
Abstract
Background
Guanfacine extended-release (GXR) is an orally administered, non-stimulant treatment for children and adolescents with attention-deficit/hyperactivity disorder (ADHD) and is primarily metabolized by the 3A4 isozyme of cytochrome P450 (CYP3A4). The results of clinical pharmacokinetic (PK) studies indicate that guanfacine is sensitive to drug–drug interactions (DDIs) perpetrated by strong inhibitors and inducers of CYP3A4.
Objective
The aim was to provide guidance on the possible requirement for GXR dose adjustment in children and adolescents with ADHD by predicting DDIs following co-administration with moderate CYP3A4 inhibitors and inducers.
Methods
A physiologically based PK model for GXR orally administered to healthy adults was developed based on physicochemical, in vitro and clinical PK data. The model was validated using clinical PK data for co-administration of GXR with ketoconazole (strong CYP3A4 inhibitor) or rifampicin (strong CYP3A4 inducer).
Results
Model predictions indicated that co-administration of GXR with the moderate CYP3A4 inhibitors erythromycin 500 mg three times a day or fluconazole 200 mg daily (q.d.) increased the guanfacine area under the plasma concentration–time curve (AUC) by 2.31-fold or 1.98-fold, respectively, compared with GXR monotherapy. The moderate CYP3A4 inducer efavirenz 400 mg or 600 mg q.d. was predicted to reduce guanfacine AUC to 58 or 33% of its value for GXR monotherapy, respectively.
Conclusion
Without the requirement for additional clinical studies, the following GXR dose recommendations were developed and approved for US labeling for use in children and adolescents with ADHD: (1) decrease GXR to 50% of the usual target dose when it is co-administered with strong or moderate CYP3A4 inhibitors; (2) consider titrating GXR up to double the usual target dose over 1–2 weeks when it is co-administered with strong or moderate CYP3A4 inducers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Guanfacine extended-release (GXR), an orally administered, non-stimulant treatment for children and adolescents with attention-deficit/hyperactivity disorder, is primarily metabolized by the 3A4 isozyme of cytochrome P450 (CYP3A4). |
Using physiologically based pharmacokinetic modeling, this study predicted the potential for GXR to be susceptible to drug–drug interactions with moderate CYP3A4 inhibitors or inducers. |
Based on the model predictions, the following dosing recommendations were approved for US labeling: (1) decrease GXR to 50% of the usual target dose when it is co-administered with strong or moderate CYP3A4 inhibitors; (2) consider titrating GXR up to double the usual target dose over 1–2 weeks when it is co-administered with strong or moderate CYP3A4 inducers. |
1 Background
US and European guidelines recommend stimulant medications for first-line use in many children and adolescents (aged 6–17 years) with attention-deficit/hyperactivity disorder (ADHD) [1,2,3]. Approximately 30% of participants in clinical studies, however, do not respond to methylphenidate (MPH), and ~ 10% do not respond to any stimulant medication (MPH or amphetamine) [4, 5]. In addition, stimulants are contraindicated in some patients with psychiatric comorbidities [6,7,8], many parents of children with ADHD express concerns about adverse effects [9], and some clinicians prefer to prescribe a non-stimulant over a stimulant [10].
Guanfacine extended-release (GXR or Intuniv®; Shire, Lexington, MA, USA) is a non-stimulant medication that acts selectively on post-synaptic α2A-adrenergic receptors in the prefrontal cortex [11, 12]. GXR is approved for use in children and adolescents aged 6–17 years with ADHD, as a monotherapy and as adjunctive therapy to stimulants in the USA [13] and Canada [14]; as a monotherapy in Japan [15]; and as a monotherapy in Europe when stimulants are not suitable, are not tolerated or have been shown to be ineffective [16]. GXR is administered orally as a modified-release matrix tablet formulation containing functional excipients that control and extend release of guanfacine in the gastrointestinal tract [17]. Its recommended therapeutic range is 0.05–0.12 mg/kg, depending on clinical response and tolerability [13, 14, 16]. GXR provides a linear, first-order guanfacine pharmacokinetic (PK) profile in children and adolescents with ADHD [17, 18] and in healthy adults [19]. Guanfacine PK parameters following GXR administration are similar in children, adolescents and adults when appropriately scaled by patient weight [18].
Metabolic clearance accounts for approximately half of total guanfacine clearance in adults, with renal clearance accounting for the remainder [20]. Guanfacine is metabolized primarily by the 3A4 isozyme of cytochrome P450 (CYP3A4) [13], and its principal circulating metabolite, 3-OH-guanfacine sulfate, lacks pharmacological activity [16]. CYP3A4, which is expressed in both the gut and liver, is involved in the metabolism of approximately half of marketed drugs [21]. Inhibitors of CYP3A4 may therefore increase exposure to sensitive drugs to toxic levels, whereas inducers may reduce exposure to below therapeutic levels. The US Food and Drug Administration (FDA) defines strong and moderate inhibitors as drugs that increase the area under the plasma concentration–time curve (AUC) of sensitive substrates of a given metabolic pathway by ≥ 5-fold and ≥ 2 to < 5-fold, respectively, while strong and moderate inducers decrease AUC by ≥ 80 and ≥ 50 to < 80%, respectively [22]. The potential for GXR to be susceptible to drug–drug interactions (DDIs) with inhibitors or inducers of CYP3A4 was indicated by the results of clinical PK studies, which led to dosing adjustments being included in the original US label. Co-administration of GXR with ketoconazole (a strong inhibitor [22]) led to a 3-fold increase in guanfacine exposure, as measured by AUC, and co-administration with rifampicin (a strong inducer [22]) led to a 70% decrease in guanfacine exposure [23].
Although clinical PK studies are the gold standard for investigating DDIs, they require participants to be exposed to investigational products for experimental rather than therapeutic purposes. Physiologically based pharmacokinetic (PBPK) models, by contrast, predict PK profiles and parameters on the basis of in vitro experimental data, the physicochemical properties of the molecule in question and available clinical PK data, and population characteristics, e.g., demographics, inter-individual variability in CYP abundance, and proportions of poor metabolizer phenotypes. For the past decade, PBPK models have been increasingly employed by the pharmaceutical industry [24]. According to the findings of a systematic review of 106 PBPK models described in publications from 2008 to 2015, they are used to simulate the properties of alternative formulations and to predict drug absorption kinetics, general clinical PK profiles, inter-individual variability, DDIs and age-related changes in PK parameters [25]. The European Medicines Agency guideline on the investigation of DDIs states that PBPK modeling may be used to estimate the potential for interactions between drugs [26], while the equivalent US FDA guideline states that PBPK models offer useful alternatives to dedicated clinical studies [27]. DDI prediction was the subject of approximately two-thirds of PBPK submissions to the FDA between 2008 and October 2014 (n = 136) [28]. Several FDA submissions for investigational new drugs or new drug applications are discussed in more detail by Zhao et al., who conclude that PBPK modeling has facilitated decision-making about labeling, study design and the need to conduct additional clinical pharmacology studies [29].
The objective of the present study was to provide guidance for possible GXR dose adjustment for US labeling in children and adolescents with ADHD by developing a PBPK model to predict DDIs when the drug is co-administered with moderate CYP3A4 perpetrators, namely the inhibitors fluconazole (an anti-fungal) or erythromycin (an antibiotic), or the inducer efavirenz (an antiretroviral).
2 Methods
2.1 Modeling Workflow
The Simcyp (version 14) population-based PBPK simulator (Symcyp Ltd, Sheffield, UK) was used to model guanfacine PK profiles and DDIs with moderate CYP3A4 inducers or inhibitors (Fig. 1a) [30]. The modeling workflow was part of a Best Practice approach, in which model development used the most robust clinical PK data available and model validation used all clinical DDI data. The workflow involved:

a Modeling workflow and b schematic representation of the Simcyp PBPK model. aInput parameters from Simcyp version 14 compound file. bInput parameters from Ke et al. [30]. Q G, Q H, Q PV, Q m and Q H,A are blood flows in the gut, liver, portal vein, mucosa and hepatic artery, respectively; F G and F H are the fractions escaping first-pass metabolism in the gut and liver, respectively; CLR is renal clearance, f a is the fraction absorbed, and k a is the first-order absorption rate constant. CYP3A4 3A4 isozyme of cytochrome P450, DDI drug–drug interaction, GXR guanfacine extended-release, PBPK physiologically based pharmacokinetic, PK pharmacokinetic, q.d. every day, t.i.d. three times a day
-
1.
Developing a fit-for-purpose PBPK model based on in vitro data and 96-h clinical guanfacine plasma PK profiles obtained following administration of a single oral dose of GXR 2 mg or 4 mg to healthy adults [19].
-
2.
Validating the PBPK model based on 96-h clinical guanfacine plasma PK profiles obtained for healthy adults following administration of a single oral dose of GXR 4 mg in the presence and absence of the strong CYP3A4 inhibitor ketoconazole or the strong CYP3A4 inducer rifampicin [13].
-
3.
Applying the PBPK model to predict the impact on guanfacine plasma PK parameters of GXR co-administration with one of the moderate CYP3A4 inhibitors fluconazole or erythromycin, or with the moderate CYP3A4 inducer efavirenz [22].
Plasma drug concentration–time profiles, clearance, distribution and absorption were simulated using populations of virtual individuals. With the exception of demographic characteristics, all parameters were the same as those of a virtual North European Caucasian population that has been described previously [31]. Unless otherwise specified, ten virtual trials, each sampling the same number of sex-matched virtual individuals with the same age range as those in a comparable clinical study, were generated to assess variability across groups.
2.2 Development of a PBPK Model for Guanfacine Exposure Following a Single Oral Dose of GXR
2.2.1 Model Parameters
A dynamic model incorporating competitive- and mechanism-based inhibition was used to simulate the effects of CYP3A4 inhibitors on the metabolism of guanfacine in the liver and gut (Fig. 1b) [32]. Unbound concentrations of inhibitor in the liver and portal vein were used to drive inhibition of guanfacine metabolism in the liver and gut, respectively. First–order absorption was assumed based on the linear PK profiles obtained from clinical studies in children and adolescents with ADHD [17] and in healthy adults [19]. The guanfacine fraction absorbed (f a) and first-order absorption rate constant (k a) were based on Caco-2 permeability (data on file, V00977–SPD503, Shire Pharmaceuticals Ltd, 2006) [33, 34]. The fraction escaping first-pass metabolism in the gut (F G) was estimated using Eq. 1, which represents a model similar to the ‘well-stirred’ liver model [35]; Q G is nominal blood flow (Fig. 1b), fuG is the free fraction of the drug within enterocytes (set at a default value of 1), and CLuG,int is the whole gut intrinsic clearance.
Caco-2 permeability (data on file, V00977–SPD503, Shire Pharmaceuticals Ltd, 2006) was used to make an initial estimate of Q G [34]. CLuG,int was initially estimated by dividing the in vitro CYP3A–mediated hepatic unbound intrinsic metabolic clearance per milligram of protein (CLuint) (data on file, V00652–SPD503–IIIG, Shire Pharmaceuticals Ltd, 2003) by the abundance of CYP3A in the liver (137 pmol/mg) and multiplying by the mean abundance of CYP3A in the whole gut (70,000 pmol) [36]. The assumption that intrinsic guanfacine clearance per picomole of CYP3A4 is identical in the liver and gut [36] is supported by in vitro comparisons of the metabolic ability of liver and gut microsomes using eight representative CYP3A4 probe substrates (including midazolam) [37]. Values used for the intrinsic turnover of hepatic and gut CYP3A4 (k deg) were 0.019 and 0.03 h−1, respectively [32, 38]. A sensitivity analysis determined the value of k a (0.459 h−1) that recovered the observed guanfacine time to maximum plasma concentration (t max) of approximately 6 h following oral administration of GXR to healthy adults [19]. The blood:plasma ratio, [B:P], was estimated to be 1.45 based on Eq. 2 and an erythrocyte:plasma ratio [E:P] of 2 [39] at a hematocrit (HCT) of 45%.
The mean renal clearance of guanfacine (CLR) (12.6 L/h) was based on observed data following intravenous administration of guanfacine in healthy adults [20] and was estimated to be 50% of total body clearance. CLuint was optimized from 7.28 µL/min/mg protein (data on file, V00652-SPD503-IIIG, Shire Pharmaceuticals Ltd, 2003) to 17 µL/min/mg protein using sensitivity analyses to ensure recovery of observed total body clearance following intravenous administration (CLIV) (24.8 L/h) [20] with equal contributions from metabolic and renal clearances. The fraction escaping first-pass metabolism in the liver (F H) was calculated to be 0.91 from hepatic blood flow (90 L/h) and hepatic clearance in blood [CLH,B = (CLIV − CLR)/[B:P] = 8.41 L/h]. The estimate for Q G was then refined to 1 L/h using Eq. 1 and the relationship F = f a·F G·F H (where F is bioavailability) to recover the observed mean value for apparent clearance (CL/F, 37.8 L/h) following oral administration of GXR to healthy adults [19]. Final input parameters for the PBPK model are shown in Table 1 [19, 20, 33, 34, 39, 40].
2.2.2 Simulation Design
The simulation of guanfacine plasma concentration–time profiles following a single oral dose of GXR was based on the results of the randomized, crossover trial by Swearingen et al. in which 52 healthy adults (aged 18–54 years; 46.2% women), 49 of whom completed the study, received a single oral dose of GXR 2 mg or 4 mg once a week for 4 weeks [19].
2.3 Validation of the GXR PBPK Model Using Data from Clinical Studies of Strong CYP3A4 Inhibitors and Inducers
Validation of GXR DDI with the strong CYP3A4 inhibitor ketoconazole was based on the results of a study of 20 healthy adults (aged 19–50 years; 65% women) who received single oral doses of GXR 4 mg in the absence of ketoconazole and on the third day of 6 days of dosing with ketoconazole [400 mg daily (q.d.)] (data on file, SPD503-106, Shire Pharmaceuticals Ltd, 2004). Simulations used the default ketoconazole input values from Simcyp version 14.
Validation of GXR DDI with the strong CYP3A4 inducer rifampicin was based on the findings of a study of 20 healthy adults (aged 18–53 years; 40% women) who received a single oral dose of GXR 4 mg in the absence of rifampicin and on the eighth day of 11 days of dosing with rifampicin (600 mg q.d.) (data on file, SPD503-108, Shire Pharmaceuticals Ltd, 2004). Two separate simulations were run, based on the results of recent literature reviews [41, 42], using the default rifampicin input values from Simcyp version 14 and a maximum fold induction (Indmax) of 16 or 8 (i.e., in the simulations, rifampicin could increase CYP3A4 production by a maximum of 16–fold or 8–fold of its production in the absence of rifampicin).
2.4 Application of the GXR PBPK Model to Predict Guanfacine Plasma Concentration–Time Profiles in the Presence of the Moderate CYP3A4 Inhibitors Fluconazole or Erythromycin
Prediction of GXR DDIs with fluconazole or erythromycin (moderate CYP3A4 inhibitors) used the default input values from the compound files in Simcyp version 14. Ten virtual trials of 20 healthy adults (aged 19–50 years; 65% women) receiving a single oral dose of GXR 4 mg were simulated in each of the following scenarios: (1) on the third day of 6 days of dosing with fluconazole (a loading dose of 400 mg followed by 200 mg q.d.); (2) on the third day of 6 days of dosing with erythromycin [500 mg three times a day (t.i.d.)].
2.5 Application of the GXR PBPK Model to Predict Guanfacine Plasma Concentration–Time Profiles in the Presence of the Moderate CYP3A4 Inducer Efavirenz
Prediction of GXR DDI with efavirenz (a moderate CYP3A4 inducer) was based on the results of ten trials of 20 virtual individuals (aged 18–50 years; 50% women) receiving a single oral dose of GXR 4 mg on day 10 of 14 days of dosing with efavirenz (600 mg q.d. or 400 mg q.d.). Efavirenz 600 mg simulations used the input parameter values recently published by Ke et al. [30]. In the context of a recent study that found that efavirenz 400 mg was virologically non-inferior to the standard 600 mg dose [43, 44], a PBPK model of efavirenz 400 mg was also developed, based on the 600 mg model. The results of clinical PK studies indicate that the dose-normalized peak plasma drug concentration (C max) and the AUC for a single dose of efavirenz 600 mg q.d. [45] are greater than those for efavirenz 400 mg q.d. in healthy adults [46]; parameter estimation was therefore used to adjust oral clearance (CLoral) from the value used by Ke et al. [30] to 17.7 L/h in order to recover the observed plasma concentrations of single-dose efavirenz 400 mg from day 1 of a multi-day study [46]. Efavirenz 400 mg model validation was based on multiple dose data from the same study, in which a cohort of 12 healthy adults were randomized 2:1 to receive efavirenz 400 mg q.d. or placebo for 10 days [46]; simulations used ten trials of eight virtual individuals (aged 19–49 years; 5% women).
3 Results
3.1 GXR Model Development: Simulation of Guanfacine Plasma Concentration–Time Profiles in Healthy Adults
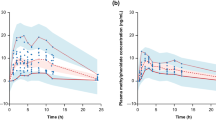
Predicted (n = 490) and observed (n = 49) plasma concentration–time profiles following a single oral dose of GXR 2 mg or 4 mg in healthy adults are shown in Fig. 2a, b, respectively. The mean observed guanfacine C max and AUC values were 1.57 ng/mL and 54.5 ng/mL·h following GXR 2 mg, and 3.58 ng/mL and 119.0 ng/mL·h following GXR 4 mg [19]. The mean predicted guanfacine C max and AUC were 1.69 ng/mL and 43.6 ng/mL·h following GXR 2 mg, and 3.37 ng/mL and 87.3 ng/mL·h following GXR 4 mg. At both doses, predicted mean C max and AUC values were within 1.3-fold of observed values, and the model was considered to have recovered the relatively linear PK profile of the clinical data.

Development and validation of a PBPK model of guanfacine exposure following administration of a single oral dose of GXR 4 mg. Model development: observed (circles) [19] and simulated (lines) mean plasma concentration–time profiles of guanfacine following a single oral dose of a GXR 2 mg or b GXR 4 mg. Gray lines represent the outcomes of 10 simulated trials (n = 49), and the black line represents mean data for the simulated population (n = 490). Model validation: observed guanfacine concentration–time profiles following administration of a single dose of GXR 4 mg in the absence (open circles) and presence (closed circles) of c ketoconazole 400 mg q.d. (on day 3 of 6 days of dosing) and d rifampicin 600 mg q.d. (on day 8 of 11 days of dosing). The solid and dashed lines represent mean data for 10 simulated trials (n = 200). AUC area under the plasma time–concentration curve, CYP3A4 3A4 isozyme of cytochrome P450, DDI drug–drug interaction, GXR guanfacine extended-release, Ind max maximum fold induction, PBPK physiologically based pharmacokinetic, q.d. every day
3.2 GXR Model Validation: Simulation of Guanfacine Plasma Concentration–Time Profiles in Healthy Adults Receiving Multiple Daily Doses of the Strong CYP3A4 Inhibitor Ketoconazole or the Strong CYP3A4 Inducer Rifampicin
Predicted (n = 200) and observed (n = 20) plasma concentration–time profiles of guanfacine following a single oral dose of GXR 4 mg in the absence and presence of ketoconazole (400 mg q.d.) are depicted in Fig. 2c. The predicted geometric mean C max ratio was equivalent to that obtained from observed data, and the predicted AUC ratio was 18% lower than observed data (Table 2).
Predicted (n = 200) and observed (n = 20) plasma concentration–time profiles of guanfacine following a single oral dose of GXR 4 mg in the absence and presence of rifampicin (600 mg q.d.) are shown in Fig. 2d. When the default values from the Simcyp version 14 rifampicin compound file were used (these include Indmax = 16), predicted geometric mean guanfacine C max and AUC ratios were 32% and 48% lower than observed values, respectively, suggesting that the model overpredicted the degree of CYP3A4 induction. After adjusting Indmax from 16 to 8 based on findings from recent literature reviews [41, 42], the predicted C max and AUC ratios were closer to the observed ratios, but were 18% and 14% higher, respectively, suggesting that this model underpredicted CYP4A4 induction (Table 2).
3.3 GXR Model Application: Prediction of Guanfacine Pharmacokinetics Following Administration of GXR in the Absence and Presence of Erythromycin or Fluconazole (Moderate CYP3A4 Inhibitors)
Predicted guanfacine plasma concentration–time profiles following administration of a single oral dose of GXR 4 mg to healthy adults in the absence and presence of steady-state erythromycin (500 mg t.i.d.; n = 200) or fluconazole (loading dose of 400 mg followed by 200 mg q.d.; n = 200) are depicted in Fig. 3a, b. Guanfacine C max and AUC were predicted to be 1.45-fold and 1.98–fold higher in the presence of fluconazole than in its absence, and to be increased by similar amounts in the presence of erythromycin (1.58–fold and 2.31–fold higher, respectively, compared with GXR monotherapy) (Table 3).

Prediction of guanfacine DDI with moderate CYP3A4 inhibitors or inducers following administration of a single oral dose of GXR 4 mg. Predicted mean plasma concentration–time profiles of guanfacine following administration of a single oral dose of GXR 4 mg in the absence (solid line) and presence (dashed line) of a erythromycin 500 mg t.i.d. (on day 3 of 6 days of dosing), b fluconazole 200 mg q.d. (on day 3 of 6 days of dosing, following a loading dose of 400 mg on day 1), c efavirenz 400 mg q.d. (on day 10 of 14 days of dosing) or d efavirenz 600 mg q.d. (on day 10 of 14 days of dosing). Lines represent mean data for 10 simulated trials (n = 200). AUC area under the plasma time–concentration curve, CYP3A4 3A4 isozyme of cytochrome P450, DDI drug–drug interaction, GXR guanfacine extended-release, q.d. every day, t.i.d. three times a day
3.4 GXR Model Application: Prediction of Guanfacine Pharmacokinetics Following Administration of GXR in the Absence and Presence of Efavirenz (Moderate CYP3A4 Inducer)
3.4.1 Efavirenz 400 mg Model Validation
Predicted and observed efavirenz plasma concentration–time profiles on day 10 of administration of efavirenz 400 mg q.d. in healthy adults are shown in Fig. 4. Predicted mean C max and AUC values (1.85 µg/mL and 24.6 µg/mL·h) were within 1.3-fold of observed values (2.20 µg/mL and 29.2 µg/mL·h) [46].

Validation of a physiologically based pharmacokinetic model of exposure to the moderate CYP3A4 inducer efavirenz 400 mg. Observed (circles) and simulated (lines) mean plasma concentration–time profiles of efavirenz on day 10 following administration of 400 mg every day for 10 days. Gray lines represent the outcomes of 10 individual simulated trials (n = 8), and the black line represents mean data for the simulated population (n = 80). CYP3A4 3A4 isozyme of cytochrome P450
3.4.2 Prediction of Guanfacine Pharmacokinetics
Predicted guanfacine plasma concentration–time profiles following administration of a single oral dose of GXR 4 mg to healthy adults in the absence and presence of efavirenz 400 mg q.d. or 600 mg q.d. are depicted in Fig. 3c, d. When GXR was co-administered with efavirenz 400 mg, guanfacine C max and AUC were reduced to 72 and 58% of their values for GXR monotherapy, respectively; co-administration with efavirenz 600 mg reduced guanfacine C max and AUC to 50 and 33% of their values for GXR monotherapy, respectively (Table 3).
4 Discussion
Here we describe the development and validation of a PBPK model that was applied to predict the impact on guanfacine PK parameters of administering GXR in the presence of moderate CYP3A4 inhibitors and inducers. As shown in Fig. 5, model predictions indicate that co-administration of GXR with the moderate CYP3A4 inhibitors fluconazole 200 mg q.d. or erythromycin 500 mg t.i.d. leads to an increase in guanfacine exposure that is slightly smaller than when GXR is co-administered with the strong CYP3A4 inhibitor ketoconazole; and that co-administration of GXR with the moderate CYP3A4 inducer efavirenz 400 or 600 mg q.d. leads to a smaller or similar reduction in guanfacine exposure, respectively, compared with co-administration with the strong CYP3A4 inducer rifampicin.

Simulated and observed guanfacine C max and AUC geometric mean ratios for moderate and strong inhibitors and inducers of CYP3A4. AUC area under the plasma concentration–time curve, CI confidence interval, C max peak plasma drug concentration, CYP3A4 3A4 isozyme of cytochrome P450, DDI drug–drug interaction, Ind max maximum fold induction, q.d. every day, t.i.d. three times a day
Several clinical PK studies have been conducted that examine potential DDIs with GXR. Results presented here show that, compared with GXR monotherapy, co-administration of GXR with the strong CYP3A4 inhibitor ketoconazole resulted in a 1.75-fold increase in plasma guanfacine C max and a 3.13-fold increase in AUC in healthy adults (Table 2). Conversely, co-administration with the strong CYP3A4 inducer rifampicin resulted in C max and AUC being reduced to 46% and 31% of their values for GXR monotherapy (Table 2). As a result of these studies, the US GXR prescribing information recommends that the dose should be decreased to half the recommended level when co-administered with a strong CYP3A4 inhibitor, and that clinicians should consider increasing the dose to double the recommended level (over 1–2 weeks) when co-administered with a strong CYP3A4 inducer [13]. In the USA and Canada, GXR is indicated for co-administration with a stimulant medication in the treatment of ADHD in addition to its use as a monotherapy. In clinical PK studies, no clinically meaningful DDIs were observed between GXR and an extended-release formulation of MPH (osmotic-release oral system MPH) [47], or the amphetamine prodrug lisdexamfetamine dimesylate [48]; hence, no additional dose adjustment is required when GXR is used adjunctive to stimulant treatment [13].
In the absence of clinical PK data when moderate CYP3A4 inhibitors or inducers are co-administered with GXR, a PBPK model was designed to predict their impact on guanfacine PK following oral administration of GXR, in order to provide appropriate dosing guidance. The model was developed based on the physicochemical properties of guanfacine, in vitro CYP metabolism, total systemic clearance, renal clearance following intravenous administration in humans and the PK profile of guanfacine following oral administration. The clinical data following administration GXR monotherapy are relatively linear across the dose range of 2–4 mg GXR (C max = 2.28-fold; AUC0–t = 2.18-fold) [19], and this linearity was recovered during model development.
The model was validated using clinical GXR DDI data for ketoconazole and rifampicin (Table 2), indicating that the intrinsic clearance of CYP3A4, estimated from a CYP3A4 fraction of metabolism of 50%, was appropriate for the purpose of predicting CYP3A4-mediated DDIs. The predicted mean C max and AUC ratios of guanfacine following a single oral 4-mg dose of GXR with and without ketoconazole (400 mg q.d.) were equivalent to and 18% lower than observed values, respectively. For co-administration with rifampicin (600 mg q.d.), however, the predicted mean C max and AUC ratios were substantially lower than the observed values (by 32 and 48%, respectively) when the default compound parameters from Simcyp (version 14) were used, including a value of 16 for Indmax. The results of a recent literature review of ten intravenous and 19 oral studies involving rifampicin and victim drugs (mainly midazolam) indicated that, although there was a decrease in bias and an increase in precision with an Indmax of 16 compared with an Indmax of 8, there was a tendency to overpredict the induction effect of rifampicin [41]. An independent analysis of FDA submissions in which a DDI with rifampicin was used for PBPK model validation showed that the predicted AUC ratio in the presence versus the absence of rifampicin was within 1.25-fold of the observed data in four out of seven models (57%) in which an Indmax of 8 was used [42]. In the current study, use of an Indmax of 8 led to a predicted AUC ratio that was more consistent with observed data than an Indmax value of 16 (Fig. 5).
After the predictive potential of the PBPK model had been validated, guanfacine PK parameters following co-administration of GXR with moderate CYP3A4 inhibitors or inducers were predicted. In prospective simulations of moderate CYP3A4 inhibitors, plasma guanfacine C max and AUC were 1.45-fold and 1.98-fold higher, respectively, in the presence of fluconazole, and 1.58-fold and 2.31-fold higher, respectively, in the presence of erythromycin, compared with their values with GXR monotherapy. Conversely, in the presence of the moderate CYP3A4 inducer efavirenz 400 mg q.d., predicted guanfacine C max and AUC were reduced to 72 and 58% of their values in the absence of efavirenz, respectively. The greater reduction in guanfacine C max and AUC observed in the presence of efavirenz 600 mg q.d. (to 50 and 33% of their values for GXR monotherapy, respectively) suggests that the effect may be related to efavirenz dose. As efavirenz is an auto-inducer of its own clearance (via the CYP2B6 pathway) [49, 50], its exposure and potential CYP3A4 induction effect would be expected to be efavirenz dose-dependent. In addition, it should be noted that efavirenz induces CYP3A4 production in the liver only [46, 51], unlike rifampicin, which induces CYP3A4 in both the liver and gut [52]. This was accounted for by Ke et al., who used sensitivity analysis to adjust the fraction unbound in the gut to recover AUC ratios for a victim drug when developing the efavirenz model used in the present study [30]. In summary, the present predictions suggest that, as with strong inhibitors and inducers of CYP3A4, DDIs between GXR and moderate inhibitors and inducers of the enzyme result in substantial changes in guanfacine exposure (Fig. 5).
In interpreting these data, three important limitations should be considered. First, GXR clinical PK data in children and adolescents (aged 6–17 years) were not used during model development. This was because a Best Practice approach was followed in which the most robust clinical PK data available were used, from an adult study (n = 49) in which plasma guanfacine concentration was measured to 96 h post-dose [19]. The half-life of guanfacine from GXR is ~ 17 h [19]; in comparison, two of the three pivotal studies providing PK data in children and adolescents ended sampling at 24 h post-dose (n = 28 [17]; n = 20 [53]), while the pivotal PK data from a placebo-controlled phase 2 pharmacodynamic study (GXR arm, n = 112 [18]) end at 8 h post-dose [53]. As an analysis of data from these three studies (n = 160) had found previously that population PK parameters were similar to mean values in healthy adults when appropriately scaled by patient weight [18], no additional analyses were conducted. Second, although model validation used all available clinical DDI data, no GXR clinical DDI studies have been conducted in children and adolescents. However, hepatic blood flow, CYP3A4 activity and renal function in this age group are similar to those in adults [54, 55], and PK parameters are similar when scaled by weight, as noted above [18]. Thus, it was judged that no prospective extrapolations of dose adjustments were required. Third, the clinical DDI studies conducted in adults used only one dose, GXR 4 mg. Recovery of the relatively linear clinical PK profile for GXR 2 mg and 4 mg [19] during model development, however, indicates that the parameters, or assignment of clearance pathways, are applicable for both doses.
As a result of this study, the following recommendations were approved for US labeling: (1) decrease GXR to 50% of the usual target dose when it is co-administered with strong or moderate CYP3A4 inhibitors; and (2) consider titrating GXR up to double the usual target dosage over 1–2 weeks when it is co-administered with strong or moderate CYP3A4 inducers [13]. It should be noted that the GXR is always titrated based on clinical judgment of response and tolerability. Titration covers a 4-fold dose range in children aged 6–12 years (1–4 mg/day) and a 4–7-fold dose range for adolescents aged 13–17 years (1–4 to 1–7 mg/day, depending on body weight) [13]. These dose ranges exceed the difference between the effects of strong and moderate perpetrators on guanfacine exposure (Fig 5), and having identical dosing recommendations for strong and moderate perpetrators simplifies their implementation in clinical practice.
Although clinical PK studies remain the gold standard for investigating the effects of DDIs on exposure to a drug of interest, recent reviews of FDA submissions of drugs in development concluded that the predictions of 15 models involving cytochrome P450 inhibitors (mainly CYP3A and CYP2D6) and 11 models involving CYP3A inducers showed good agreement with observed data [42, 56]. The present data confirm that PBPK modeling is a powerful tool in the extrapolation of existing preclinical and clinical data to generate recommendations for treatment regimens without the need for individuals to be exposed to investigational products for experimental rather than therapeutic purposes.
5 Conclusions
Using data from clinical PK studies in which GXR was administered as a monotherapy or co-administered with strong CYP3A4 inhibitors or inducers, PBPK modeling was employed to evaluate the potential for DDIs when GXR is co-administered with moderate CYP3A4 inhibitors or inducers. Based on these predictions, dosing recommendations for GXR were approved by the FDA as US labeling recommendations without the need to conduct further clinical studies [13].
In summary, the recommendations are to (1) decrease GXR to 50% of the usual target dose when it is co-administered with strong or moderate CYP3A4 inhibitors and (2) consider titrating GXR up to double the usual target dosage over 1–2 weeks when it is co-administered with strong or moderate CYP3A4 inducers [13]. Thus, prescribers can be given specific dosing guidance on the use of GXR with moderate CYP3A4 inhibitors or inducers in the absence of clinical PK data on these interactions.
Change history
28 December 2017
The article “Development of Guanfacine Extended-Release Dosing Strategies in Children and Adolescents with ADHD Using a PhysiologicallyBased Pharmacokinetic Model to Predict Drug–Drug Interactions with Moderate CYP3A4 Inhibitors or Inducers”, written by Aiqun Li, Karen Yeo, Devin Welty, Haojing Rong, was originally published electronically on the publisher’s internet portal (currently SpringerLink) on 02nd November, 2017 without open access.
References
Taylor E, Dopfner M, Sergeant J, Asherson P, Banaschewski T, Buitelaar J, et al. European clinical guidelines for hyperkinetic disorder—first upgrade. Eur Child Adolesc Psychiatry. 2004;13(Suppl 1):I7–30. https://doi.org/10.1007/s00787-004-1002-x.
Subcommittee on Attention-Deficit/Hyperactivity Disorder, Steering Committee on Quality Improvement and Management, Wolraich M, Brown L, Brown RT et al. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2011;128(5):1007–22. https://doi.org/10.1542/peds.2011-2654.
National Institute for Health and Care Excellence. Attention deficit hyperactivity disorder: diagnosis and management (CG72). 2008 [updated February 2016].
Arnold LE. Methyiphenidate vs. amphetamine: comparative review. J Atten Disord. 2000;3(4):200–11. https://doi.org/10.1177/108705470000300403.
Hodgkins P, Shaw M, Coghill D, Hechtman L. Amfetamine and methylphenidate medications for attention-deficit/hyperactivity disorder: complementary treatment options. Eur Child Adolesc Psychiatry. 2012;21(9):477–92. https://doi.org/10.1007/s00787-012-0286-5.
Janssen Pharmaceuticals Inc. Concerta US prescribing information. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021121s038lbl.pdf. Accessed 3 Oct 2017.
Shire Pharmaceuticals Ltd. Equasym XL UK summary of product characteristics. 2014. https://www.medicines.org.uk/emc/medicine/15804/SPC/. Accessed 3 Oct 2017.
Shire Pharmaceuticals Ltd. Elvanse UK summary of product characteristics. 2016. http://www.medicines.org.uk/emc/medicine/27442/SPC/. Accessed 3 Oct 2017.
Schatz NK, Fabiano GA, Cunningham CE, dosReis S, Waschbusch DA, Jerome S, et al. Systematic review of patients’ and parents’ preferences for ADHD treatment options and processes of care. Patient. 2015;8(6):483–97. https://doi.org/10.1007/s40271-015-0112-5.
Stockl KM, Hughes TE, Jarrar MA, Secnik K, Perwien AR. Physician perceptions of the use of medications for attention deficit hyperactivity disorder. J Manag Care Pharm. 2003;9(5):416–23. https://doi.org/10.18553/jmcp.2003.9.5.416.
Uhlen S, Porter AC, Neubig RR. The novel alpha-2 adrenergic radioligand [3H]-MK912 is alpha-2C selective among human alpha-2A, alpha-2B and alpha-2C adrenoceptors. J Pharmacol Exp Ther. 1994;271(3):1558–65.
Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129(2):397–410. https://doi.org/10.1016/j.cell.2007.03.015.
Shire US Inc. Intuniv US prescribing information. Shire US Inc. 2016. http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/022037s013lbl.pdf. Accessed 3 Oct 2017.
Shire Pharma Canada ULC. Intuniv XR Canadian product monograph. Shire Pharma Canada ULC. 2017. http://www.shirecanada.com/-/media/shire/shireglobal/shirecanada/pdffiles/product%20information/intuniv-xr-pm-en.pdf. Accessed 3 Oct 2017.
Shinonogi & Co. Ltd. Intuniv Japanese prescribing information. 2017. http://www.info.pmda.go.jp/go/pack/1179057G1021_1_03/1179057G1021_1_03?view=body. Accessed 3 Oct 2017.
Shire Pharmaceuticals Ltd. Intuniv EMA summary of product characteristics. Shire Pharmaceuticals Ltd. Jun 2017. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003759/WC500195130.pdf. Accessed 3 Oct 2017.
Boellner SW, Pennick M, Fiske K, Lyne A, Shojaei A. Pharmacokinetics of a guanfacine extended-release formulation in children and adolescents with attention-deficit-hyperactivity disorder. Pharmacotherapy. 2007;27(9):1253–62. https://doi.org/10.1592/phco.27.9.1253.
Knebel W, Corcoran M, Ermer J, Gastonguay MR. Population pharmacokinetic modeling of guanfacine in pediatric patients. Clin Pharmacokinet. 2015;54(8):875–83. https://doi.org/10.1007/s40262-015-0245-7.
Swearingen D, Pennick M, Shojaei A, Lyne A, Fiske K. A phase I, randomized, open-label, crossover study of the single-dose pharmacokinetic properties of guanfacine extended-release 1-, 2-, and 4-mg tablets in healthy adults. Clin Ther. 2007;29(4):617–25. https://doi.org/10.1016/j.clinthera.2007.04.016.
Carchman SH, Crowe JTJ, Wright GJ. The bioavailability and pharmacokinetics of guanfacine after oral and intravenous administration to healthy volunteers. J Clin Pharmacol. 1987;27(10):762–7.
Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. https://doi.org/10.1146/annurev.pharmtox.39.1.1.
U.S. Food and Drug Administration. Drug development and drug interactions: table of substrates, inhibitors and inducers. US Food and Drug Administration. 2016. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed 3 Oct 2017.
Shire US Inc. Intuniv US prescribing information. Shire US Inc. 2009. http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022037lbl.pdf. Accessed 3 Oct 2017.
Poggesi I, Snoeys J, Van Peer A. The successes and failures of physiologically based pharmacokinetic modeling: there is room for improvement. Expert Opin Drug Metab Toxicol. 2014;10(5):631–5. https://doi.org/10.1517/17425255.2014.888058.
Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos. 2015;43(11):1823–37. https://doi.org/10.1124/dmd.115.065920.
European Medicines Agency. Guideline on the investigation of drug interactions. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. Accessed 3 Oct 2017.
U.S. Food and Drug Administration. Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. U.S. Food and Drug Administration. 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed 3 Oct 2017.
Grillo JA. Pediatric applications of PBPK modeling and simulation in drug regulatory science: where are we now? Presented at the American Association of Pharmaceutical Scientists (AAPS) Annual Meeting; San Diego, CA, USA; 2–6 November 2014. Available from: https://zerista.s3.amazonaws.com/item_files/1f71/attachments/32090/original/186.pdf. Accessed: 23 Jan 2017.
Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89(2):259–67. https://doi.org/10.1038/clpt.2010.298.
Ke A, Barter Z, Rowland-Yeo K, Almond L. Towards a best practice approach in PBPK modeling: case example of developing a unified efavirenz model accounting for induction of CYPs 3A4 and 2B6. CPT Pharmacomet Syst Pharmacol. 2016;5(7):367–76. https://doi.org/10.1002/psp4.12088.
Howgate EM, Rowland Yeo K, Proctor NJ, Tucker GT, Rostami-Hodjegan A. Prediction of in vivo drug clearance from in vitro data. I: impact of inter-individual variability. Xenobiotica. 2006;36(6):473–97. https://doi.org/10.1080/00498250600683197.
Rowland Yeo K, Jamei M, Yang J, Tucker GT, Rostami-Hodjegan A. Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut—the effect of diltiazem on the time-course of exposure to triazolam. Eur J Pharm Sci. 2010;39(5):298–309. https://doi.org/10.1016/j.ejps.2009.12.002.
Sun D, Lennernas H, Welage LS, Barnett JL, Landowski CP, Foster D, et al. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm Res. 2002;19(10):1400–16.
Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8(7):676–84.
Rostami-Hodjegan A, Tucker G. ‘In silico’ simulations to assess the ‘in vivo’ consequences of ‘in vitro’ metabolic drug-drug interactions. Drug Discov Today Technol. 2004;1(4):441–8. https://doi.org/10.1016/j.ddtec.2004.10.002.
Yang J, Tucker GT, Rostami-Hodjegan A. Cytochrome P450 3A expression and activity in the human small intestine. Clin Pharmacol Ther. 2004;76(4):391. https://doi.org/10.1016/j.clpt.2004.07.001.
Galetin A, Houston JB. Intestinal and hepatic metabolic activity of five cytochrome P450 enzymes: impact on prediction of first-pass metabolism. J Pharmacol Exp Ther. 2006;318(3):1220–9. https://doi.org/10.1124/jpet.106.106013.
Yang J, Liao M, Shou M, Jamei M, Yeo KR, Tucker GT, et al. Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab. 2008;9(5):384–94.
Kiechel JR. Pharmacokinetics and metabolism of guanfacine in man: a review. Br J Clin Pharmacol. 1980;(10 Suppl 1):25S–32S.
National Center for Biotechnology Information. PubChem Compound Database; CID = 3519. National Center for Biotechnology Information. 2016. https://pubchem.ncbi.nlm.nih.gov/compound/3519 Accessed 3 Oct 2017.
Almond LM, Mukadam S, Gardner I, Okialda K, Wong S, Hatley O, et al. Prediction of drug-drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab Dispos. 2016;44(6):821–32. https://doi.org/10.1124/dmd.115.066845.
Wagner C, Pan Y, Hsu V, Sinha V, Zhao P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the U.S. FDA. Clin Pharmacokinet. 2016;55(4):475–83. https://doi.org/10.1007/s40262-015-0330-y.
Puls R, Amin J, Losso M, Phanuphak P, Nwizu C, Orrell C, et al. Efficacy of 400 mg efavirenz versus standard 600 mg dose in HIV-infected, antiretroviral-naive adults (ENCORE1): a randomised, double-blind, placebo-controlled, non-inferiority trial. Lancet. 2014;383(9927):1474–82. https://doi.org/10.1016/s0140-6736(13)62187-x.
Dickinson L, Amin J, Else L, Boffito M, Egan D, Owen A, et al. Pharmacokinetic and pharmacodynamic comparison of once-daily efavirenz (400 mg vs. 600 mg) in treatment-naive HIV-infected patients: results of the ENCORE1 study. Clin Pharmacol Ther. 2015;98(4):406–16. https://doi.org/10.1002/cpt.156.
Huang L, Parikh S, Rosenthal PJ, Lizak P, Marzan F, Dorsey G, et al. Concomitant efavirenz reduces pharmacokinetic exposure to the antimalarial drug artemether-lumefantrine in healthy volunteers. J Acquir Immune Defic Syndr. 2012;61(3):310–6. https://doi.org/10.1097/QAI.0b013e31826ebb5c.
Mouly S, Lown KS, Kornhauser D, Joseph JL, Fiske WD, Benedek IH, et al. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin Pharmacol Ther. 2002;72(1):1–9. https://doi.org/10.1067/mcp.2002.124519.
Roesch B, Corcoran M, Haffey M, Stevenson A, Wang P, Purkayastha J, et al. Pharmacokinetics of coadministration of guanfacine extended release and methylphenidate extended release. Drugs R D. 2013;13(1):53–61. https://doi.org/10.1007/s40268-013-0009-5.
Roesch B, Corcoran ME, Fetterolf J, Haffey M, Martin P, Preston P, et al. Pharmacokinetics of coadministered guanfacine extended release and lisdexamfetamine dimesylate. Drugs R D. 2013;13(2):119–28. https://doi.org/10.1007/s40268-013-0014-8.
Kharasch ED, Whittington D, Ensign D, Hoffer C, Bedynek PS, Campbell S, et al. Mechanism of efavirenz influence on methadone pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2012;91(4):673–84. https://doi.org/10.1038/clpt.2011.276.
Zhu M, Kaul S, Nandy P, Grasela DM, Pfister M. Model-based approach to characterize efavirenz autoinduction and concurrent enzyme induction with carbamazepine. Antimicrob Agents Chemother. 2009;53(6):2346–53. https://doi.org/10.1128/AAC.01120-08.
Oswald S, Meyer zu Schwabedissen HE, Nassif A, Modess C, Desta Z, Ogburn ET, et al. Impact of efavirenz on intestinal metabolism and transport: insights from an interaction study with ezetimibe in healthy volunteers. Clin Pharmacol Ther. 2012;91(3):506–13. https://doi.org/10.1038/clpt.2011.255.
Chen J, Raymond K. Roles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob. 2006;5:3. https://doi.org/10.1186/1476-0711-5-3.
U.S. Food and Drug Administration. Clinical pharmacology review NDA 22-037, guanfacine U.S. Food and Drug Administration. 2007. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM226551.pdf. Accessed 3 Oct 2017.
Alcorn J, McNamara PJ. Ontogeny of hepatic and renal systemic clearance pathways in infants: part I. Clin Pharmacokinet. 2002;41(12):959–98. https://doi.org/10.2165/00003088-200241120-00003.
de Wildt SN, Kearns GL, Leeder JS, van den Anker JN. Cytochrome P450 3A: ontogeny and drug disposition. Clin Pharmacokinet. 1999;37(6):485–505. https://doi.org/10.2165/00003088-199937060-00004.
Wagner C, Pan Y, Hsu V, Grillo JA, Zhang L, Reynolds KS, et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54(1):117–27. https://doi.org/10.1007/s40262-014-0188-4.
Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24(5):918–33. https://doi.org/10.1007/s11095-006-9210-3.
Acknowledgements
Under the direction of the authors, Dr. Heather Lang and Dr. Eric Southam, employees of Oxford PharmaGenesis, Oxford, UK, provided writing assistance; editorial assistance in formatting, proofreading, copy editing, and fact checking was also provided by Oxford PharmaGenesis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Shire. Shire develops and markets medications for attention-deficit/hyperactivity disorder, including guanfacine extended-release (GXR or Intuniv®). Simcyp Ltd (a Certara company) received funding from Shire for designing and conducting this study and analyzing and reporting the results. Oxford PharmaGenesis, Oxford, UK received funding from Shire International GmbH for writing and editing support.
Conflict of interest
Haojing Rong and Devin Welty are employees of Shire and own stock or stock options. Aiqun Li is a former employee of Shire. Karen Yeo is an employee of Certara.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Additional information
The original version of this article was revised due to retrospective open access order.
This was Aiqun Li's employment address when this work was performed.
A correction to this article is available online at https://doi.org/10.1007/s40272-017-0275-8.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license and any changes made are indicated.
About this article

Cite this article
Li, A., Yeo, K., Welty, D. et al. Development of Guanfacine Extended-Release Dosing Strategies in Children and Adolescents with ADHD Using a Physiologically Based Pharmacokinetic Model to Predict Drug–Drug Interactions with Moderate CYP3A4 Inhibitors or Inducers. Pediatr Drugs 20, 181–194 (2018). https://doi.org/10.1007/s40272-017-0270-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-017-0270-0