
Abstract
Health economic analyses are essential for health services research, providing decision-makers and payers with evidence about the value of interventions relative to their opportunity cost. However, many health economic approaches are still limited, especially regarding the primary prevention of cardiovascular disease (CVD). In this article, we discuss some limitations to current health economic models and then outline an approach to address these via the incorporation of genomics into the design of health economic models for CVD. We propose that when a randomised clinical trial is not possible or practical, health economic models for primary prevention of CVD can be based on Mendelian randomisation analyses, a technique to assess causality in observational data. We discuss the advantages of this approach, such as integrating well-known disease biology into health economic models and how this may overcome current statistical approaches to assessing the benefits of interventions. We argue that this approach may provide the economic argument for integrating genomics into clinical practice and the efficient targeting of newer therapeutics, transforming our approach to the primary prevention of CVD, thereby moving from reactive to preventive healthcare. We end by discussing some limitations and potential pitfalls of this approach.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Current health economic models for primary prevention of atherosclerotic cardiovascular disease (ASCVD) are at risk of significantly underestimating benefits of existing and newer therapeutics by neglecting well-known disease biology. |
We propose that when a randomised clinical trial is not possible or practical, health economics models for primary prevention of ASCVD should be based on the results of naturally randomised trials informed by Mendelian randomisation analyses, because these incorporate well-known disease biology and account for the cumulative effects of modifying causal risk factors for ASCVD. |
We argue adopting our proposed approach could provide the health economic argument for integrating genomics into clinical practice and shifting ASCVD treatment from a reactive to a preventive approach. |
1 Introduction
Health economic analyses are essential for health services research. Because healthcare resources are limited, health interventions and therapeutics need to be shown to be cost-effective before payers (principally governments and insurers) will agree to provide them to, or subsidise them for, healthcare consumers. In other words, new interventions need to demonstrate that the benefits come at an acceptable cost. Health economic models are the primary tool via which cost-effectiveness is shown, providing decision-makers and payers with evidence about the value of interventions relative to their opportunity cost.
The reason to use health economic models is that randomised clinical trials (RCTs) have limited follow-up, may not be representative of the general population [1] and often do not collect the necessary data for health economic evaluations (such as quality of life and costs). Furthermore, data from RCTs alone may be insufficient to inform practice and policy as they cannot provide estimates of real-world or long-term costs and benefits of interventions. Importantly, inaccurate estimates of costs and outcomes of newer interventions may lead to significant misallocation of resources and risks denying the public access to cost-effective interventions or inappropriately funding interventions that are not cost-effective.
The approach to health economic modelling continues to evolve. Indeed, novel approaches are continually developed to project the economic impact and the effects of newer therapies [2, 3]. Nevertheless, health economic approaches for primary prevention of atherosclerotic cardiovascular disease (ASCVD) are still limited [4, 5]. Most importantly, the majority of health economic models for ASCVD use age-related risk estimates [4], which assume that interventions can only influence ASCVD on an instantaneous basis and thus do not account for the cumulative benefits of modifying risk factors causal for ASCVD. This approach is largely ignorant of disease biology, and usually restricts treatment to those where near-term risk of ASCVD is high. Moreover, these limitations are becoming increasingly important as healthcare costs rise and more complex (and thus often expensive) technologies are introduced. Thus, novel modelling approaches that can handle complexities beyond the capabilities of current methods are required to predict health economic impact and assess existing and new therapies.
The decreasing cost and availability of genetic data has led to a marked increase in the use of genetics to answer questions of fundamental biology, clinical medicine, and epidemiology. Despite this, uptake of genomics into health economics has been limited. The field so far has primarily investigated whether genomics can be used to understand the effects of disease on quality adjusted life-years or healthcare spending [6,7,8]. Moreover, while there are some studies in other diseases [8], little attention has been paid to whether genetics can improve the design of health economic models for ASCVD. This is a deficiency that needs addressing, as there is accumulating genetic evidence that major risk factors for ASCVD affect the disease proportional to cumulative exposure [9], suggesting that earlier detection and intervention to prevent ASCVD may be more beneficial than current health economic approaches (which assume risk is proportional to instantaneous exposure) suggest.
In this proposal, we discuss limitations of current health economic approaches, then propose an approach to integrate genomics into the epidemiology of the decision health economic modelling for ASCVD via the use of Mendelian randomisation (MR), how this can address some fundamental limitations to current approaches, and finally key potential pitfalls to our proposed approach. To illustrate our approach, we focus on the example of lowering low-density lipoprotein-cholesterol (LDL-C) to reduce ASCVD.
2 Limitations to Current Health Economic Approaches
Models derived from trial populations have the advantage of evaluating outcomes and costs in a group that has been proven to benefit from the intervention and thus do not rely on assumptions about the efficacy of that intervention outside of the trial population. However, this can also be considered a disadvantage, as trial populations are usually not representative of the target population for an intervention [1], which is likely to lead to either systematic overestimation or underestimation of the benefits of the intervention. Indeed, in models based on RCTs alone, transition probabilities are usually drawn straight from the trial [4, 10], which is likely to underestimate the absolute event rate of the disease outcomes relative to the general population (and thus underestimate the benefits of the intervention) as people who enrol in trials are usually healthier than the broader populations they are supposed to represent [1]. Conversely, for interventions that are influenced by compliance, higher compliance in the trial population than the general population has the potential to overestimate the benefit of the intervention (statins are an example [11]). Moreover, trial follow-up periods may be shorter than the intended duration of therapy. Thus, models with short-term follow-up for interventions that may have more long-term benefits or interventions that become more effective over time could systematically underestimate the benefit of interventions.
These disadvantages are partially overcome by selecting model populations on broader and more representative populations (such as specific disease cohorts or the general population) and conducting health economic analyses over a longer time horizon. One obvious disadvantage to this strategy is applying effect estimates from RCTs to populations that differ from the trial population, even if they would have met the selection criteria (although it is also common to generalise trial effect estimates to people who would not have met trial selection criteria). This limitation can be partially accounted for if there is real-world evidence of the effectiveness of the intervention, but this is usually lower quality evidence and more susceptible to bias than the original trial.
The transition probabilities underlying models based on broader populations are usually from one of two sources: age-specific estimates of disease risk or estimates of disease risk derived from risk scores (which are heavily influenced by age) [4]. However, both can be problematic, especially for primary prevention of ASCVD. While age-specific risk estimates are generally representative of the whole population, they consequently do not target individuals who are most likely to benefit from early treatment. Thus, conducting analyses on the entire population will systematically overestimate the benefit for low-risk sub-groups of the population and underestimate the benefit for high-risk sub-groups. It is thus attractive to stratify the population into groups based on their perceived risk of an outcome. This is relatively straightforward for populations with existing ASCVD (secondary prevention) because they have a high risk for repeat events or other chronic diseases. For primary prevention (i.e. people who have not had an ASCVD event), the most common approach to population stratification is the use of risk scores [4]. However, most risk scores for ASCVD use statistical approaches to select the most predictive risk factors for an event as measured at a single point in time [12, 13]. Consequently, most risk scores are primarily driven by age and sex, with modifiable risk factors usually only marginally improving prediction [14,15,16]. By allowing statistical methods to drive the selection of predictors, such estimates may be effectually ignorant of the biology of disease. More importantly, models based on these risk scores often rely on changes in surrogate outcomes (such as LDL-C) to model changes in clinical outcomes and thus do not account for the cumulative effects of changing risk factors, as ASCVD risk estimates are based on instantaneous risk at current risk factor levels [14,15,16,17,18]. Therefore, models that assume that risk factors only influence outcomes on an instantaneous basis could systematically underestimate the benefits of primary prevention by ignoring the biology of the disease process (and thus ignoring the opportunity to modify earlier trajectories of disease when instantaneous risk is lower) and instead only allowing treatment when the disease has progressed to the point that short-term risk is high [19].
Indeed, it is well-established that both the duration and magnitude of exposure to LDL-C drives the risk of ASCVD, rather than instantaneous levels [9, 19, 20], but this is not reflected in current risk scores or epidemiological models that are based on them [14,15,16,17,18]. For example, if a 60-year-old female who has had an LDL-C of 4 mmol/L for decades initiates lipid-lowering therapy and within 1 month lowers her LDL-C from 4 to 2 mmol/L, because her current LDL-C is 2 mmol/L, most risk scores would estimate her 10-year risk of an event to be lower than the equivalent female with lifelong LDL-C of 2.5 mmol/L. Given our understanding of the biology of ASCVD [19], this risk estimate is nonsensical. This example generalises to other risk factors, such as blood pressure, which also has a cumulative effect on ASCVD [9]. This has important implications for models based on risk scores, as the benefits of intervening earlier versus later in life or primary versus secondary prevention could be systematically underestimated in such health economic models.
3 Integrating Genomics into Health Economic Decision Modelling
Thus, there is a need to improve health economic models for the primary prevention of ASCVD. This need has increased with the advent of more complex (and typically expensive) biologic therapeutics [21], which are usually only approved for primary prevention of ASCVD in the most severe cases, such as for people with familial hypercholesterolaemia (FH) (e.g. evolocumab in Australia [22] and the UK [23]). Without improving our approach to health economic analysis, we risk forgoing the benefits of these therapies in primary prevention. We suggest that integrating genomics into health economic analyses is a practical and potentially beneficial way of overcoming the limitations to current health economic analyses outlined above.
RCTs are considered the ‘gold standard’ in terms of information about optimal interventions, but data from RCTs alone are insufficient. In this context, there is a strong demand for real-world data to quantify the value of complex therapeutic approaches to disease management, and to handle complexity that is beyond the capability of current conventional methods. We propose that when an RCT that would produce the results required to design a health economic analysis is not possible or practical, health economic models for primary prevention of ASCVD can be based on the results of MR analyses [24,25,26,27]. MR is an epidemiological technique that exploits the fact that the inheritance of alleles from parents is random to deal with confounding inherent to other epidemiological study designs (Box 1)—a random assortment of alleles ensures that they are unlikely to be associated with other genetic or environmental factors that traditionally bias observational studies [27]. By assuming that the random assortment of alleles from parents to offspring generalises at the population level, when grouping individuals by their genotype, it can be assumed that all other confounders are equal between groups, particularly when the size of the groups are very large. This is analogous to an RCT, in which randomisation theoretically balances measured and unmeasured confounders, such that the only difference between the groups is the intervention (Fig. 1). In MR, instead of the intervention of an RCT, the only difference between groups is assumed to be their alleles. Therefore, if individuals with an allele that predisposes them to both higher levels of a risk factor (and nothing else of relevance) and a higher risk of a disease outcome, this provides strong evidence of causality for that risk factor causing that outcome via this randomised and thus (theoretically) unconfounded evidence [27]. For example, variants in the HMG-CoA reductase gene (the target of statins) are associated both with levels of LDL-C in the blood and the risk of ASCVD [28], providing strong evidence of causality for the association between LDL-C and ASCVD.

Mendelian randomisation is analogous to a randomised clinical trial
Notably, one can also use MR to derive estimates of the magnitude of effect per unit change in the risk factor on the outcome [24,25,26,27]. While these need to be interpreted with caution and are primarily underpinned by an understanding of disease biology, these estimated effects can be used to conduct naturally randomised trials [26]. In these naturally randomised trials, individuals in a microsimulation model can be allocated to both treatment and control (in separate simulations), with the effects of the intervention on outcomes modelled via changes in the risk factor by unit time using effect estimates from MR. These individuals are then followed up for any amount of time, and the results provide estimates of the benefits of targeting that risk factor. Importantly, effect estimates from MR give a measure of the effect of higher levels of exposure to the risk factor over the lifetime, so these will need to be adjusted to an effective impact of the risk factor by unit time (for example, LDL-C years or smoking pack-years) for use in a naturally randomised trial [26]. It should be noted that MR is not a substitute for a randomised trial, as interventions can have unexpected effects on the primary outcome or adverse safety outcomes (discussed more below). Nevertheless, naturally randomised trials will be the best available evidence for situations where conducting an RCT is not possible or practical. For example, testing the effect of lowering LDL-C from 30 years of age on the lifetime risk of cardiovascular disease would involve recruiting a high number of participants, followed for decades to accrue enough events to have power on any outcome, with likely a high rate of dropout and treatment noncompliance. Consequently, such an RCT is unlikely to be run, while a naturally randomised trial does not have the same limitations.
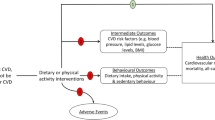
These naturally randomised trials can then form the basis of a health economic model (Fig. 2). These models can be like other health economic models in that quality of life and costs can be sourced from the best available sources, with the fundamental change being only to the underlying transition probabilities and effect estimates. In this situation, the only change would be using MR to incorporate the biology of disease into the underlying structure of the model – the remainder of the health economic analysis remains the same (including costs and quality of life). However, MR can also generate causal effect estimates of the association between risk factors and quality of life or healthcare costs. Indeed, Dixon and colleagues have demonstrated a causal effect of adiposity on hospital costs [6], and Harrison et al. used MR to estimate the causal effect of body mass index (BMI) on predicted quality of life and healthcare costs, and integrated them into a simulation model to conduct a health economic analysis on obesity interventions [8]. Thus, it is possible (but not necessary for our proposed approach) to also use MR to enhance the generation of quality of life and costs for health economic models.

Current approach to health economic models and proposed approach to integrate genomics into health economic analyses. In the proposed approach, health economic outcomes (quality of life and costs) can be sourced via traditional methods, or from Mendelian randomisation (MR), if available
There are several advantages to the MR-based approach. Because naturally randomised trials are simulated, they can be conducted over a lifetime (or any length of time). Like a model based on an RCT, they are still underpinned by solid evidence of causality [26]. However, it is worth noting that the goal of a naturally randomised trial differs from an RCT. The goal of an RCT is to determine whether, and to what degree, a particular intervention is effective in the population under study. By contrast, the goal of a naturally randomised trial is to use understanding of the causal structure of the disease to determine the optimal time to intervene on the risk factor to maximise benefits, and, by extension, determine which interventions produce clinically relevant benefits in different groups and at different times. The goal therefore of MR-based health economic models will be to extend this by determining whether, when in the disease trajectory, and for whom it is cost-effective to intervene, and how this varies for different interventions.
The simulated nature of the naturally randomised trial also overcomes the lack of generalisability of RCT estimates because the investigator can modify the specific population under study to estimate the absolute benefits of modifying the causal risk factor in the population of interest. The simulated nature of these studies also means that the investigator can modify the strength, level of compliance to, and timing of the intervention to find the optimal age or dose at which an intervention becomes cost-effective (using estimates from the best available literature). Indeed, the inputs to naturally randomised trials will necessarily be informed by shorter RCTs measuring the efficacy, dose-response, side effects, and compliance to therapies (as distinct from the desired long-term, but impossible or impractical, RCT that the naturally randomised trial is simulating). The investigator can also compare separate interventions directly in the same population under the same conditions, overcoming the limitations of contrasting RCT estimates that may not be directly comparable. Importantly, unlike models based on ‘classical epidemiology’ (epidemiology without the use of genomic data), using causal effect estimates of changing the risk factor by unit of time accounts for the cumulative benefit of decreasing exposure to a risk factor over time [19].
Another key advantage to MR is the ability to stratify the population not just by the risk of the outcome but based on the risk of the outcome via a particular causal pathway (thus stratifying the population by amenity to a particular intervention that acts via that causal pathway). For example, individuals with genetically predicted high LDL-C but low genetically predicted systolic blood pressure may have a lifetime risk of ASCVD that approximates the average (because the two risk factors ‘cancel’ each other out), so a health economic analysis that does not account for this may underestimate the benefit of lowering LDL-C and overestimate the benefit of lowering blood pressure in this group, potentially denying these individuals access to beneficial therapies based on incorrect estimates of cost-effectiveness.
However, a model based on MR would, appropriately, identify the greater benefit of lowering LDL-C and minimal benefit of lowering blood pressure in this group and thus increase access to therapies targeted at lowering LDL-C [9]. Thus, genetic risk stratification via MR could improve the selection of populations who are most likely to benefit from interventions, consequently providing real-world cost/benefit ratios in cost-effectiveness analyses. Indeed, if we are to incorporate expensive therapies into clinical practice in a cost-effective manner, targeting the interventions to those most in need will be required, thereby maximising the population-level benefit of newer and existing therapies.
This approach could also underpin the uptake of prescriptive polygenic scores into clinical practice—by identifying and treating individuals at high risk of disease via a particular pathophysiological pathway using their genetics, the genome is effectively being used to prescribe an action. Thus, not only could MR-based health economic models improve access to therapeutics for high-risk groups, but they may also underpin the economic argument for integrating genetics into clinical practice (Box 2). Nevertheless, it remains to be seen whether prescriptive polygenic scores would be a superior approach to simple stratification by readily available clinical measures such as LDL-C or blood pressure, especially given the practical and cost considerations of implementing wide-scale genomic testing. The MR-based approach to health economics we propose does not rely on this; thus, we suggest that MR-based health economic evaluations compare the costs and benefits of widespread genomic uptake in clinical practice into their models against simple clinical measurements of the risk factor, if available.
MR-based models could also be useful for drug development [24]. Because MR-based models are based on causal risk factors, they minimise the likelihood that drugs will fail in phase III RCTs for lack of efficacy. This works in two ways: first, drugs targeted at non-causal risk factors will not be pursued; and second, drugs that do not change a causal risk factor by a magnitude sufficient to produce effective clinical benefits as estimated from naturally randomised trials will not be pursued. Moreover, MR-based models could also efficiently identify cost-effective drugs for repurposing, as well as detecting potential adverse impacts from repurposing [29, 30]. Attaching costs to these models will further invigorate the drug development process. Using naturally randomised trials as the basis for health economic models will provide knowledge about expected benefit/cost ratios for interventions in the early stages of drug development, further streamlining cost-effective drug development by identifying drug candidates most likely to be cost-effective at scale. MR-based models will have utility not only in drug development and determining the cost-effectiveness of drug-based interventions, but also can be used to model the effects of public health interventions on ASCVD.
Notably, we have already demonstrated the utility of an MR-based approach to health-economics in a study testing the cost-effectiveness of cascade screening for heterozygous FH in children [31]. In this study, we used MR-based estimates of the impact of LDL-C lowering to model the effects (on ASCVD) of cascade screening to identify and treat people with heterozygous FH from 10 years of age, with follow-up over the lifetime horizon. We found that this strategy was likely cost-saving from a healthcare perspective. This is a situation where an RCT will never be undertaken, and a model based only on age-specific estimates of risk and ASCVD risk reductions from RCTs could have significantly underestimated the cumulative benefit of LDL-C lowering of such a strategy in children [28]. However, the most important outcome of this study is that the results of this analysis were directly influential in persuading the Australian Government to approve and subsidise cascade genetic screening for FH in clinical practice, thereby demonstrating the acceptability of this approach to a payer. Proposed steps for integrating Mendelian randomisation (MR) into the design of health economic models for atherosclerotic cardiovascular disease are shown in Box 2.
4 Potential Pitfalls
This approach is not without limitations. Foremost is that a naturally randomised trial is not a substitute for an RCT. Interventions, mainly pharmacological interventions, can lead to unexpected adverse events or unexpected effects on the primary and secondary outcomes. These can be estimated from MR if these effects are a result of on-target effects (i.e. if they are direct consequences of modifying the risk factor), such as MR analyses that have also reported the increased risk of diabetes with 3-hydroxy-3-methylglutaryl-CoA reductase inhibition with statins [32, 33]. However, if the intervention has off-target effects or other unexpected side effects unrelated to the intended mechanism of action, a naturally randomised trial will not be able to detect these.
Furthermore, a reasonable level of understanding of the biology of the disease of interest is required to extrapolate MR effect estimates and conduct naturally randomised trials because there are several reasons that effect estimates derived from MR will not accurately reflect the effect of modifying the risk factor in an RCT [25]. First, MR effect estimates represent the average effect of the risk factor on the outcome over the lifetime and, thus, must be converted into a per-unit-time effect; however, this will only be appropriate if the risk factor has an irreversible cumulative effect on the outcome over time. For example, while both systolic blood pressure and LDL-C show a dose-dependent, cumulative effect on the risk of ASCVD [9], some risk factors do not have a cumulative effect on disease, such as the relationship between BMI and insulin resistance [34]. Thus, while our proposal is specifically for ASCVD, which does have a causal structure amenable to this kind of modelling, approaches to other diseases will vary depending on their causal structure.
Second, even for cumulative risk factors, risk factor effects may vary by disease stage. In these cases, the effects of intervening on the risk factor will vary by disease stage, but this is not accounted for in MR effect estimates. Indeed, extrapolation between a lifelong effect of the risk factor to treatment effects at different ages will involve several assumptions specific to the disease under study, some of which may be violated in reality. While shorter-term estimates from RCTs can inform this process, these may not be available for every risk factor and outcome, and will not cover the entire age-range or disease staging of the naturally randomised trial.
Third, converting MR effect estimates into effect estimates from interventions requires substantial extrapolation as genetic variation is usually much less than the proposed intervention. For example, variation in LDL-C via genetic variants is generally in the order of 2–5% per allele, while statins reduce LDL-C by 30–50%, meaning that extrapolation of MR effect estimates is reliant on the assumption that LDL-C does have a linear or log-linear relationship with ASCVD risk across all values of LDL-C [25]. Fourth, it is possible that large changes in risk factor levels lead to compensation via other pathways (for example, in inflammatory pathways [35]), which may produce differences between MR effect estimates and results in interventions.
Many of these issues often arise because it is not always possible to generate evidence of a causal effect using variants in the protein encoding gene that is the target of a drug (for example, the strongest evidence linking the effect of HMG-CoA reductase inhibition via statins to the genetic effect leverages natural variation in the gene encoding HMG-CoA reductase [28]), meaning the strength of the evidence for each pathway will vary depending on what natural biological variation is present. Therefore, a robust understanding of the biology underlying the disease process and the relationship between the risk factor and outcome will be required to guide the development of naturally randomised trials and to factor these effects into models accordingly. Understanding of disease biology is likely to come from many sources including RCTs, clinical studies, and basic science. We encourage health economists to engage expert scientists and clinicians in the disease of interest in the early stages of designing MR-based health economic models, as it is important to understand the strengths and limitations of model inputs and show the potential consequences of these in their results. In general, the job of the health economist would be to enhance our understanding of the causal structure of disease and apply these principles to improve the design of health economic models. For example, health economists have used MR to understand the association of risk factors with quality of life and costs [7], as well as educational outcomes [36].
Nevertheless, naturally randomised trials will never perfectly substitute RCTs, and when extrapolating results from MR, we must carefully consider the similarity of the population studied to the target population, differences in the intended duration and intensity of therapy to MR, and the goals of the interventions [37]. Cost-effectiveness analyses based on naturally randomised trials should therefore be interpreted with these important limitations in mind.
Another limitation to this approach is the requirement of a suitable data source to conduct the naturally randomised trial. To conduct the best possible naturally randomised trial, individual-level data on genetics, the risk factor, and the outcome need to be available for everyone in the microsimulation. Few datasets meet these requirements. Fortunately, the largest of these datasets, the UK Biobank [38], is relatively simple to access (and is becoming increasingly so), making this limitation less of a concern. There are several other large biobanks with varying degrees of availability to researchers [39]. Moreover, it is possible to use published MR effect estimates (with the appropriate adjustment from lifetime effect to by unit time effect) in models with risk factor levels and transition probabilities derived from other sources. This approach will likely be most useful when the disease of interest is rare (such as FH), and thus will not be well-represented in non-specific datasets such as the UK Biobank [31]. However, this approach is not preferred when individual-level data are available [40]. With increasing access to a larger number of cohorts from more ethnically diverse populations, the value of these studies in terms of their implications for more geographical settings will continue to improve.
Nevertheless, health economists will not always have the option to limit themselves to disease and interventions where MR-based analyses are feasible or practical, such as ASCVD. Thus, our approach must be considered on a case-by-case basis as to whether MR can or will improve approaches to health economic evaluation. We have focused on the case of ASCVD, as we believe there is considerable evidence supporting the causality of several risk factors on the development of ASCVD that are overlooked in current health economic approaches. Other less well-understood diseases may not have these advantages, while others are further ahead—there has already been a study on obesity (discussed above) that leveraged MR to conduct a health economic evaluation.
Finally, we have not discussed some of the many limitations of MR itself, which have been reviewed elsewhere [41, 42]. Indeed, there is concern that increasing ease and availability of MR has led to an increase in the publication of incorrect or spurious ‘causal’ effect estimates [40]. It is therefore important to be familiar with how the effects of phenomena like horizontal pleiotropy (when a genetic variant influences the outcome through a pathway unrelated to the risk factor) can bias traditional effect estimates in MR studies, and how these may be accounted for [43]. Indeed, MR-based models are less useful for secondary prevention, as selecting diseased populations is likely to introduce collider bias [44]. We have not discussed the possibility of using genetic liability to disease (vs. simple risk estimates) to enhance health economic analysis [45]. It is also worth noting that the generalisability of MR effect estimates is not always a given. For example, there is strong evidence of selection bias in the UK Biobank [46], and most genetic associations have been performed in people of European ancestry [47]. We therefore recommend engaging with experts in MR if novel or contentious causal pathways are to be targets of MR-based health economic analyses. The reader is directed to other reviews on MR [27], a guide to reading MR for non-specialists [48], and a checklist to assess the quality of MR studies [49] for further reading. As usual, sensitivity and scenario analyses will be important, but especially so in this case as the protocols and procedures surrounding MR-based health economic analysis are established in coming years. Moreover, other sources of uncertainty will continue to exist in decision modelling, and their sources and consequences should be made available to decision makers; health economic models are only one aspect of decision making; other aspects, such as legal, ethical, cultural, political, and structural factors will also continue to play a role.
5 Conclusions
We have outlined a proposal to use naturally randomised trials to enhance the design of health economic models for ASCVD. We believe that integrating genomics into health economics will be a powerful way to improve primary prevention of ASCVD, especially as healthcare continues to become more expensive and efficient allocation of resources becomes more of a priority. Indeed, continuing the current approach to primary prevention of ASCVD will greatly limit the population benefits of newer therapeutics by hindering access to these therapies, curbing their use in those patients who stand to benefit most.
References
Kennedy-Martin T, Curtis S, Faries D, Robinson S, Johnston J. A literature review on the representativeness of randomized controlled trial samples and implications for the external validity of trial results. Trials. 2015;16:495.
Reed SD, Dubois RW, Johnson FR, Caro JJ, Phelps CE. Novel approaches to value assessment beyond the cost-effectiveness framework. Value Health. 2019;22(6):S18–23.
Garrison LP Jr, Jansen JP, Devlin NJ, Griffin S. Novel approaches to value assessment within the cost-effectiveness framework. Value Health. 2019;22(6):S12–7.
Marquina C, Zomer E, Vargas-Torres S, Zoungas S, Ofori-Asenso R, Liew D, et al. Novel treatment strategies for secondary prevention of cardiovascular disease: a systematic review of cost-effectiveness. Pharmacoeconomics. 2020;38(10):1095–113.
Schwappach DLB, Boluarte TA, Suhrcke M. The economics of primary prevention of cardiovascular disease – a systematic review of economic evaluations. Cost Eff Resour Alloc. 2007;5(1):5.
Dixon P, Hollingworth W, Harrison S, Davies NM, Davey SG. Mendelian randomization analysis of the causal effect of adiposity on hospital costs. J Health Econ. 2020;70: 102300.
Dixon P, Davey Smith G, von Hinke S, Davies NM, Hollingworth W. Estimating marginal healthcare costs using genetic variants as instrumental variables: mendelian randomization in economic evaluation. Pharmacoeconomics. 2016;34(11):1075–86.
Harrison S, Dixon P, Jones HE, Davies AR, Howe LD, Davies NM. Long-term cost-effectiveness of interventions for obesity: a mendelian randomisation study. PLoS Med. 2021;18(8): e1003725.
Ference BA, Bhatt DL, Catapano AL, Packard CJ, Graham I, Kaptoge S, et al. Association of genetic variants related to combined exposure to lower low-density lipoproteins and lower systolic blood pressure with lifetime risk of cardiovascular disease. JAMA. 2019;322(14):1381–91.
Abushanab D, Al-Badriyeh D, Marquina C, Bailey C, Jaam M, Liew D, et al. A systematic review of cost-effectiveness of non-statin lipid-lowering drugs for primary and secondary prevention of cardiovascular disease in patients with type 2 diabetes mellitus. Curr Probl Cardiol. 2022:101211.
Vonbank A, Agewall S, Kjeldsen KP, Lewis BS, Torp-Pedersen C, Ceconi C, et al. Comprehensive efforts to increase adherence to statin therapy. Eur Heart J. 2017;38(32):2473–9.
Lloyd-Jones DM, Braun LT, Ndumele CE, Smith SC, Sperling LS, Virani SS, et al. Use of risk assessment tools to guide decision-making in the primary prevention of atherosclerotic cardiovascular disease: a special report from the American Heart Association and American College of Cardiology. Circulation. 2019;139(25):e1162–77.
Noble D, Mathur R, Dent T, Meads C, Greenhalgh T. Risk models and scores for type 2 diabetes: systematic review. BMJ. 2011;343: d7163.
Conroy RM, Pyörälä K, Fitzgerald A, Sans S, Menotti A, De Backer G, et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J. 2003;24(11):987–1003.
D’Agostino RB Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117(6):743–53.
Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297(6):611–9.
Hippisley-Cox J, Coupland C, Robson J, Brindle P. Derivation, validation, and evaluation of a new QRISK model to estimate lifetime risk of cardiovascular disease: cohort study using QResearch database. BMJ. 2010;341: c6624.
Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, May M, Brindle P. Derivation and validation of QRISK, a new cardiovascular disease risk score for the United Kingdom: prospective open cohort study. BMJ. 2007;335(7611):136.
Ference BA, Graham I, Tokgozoglu L, Catapano AL. Impact of lipids on cardiovascular health: JACC health promotion series. J Am Coll Cardiol. 2018;72(10):1141–56.
Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459–72.
Pothier K, Gustavsen G. Combating complexity: partnerships in personalized medicine. Pers Med. 2013;10(4):387–96.
The Pharmaceutical Benefits Scheme. Evolocumab. https://www.pbs.gov.au/medicine/item/10958R-11193D-11484K-11485L-11972D-11977J-11985T-11986W. Accessed 9 Aug 22.
National Institute for Health and Care Excellence. Evolocumab for treating primary hypercholesterolaemia and mixed dyslipidaemia. 2016. https://www.nice.org.uk/guidance/ta394. Accessed 9 Aug 22.
Holmes MV, Richardson TG, Ference BA, Davies NM, Davey SG. Integrating genomics with biomarkers and therapeutic targets to invigorate cardiovascular drug development. Nat Rev Cardiol. 2021;18(6):435–53.
Burgess S, Butterworth A, Malarstig A, Thompson SG. Use of Mendelian randomisation to assess potential benefit of clinical intervention. BMJ Br Med J. 2012;345: e7325.
Ference BA, Holmes MV, Smith GD. Using Mendelian randomization to improve the design of randomized trials. Cold Spring Harbor Perspect Med. 2021;11(7).
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey SG. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Ference Brian A, Yoo W, Alesh I, Mahajan N, Mirowska Karolina K, Mewada A, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease. J Am Coll Cardiol. 2012;60(25):2631–9.
Zheng J, Xu M, Walker V, Yuan J, Korologou-Linden R, Robinson J, et al. Evaluating the efficacy and mechanism of metformin targets on reducing Alzheimer’s disease risk in the general population: a Mendelian randomization study. medRxiv. 2022:2022.04.09.22273625.
Ryan DK, Karhunen V, Su B, Traylor M, Richardson TG, Burgess S, Tzoulaki I, et al. Genetic evidence for protective effects of angiotensin converting enzyme against Alzheimer’s disease but not other neurodegenerative diseases in European populations. Neurol Genet. 2022. https://doi.org/10.17863/CAM.85694.
Ademi Z, Norman R, Pang J, Liew D, Zoungas S, Sijbrands E, et al. Health economic evaluation of screening and treating children with familial hypercholesterolemia early in life: Many happy returns on investment? Atherosclerosis. 2020;304:1–8.
Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385(9965):351–61.
Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375(9716):735–42.
Campbell PJ, Gerich JE. Impact of obesity on insulin action in volunteers with normal glucose tolerance: demonstration of a threshold for the adverse effect of obesity*. J Clin Endocrinol Metab. 1990;70(4):1114–8.
Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339(6116):166–72.
von Hinke Kessler Scholder S, Wehby GL, Lewis S, Zuccolo L. Alcohol exposure in utero and child academic achievement. Econ J. 2014;124(576):634–67.
Sobczyk MK, Zheng J, Smith GD, Gaunt TR. Systematic comparison of Mendelian randomization studies and randomized controlled trials using electronic databases. medRxiv. 2022:2022.04.11.22273633.
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3): e1001779.
Yaghoobi H, Hosseini SA. History of the largest global biobanks, ethical challenges, registration, and biological samples ownership. J Public Health. 2021.
Hartwig FP, Davies NM, Hemani G, Davey SG. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol. 2016;45(6):1717–26.
Holmes MV, Ala-Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol. 2017;14(10):577–90.
Davey Smith G, Holmes MV, Davies NM, Ebrahim S. Mendel’s laws, Mendelian randomization and causal inference in observational data: substantive and nomenclatural issues. Eur J Epidemiol. 2020;35(2):99–111.
Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2020;4:186.
Gkatzionis A, Burgess S. Contextualizing selection bias in Mendelian randomization: how bad is it likely to be? Int J Epidemiol. 2019;48(3):691–701.
Dixon P, Harrison S, Hollingworth W, Davies NM, Davey SG. Estimating the causal effect of liability to disease on healthcare costs using Mendelian randomization. Econ Hum Biol. 2022;46: 101154.
Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, et al. Comparison of sociodemographic and health-related characteristics of uk biobank participants with those of the general population. Am J Epidemiol. 2017;186(9):1026–34.
Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177(1):26–31.
Davies NM, Holmes MV, Davey SG. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362: k601.
Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. JAMA. 2021;326(16):1614–21.
Acknowledgements
The constructive comments of the editor and three anonymous reviewers are gratefully acknowledged.
Funding
This work was supported by the National Health and Medical Research Council Ideas Grants Application ID: 2012582. ZA and JIM are supported by the National Health and Medical Research Council Ideas Grants Application ID: 2012582.
Author contributions
ZA and JIM drafted the manuscript for intellectual content. All authors provided feedback on early drafts and revised the manuscript for intellectual content.
Conflict of interest
ZA and JIM declare no conflict of interest. DL declares grants from AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Pfizer, and Sanofi, and past participation in advisory boards and/or receipt of honoraria from AbbVie, Amgen, Astellas, AstraZeneca, Bristol-Myers Squibb, Edwards Lifesciences, Novartis, Pfizer, Sanofi, and Shire, outside the submitted work. SJN has received research support and/or honoraria from Amgen, AstraZeneca, Eli Lilly, Esperion, Novartis, Merck, Pfizer, Iowa, and Sanofi-Regeneron. SZ declares participation in the advisory boards of Eli Lily, Boehringer-Ingelheim, MSD Australia, Sanofi, Novo Nordisk, and Astra Zeneca, outside the submitted work. BAF declares honoraria and/or research grants from Amgen, Merck & Co, Regeneron, Sanofi, Pfizer, Mylan, Novo Nordisk, Eli Lilly, Silence Therapeutics, Ionis, KrKa Pharmaceuticals, adICOR, CiViPharma, American College of Cardiology, European Society of Cardiology, and European Atherosclerosis Society, outside the submitted work.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ademi, Z., Morton, J.I., Liew, D. et al. Integrating the Biology of Cardiovascular Disease into the Epidemiology of Economic Decision Modelling via Mendelian Randomisation. PharmacoEconomics 40, 1033–1042 (2022). https://doi.org/10.1007/s40273-022-01183-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-022-01183-1