. J Gerontol A Biol Sci Med Sci. 2015;70(11):1379–85.
Broskey NT, et al. Exercise efficiency relates with mitochondrial content and function in older adults. Physiol Rep. 2015;3(6):e12418.
Protasi F, Pietrangelo L, Boncompagni S. Improper remodeling of organelles deputed to Ca(2+) handling and aerobic ATP production underlies muscle dysfunction in ageing. Int J Mol Sci. 2021;22(12):6195.
Ogata T, Yamasaki Y. Scanning electron-microscopic studies on the three-dimensional structure of mitochondria in the mammalian red, white and intermediate muscle fibers. Cell Tissue Res. 1985;241(2):251–6.
Murgia M, et al. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015;16(3):387–95.
Gouspillou G, et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. Faseb j. 2014;28(4):1621–33.
Wyckelsma VL, et al. Preservation of skeletal muscle mitochondrial content in older adults: relationship between mitochondria, fibre type and high-intensity exercise training. J Physiol. 2017;595(11):3345–59.
Johannsen DL, et al. Ectopic lipid accumulation and reduced glucose tolerance in elderly adults are accompanied by altered skeletal muscle mitochondrial activity. J Clin Endocrinol Metab. 2012;97(1):242–50.
Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526 Pt 1(Pt 1):203–10.
Hebert SL, et al. Mitochondrial aging and physical decline: insights from three generations of women. J Gerontol A Biol Sci Med Sci. 2015;70(11):1409–17.
Short KR, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102(15):5618–23.
Furrer R, Hawley JA, Handschin C. The molecular athlete: exercise physiology from mechanisms to medals. Physiol Rev. 2023;103(3):1693–787.
Smith JAB, et al. Exercise metabolism and adaptation in skeletal muscle. Nat Rev Mol Cell Biol. 2023;24(9):607–32.
Iversen N, et al. Mitochondrial biogenesis and angiogenesis in skeletal muscle of the elderly. Exp Gerontol. 2011;46(8):670–8.
Gollnick PD, et al. Enzyme activity and fiber composition in skeletal muscle of untrained and trained men. J Appl Physiol. 1972;33(3):312–9.
Cobley JN, et al. PGC-1α transcriptional response and mitochondrial adaptation to acute exercise is maintained in skeletal muscle of sedentary elderly males. Biogerontology. 2012;13(6):621–31.
Lanza IR, et al. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57(11):2933–42.
Grevendonk L, et al. Impact of aging and exercise on skeletal muscle mitochondrial capacity, energy metabolism, and physical function. Nat Commun. 2021;12(1):4773.
Cobley JN, et al. Exercise improves mitochondrial and redox-regulated stress responses in the elderly: better late than never! Biogerontology. 2015;16(2):249–64.
Tonkonogi M, et al. Reduced oxidative power but unchanged antioxidative capacity in skeletal muscle from aged humans. Pflugers Arch. 2003;446(2):261–9.
Porter C, et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am J Physiol Endocrinol Metab. 2015;309(3):E224–32.
Boffoli D, et al. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim Biophys Acta. 1994;1226(1):73–82.
Holloway GP, et al. Age-associated impairments in mitochondrial ADP sensitivity contribute to redox stress in senescent human skeletal muscle. Cell Rep. 2018;22(11):2837–48.
Hütter E, et al. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell. 2007;6(2):245–56.
Larsen S, et al. The influence of age and aerobic fitness: effects on mitochondrial respiration in skeletal muscle. Acta Physiol (Oxf). 2012;205(3):423–32.
Rasmussen UF, et al. Human skeletal muscle mitochondrial metabolism in youth and senescence: no signs of functional changes in ATP formation and mitochondrial oxidative capacity. Pflugers Arch. 2003;446(2):270–8.
Trounce I, Byrne E, Marzuki S. Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet. 1989;1(8639):637–9.
Picard M, et al. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell. 2010;9(6):1032–46.
Distefano G, et al. Chronological age does not influence ex-vivo mitochondrial respiration and quality control in skeletal muscle. J Gerontol A Biol Sci Med Sci. 2017;72(4):535–42.
Distefano G, et al. Physical activity unveils the relationship between mitochondrial energetics, muscle quality, and physical function in older adults. J Cachexia Sarcopenia Muscle. 2018;9(2):279–94.
Gouspillou G, et al. Alteration of mitochondrial oxidative phosphorylation in aged skeletal muscle involves modification of adenine nucleotide translocator. Biochim Biophys Acta. 2010;1797(2):143–51.
Collins Y, et al. Mitochondrial redox signalling at a glance. J Cell Sci. 2012;125(Pt 4):801–6.
Jackson MJ. Reactive oxygen species in sarcopenia: Should we focus on excess oxidative damage or defective redox signalling? Mol Aspects Med. 2016;50:33–40.
Jackson MJ, McArdle A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J Physiol. 2011;589(Pt 9):2139–45.
Hepple RT. Impact of aging on mitochondrial function in cardiac and skeletal muscle. Free Radic Biol Med. 2016;98:177–86.
Capel F, et al. Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech Ageing Dev. 2005;126(4):505–11.
Picard M, et al. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE. 2011;6(3): e18317.
Gram M, et al. Skeletal muscle mitochondrial H2 O2 emission increases with immobilization and decreases after aerobic training in young and older men. J Physiol. 2015;593(17):4011–27.
Pileggi CA, et al. Exercise recovery increases skeletal muscle H(2)O(2) emission and mitochondrial respiratory capacity following two-weeks of limb immobilization. Free Radic Biol Med. 2018;124:241–8.
Konopka AR, et al. Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training. J Gerontol A Biol Sci Med Sci. 2014;69(4):371–8.
Bori Z, et al. The effects of aging, physical training, and a single bout of exercise on mitochondrial protein expression in human skeletal muscle. Exp Gerontol. 2012;47(6):417–24.
Drummond MJ, et al. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison. J Gerontol A Biol Sci Med Sci. 2014;69(8):1040–8.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Broome SC, Woodhead JST, Merry TL. Mitochondria-targeted antioxidants and skeletal muscle function. Antioxidants (Basel), 2018. 7(8).
Bolt J, Sandhu S, Mohammadi A. Effect of coenzyme Q10 supplementation on sarcopenia, frailty, and falls: a scoping review. J Nutr Health Aging. 2023;27(7):586–92.
Zaki NM. Strategies for oral delivery and mitochondrial targeting of CoQ10. Drug Deliv. 2016;23(6):1868–81.
Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–56.
Robb EL, et al. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem. 2018;293(25):9869–79.
Rossman MJ, et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension. 2018;71(6):1056–63.
Snow BJ, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25(11):1670–4.
Pham T, et al. MitoQ and CoQ10 supplementation mildly suppresses skeletal muscle mitochondrial hydrogen peroxide levels without impacting mitochondrial function in middle-aged men. Eur J Appl Physiol. 2020;120(7):1657–69.
Williamson J, et al. The mitochondria-targeted antioxidant MitoQ, attenuates exercise-induced mitochondrial DNA damage. Redox Biol. 2020;36: 101673.
Bispham NZ, et al. MitoQ supplementation improves leg-extension power in healthy late middle-aged and older adults. The FASEB J. 2017;31(S1):lb852.
Reid MB. Invited review: redox modulation of skeletal muscle contraction: what we know and what we don’t. J Appl Physiol (1985). 2001;90(2):724–31.
Cobley JN, et al. Lifelong training preserves some redox-regulated adaptive responses after an acute exercise stimulus in aged human skeletal muscle. Free Radic Biol Med. 2014;70:23–32.
Kumar V, et al. Age-related differences in the dose-response relationship of muscle protein synthesis to resistance exercise in young and old men. J Physiol. 2009;587(1):211–7.
Jackson MJ. On the mechanisms underlying attenuated redox responses to exercise in older individuals: a hypothesis. Free Radic Biol Med. 2020;161:326–38.
Dimauro I, et al. In vitro susceptibility of thioredoxins and glutathione to redox modification and aging-related changes in skeletal muscle. Free Radic Biol Med. 2012;53(11):2017–27.
Pugh JN, et al. Exercise stress leads to an acute loss of mitochondrial proteins and disruption of redox control in skeletal muscle of older subjects: an underlying decrease in resilience with aging? Free Radic Biol Med. 2021;177:88–99.
Broome SC, et al. MitoQ supplementation augments acute exercise-induced increases in muscle PGC1α mRNA and improves training-induced increases in peak power independent of mitochondrial content and function in untrained middle-aged men. Redox Biol. 2022;53: 102341.
Espín JC, et al. Biological significance of urolithins, the gut microbial ellagic Acid-derived metabolites: the evidence so far. Evid Based Complement Alternat Med. 2013;2013: 270418.
González-Barrio R, et al. Bioavailability of anthocyanins and ellagitannins following consumption of raspberries by healthy humans and subjects with an ileostomy. J Agric Food Chem. 2010;58(7):3933–9.
Singh A, et al. Direct supplementation with Urolithin A overcomes limitations of dietary exposure and gut microbiome variability in healthy adults to achieve consistent levels across the population. Eur J Clin Nutr. 2022;76(2):297–308.
Andreux PA, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1(6):595–603.
Liu S, et al. Effect of urolithin A supplementation on muscle endurance and mitochondrial health in older adults: a randomized clinical trial. JAMA Netw Open. 2022;5(1): e2144279.
Singh A, et al. Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell Rep Med. 2022;3(5): 100633.
Reid AL, Alexander MS. The Interplay of Mitophagy and Inflammation in Duchenne Muscular Dystrophy. Life (Basel). 2021;11(7).
Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27(7):728–35.
Sandri M, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. 2006;103(44):16260–5.
Herbst EA, et al. Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J Physiol. 2014;592(6):1341–52.
Gerling CJ, et al. Incorporation of omega-3 fatty acids into human skeletal muscle sarcolemmal and mitochondrial membranes following 12 weeks of fish oil supplementation. Front Physiol. 2019;10:348.
McGlory C, et al. Temporal changes in human skeletal muscle and blood lipid composition with fish oil supplementation. Prostaglandins Leukot Essent Fatty Acids. 2014;90(6):199–206.
Ferguson EJ, Seigel JW, McGlory C. Omega-3 fatty acids and human skeletal muscle. Curr Opin Clin Nutr Metab Care. 2021;24(2):114–9.
Smith GI, et al. Omega-3 polyunsaturated fatty acids augment the muscle protein anabolic response to hyperinsulinaemia-hyperaminoacidaemia in healthy young and middle-aged men and women. Clin Sci (Lond). 2011;121(6):267–78.
Smith GI, et al. Dietary omega-3 fatty acid supplementation increases the rate of muscle protein synthesis in older adults: a randomized controlled trial. Am J Clin Nutr. 2011;93(2):402–12.
Da Boit M, et al. Sex differences in the effect of fish-oil supplementation on the adaptive response to resistance exercise training in older people: a randomized controlled trial. Am J Clin Nutr. 2017;105(1):151–8.
Smith GI, et al. Fish oil-derived n-3 PUFA therapy increases muscle mass and function in healthy older adults. Am J Clin Nutr. 2015;102(1):115–22.
Kunz HE, et al. A randomized trial of the effects of dietary n3-PUFAs on skeletal muscle function and acute exercise response in healthy older adults. Nutrients. 2022;14(17):3537.
McGlory C, et al. Omega-3 fatty acid supplementation attenuates skeletal muscle disuse atrophy during two weeks of unilateral leg immobilization in healthy young women. Faseb j. 2019;33(3):4586–97.
Murphy CH, et al. Does supplementation with leucine-enriched protein alone and in combination with fish-oil-derived n-3 PUFA affect muscle mass, strength, physical performance, and muscle protein synthesis in well-nourished older adults? A randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2021;113(6):1411–27.
Bischoff-Ferrari HA, et al. Effect of vitamin D supplementation, omega-3 fatty acid supplementation, or a strength-training exercise program on clinical outcomes in older adults: the DO-HEALTH Randomized Clinical Trial. JAMA. 2020;324(18):1855–68.
Lalia AZ, et al. Influence of omega-3 fatty acids on skeletal muscle protein metabolism and mitochondrial bioenergetics in older adults. Aging (Albany NY). 2017;9(4):1096–129.
Miotto PM, et al. Supplementation with dietary ω-3 mitigates immobilization-induced reductions in skeletal muscle mitochondrial respiration in young women. Faseb j. 2019;33(7):8232–40.
Calvani R, et al. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem. 2013;394(3):393–414.
Cartee GD, et al. Exercise promotes healthy aging of skeletal muscle. Cell Metab. 2016;23(6):1034–47.
Molenaars M, et al. A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metab. 2020;31(3):549-563.e7.
Sekhar RV, et al. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am J Clin Nutr. 2011;94(3):847–53.
Lizzo G, et al. A randomized controlled clinical trial in healthy older adults to determine efficacy of glycine and N-acetylcysteine supplementation on glutathione redox status and oxidative damage. Front Aging. 2022;3: 852569.
Kumar P, et al. Supplementing glycine and N-Acetylcysteine (GlyNAC) in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, physical function, and aging hallmarks: a randomized clinical trial. J Gerontol A Biol Sci Med Sci. 2023;78(1):75–89.
Kumar P, et al. Glycine and N-acetylcysteine (GlyNAC) supplementation in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, insulin resistance, endothelial dysfunction, genotoxicity, muscle strength, and cognition: Results of a pilot clinical trial. Clin Transl Med. 2021;11(3): e372.
Kumar P, et al. Supplementing Glycine and N-acetylcysteine (GlyNAC) in Aging HIV patients improves oxidative stress, mitochondrial dysfunction, inflammation, endothelial dysfunction, insulin resistance, genotoxicity, strength, and cognition: results of an open-label clinical trial. Biomedicines. 2020;8(10):390.
Sekhar RV, GlyNAC (Glycine and N-Acetylcysteine) supplementation improves impaired mitochondrial fuel oxidation and lowers insulin resistance in patients with type 2 diabetes: results of a pilot study. Antioxidants (Basel). 2022;11(1):154.
Covarrubias AJ, et al. NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021;22(2):119–41.
Damgaard MV, Treebak JT. What is really known about the effects of nicotinamide riboside supplementation in humans. Sci Adv. 2023;9(29):eadi4862.
Kropotov A, et al. Equilibrative nucleoside transporters mediate the import of nicotinamide riboside and nicotinic acid riboside into human cells. Int J Mol Sci. 2021;22(3):1391.
Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117(4):495–502.
Belenky P, et al. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J Biol Chem. 2009;284(1):158–64.
Yaku K, et al. BST1 regulates nicotinamide riboside metabolism via its glycohydrolase and base-exchange activities. Nat Commun. 2021;12(1):6767.
Chellappa K, et al. NAD precursors cycle between host tissues and the gut microbiome. Cell Metab. 2022;34(12):1947-1959.e5.
Shats I, et al. Bacteria boost mammalian host NAD metabolism by engaging the deamidated biosynthesis pathway. Cell Metab. 2020;31(3):564-579.e7.
Liu L, et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 2018;27(5):1067-1080.e5.
Lapatto HAK, et al. Nicotinamide riboside improves muscle mitochondrial biogenesis, satellite cell differentiation, and gut microbiota in a twin study. Sci Adv. 2023;9(2):eadd5163.
Pencina KM, et al. Nicotinamide adenine dinucleotide augmentation in overweight or obese middle-aged and older adults: a physiologic study. J Clin Endocrinol Metab. 2023;108(8):1968–1980.
Yoshino M, et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science. 2021;372(6547):1224–9.
Trammell SA, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948.
Martens CR, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat Commun. 2018;9(1):1286.
Dolopikou CF, et al. Acute nicotinamide riboside supplementation improves redox homeostasis and exercise performance in old individuals: a double-blind cross-over study. Eur J Nutr. 2020;59(2):505–15.
Elhassan YS, et al. Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 2019;28(7):1717-1728.e6.
Dollerup OL, et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J Physiol. 2020;598(4):731–54.
Remie CME, et al. Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. Am J Clin Nutr. 2020;112(2):413–26.
Stocks B, et al. Nicotinamide riboside supplementation does not alter whole-body or skeletal muscle metabolic responses to a single bout of endurance exercise. J Physiol. 2021;599(5):1513–31.
Lamb GD, Westerblad H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J Physiol. 2011;589(Pt 9):2119–27.
Igarashi M, et al. Chronic nicotinamide mononucleotide supplementation elevates blood nicotinamide adenine dinucleotide levels and alters muscle function in healthy older men. NPJ Aging. 2022;8(1):5.
Mori V, et al. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE. 2014;9(11): e113939.
Pirinen E, et al. Niacin cures systemic NAD(+) deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metab. 2020;31(6):1078-1090.e5.
McKendry J, et al. Muscle morphology and performance in master athletes: a systematic review and meta-analyses. Ageing Res Rev. 2018;45:62–82.
Zampieri S, et al. Lifelong physical exercise delays age-associated skeletal muscle decline. J Gerontol A Biol Sci Med Sci. 2015;70(2):163–73.
Striegel H, et al. The use of nutritional supplements among master athletes. Int J Sports Med. 2006;27(3):236–41.
Harnett J, et al. The use of medications and dietary supplements by masters athletes—a review. Curr Nutr Rep. 2022;11(2):253–62.
Doering TM, et al. Lower integrated muscle protein synthesis in masters compared with younger athletes. Med Sci Sports Exerc. 2016;48(8):1613–8.
Easthope CS, et al. Effects of a trail running competition on muscular performance and efficiency in well-trained young and master athletes. Eur J Appl Physiol. 2010;110(6):1107–16.
Lewis NA, et al. Are there benefits from the use of fish oil supplements in athletes? A systematic review. Adv Nutr. 2020;11(5):1300–14.
Broome SC, et al. Mitochondria-targeted antioxidant supplementation improves 8 km time trial performance in middle-aged trained male cyclists. J Int Soc Sports Nutr. 2021;18(1):58.
Jäger S, et al. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104(29):12017–22.
Akimoto T, et al. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. 2005;280(20):19587–93.
Schreiber SN, et al. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A. 2004;101(17):6472–7.
Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A. 1994;91(4):1309–13.
Baar K. Involvement of PPAR gamma co-activator-1, nuclear respiratory factors 1 and 2, and PPAR alpha in the adaptive response to endurance exercise. Proc Nutr Soc. 2004;63(2):269–73.
Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79(2):208–17.
Handschin C. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha in muscle links metabolism to inflammation. Clin Exp Pharmacol Physiol. 2009;36(12):1139–43.
Handschin C, et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J Clin Invest. 2007;117(11):3463–74.
St-Pierre J, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278(29):26597–603.
Valle I, et al. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66(3):562–73.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
In the interest of full disclosure, SCB reports gifts from MitoQ Ltd. who manufacture MitoQ. JAH reports research funding received from Amazentis Inc. who manufacture the urolithin A supplement Mitopure. JW has received conference and travel support from Amazentis Inc. LGK was an employee of Nestlé Research and Nestlé Health Science and reports personal fees from Nestlé Health Science who manufacture n3-PUFAs, urolithin A, GlyNAC, and nicotinamide riboside including multivitamins such as B6 and B3.
Author contributions
SCB, JW, LGK and JAH formulated the research questions. SCB and JW conducted the literature search. Figures and tables were prepared by SCB, JW and JAH. SCB wrote the manuscript with critical input from JW, LGK, and JAH. All authors read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Broome, S.C., Whitfield, J., Karagounis, L.G. et al. Mitochondria as Nutritional Targets to Maintain Muscle Health and Physical Function During Ageing. Sports Med (2024). https://doi.org/10.1007/s40279-024-02072-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s40279-024-02072-7