
Abstract
Background and Objective
Abiraterone acetate tablet is an inhibitor of androgen synthesis, primarily for the treatment of metastatic castration-resistant prostate cancer (mCRPC). This study evaluated the bioequivalence and pharmacokinetics of the reference and test formulations of abiraterone acetate tablets in healthy Chinese volunteers.
Methods
A single-center, open, single-dose, randomized, three-period, three-sequence, semi-repeat (only repeated reference formulations), and reference formulation-corrected fasting reference-scaled average bioequivalence test was conducted in 36 healthy volunteers included in this study. Volunteers were randomly assigned to one of three groups in a 1:1:1 ratio. There was a minimum 7-day washout period between each dose. Blood samples were collected at prescribed time intervals, the plasma concentration of abiraterone acetate tablets was determined by liquid chromatography-tandem mass spectrometry, and adverse events were recorded.
Results
Under fasting conditions, the maximum plasma concentration (Cmax) was 27.02 ± 14.21 ng/mL, area under the concentration-time curve from time zero to time t (AUCt) was 125.30 ± 82.41 h·ng/mL, and AUC from time zero to infinity (AUC∞) was 133.70 ± 83.99 h·ng/mL. The 90% confidence intervals (CIs) of the geometric mean ratio (GMR) of AUCt and AUC∞ were in the range of 0.8000–1.2500, and the coefficient of variation (CVWR) of Cmax was more than 30%. The Critbound result was − 0.0522, and the GMR was between 0.8000 and 1.2500.
Conclusion
Both test and reference formulations of abiraterone acetate tablets were bioequivalent in healthy Chinese subjects under fasting conditions.
Trial registration
ClinicalTrials.gov identifier NCT04863105, registered 26 April 2021—retrospectively registered (https://register.clinicaltrials.gov/prs/app/action/SelectProtocol?sid=S000ARAA&selectaction=Edit&uid=U00050YQ&ts=2&cx=-vbtjri
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Both test and reference formulations of abiraterone acetate tablets were bioequivalent under fasting conditions in healthy Chinese subjects, and both were well tolerated and safe. |
The availability of this drug may help increase access to abiraterone acetate for prostate cancer patients and reduce the burden on the Chinese health care system. |
1 Introduction
Prostate cancer is the second most common malignancy among men worldwide [1] and the fifth most common cause of death due to cancer worldwide [2]. In 2018, there were nearly 1,276,106 new cases of prostate cancer and 358,989 related deaths globally [1]. The etiology of prostate cancer is the subject of numerous studies and remains largely unknown compared with other common cancers [3]. Prostate cancer has considerable regional and age differences. It has also shown significant differences in morbidity and mortality among racial and ethnic groups [2].
Death due to prostate cancer is usually the result of metastatic castration-resistant prostate cancer (mCRPC), with a median survival of < 2 years historically [4]. MCRPC refers to the fact that prostate cancer continues to develop even when testosterone levels are reduced to very low levels [5]. Castration resistance in prostate cancer patients may have a number of different mechanisms. One of these is androgen receptor (AR) mutations, which are occasionally observed in patients with advanced disease [6]. The abiraterone acetate tablet, as an inhibitor of androgen synthesis, provides a novel approach to targeting AR pathways in men with CRPC. It significantly reduces androgen production by blocking the enzyme cytochrome P450 17 α-hydroxylase (CYP17) [6]. Oral abiraterone acetate tablets combined with prednisone are an important first-line choice for the treatment of mCRPC [7]. Although the abiraterone acetate tablet has been marketed in China, it is difficult for average patients to afford because of its high price. Therefore, the development of generic formulations has become an urgent need. The primary objective of this study was to evaluate the bioequivalence of abiraterone acetate tablets developed by Qilu Pharmaceutical Co., Ltd and abiraterone acetate tablets (Zecke®) produced by Patheon Inc., and to investigate its pharmacokinetics after a single fasting administration in healthy male volunteers.
2 Methods
2.1 Ethics
The ethical approval process of this trial was in accordance with the requirements of Good Clinical Practice (GCP) [8], the Helsinki Declaration [9], and relevant domestic laws and regulations. The study plan and informed consent were approved by the Medical Ethics Committee of the Affiliated Hospital of Qingdao University. All volunteers signed informed consent to be involved in the study before receiving the study drug.
2.2 Study Drugs
The abiraterone acetate tablet test product was obtained from Qilu Pharmaceutical Co., Ltd (batch number 17F0023DD9), and the reference product Zecke® (abiraterone acetate tablet) was purchased from Patheon Inc. (batch number VYCB).
2.3 Study Participants
The selection criteria for volunteers were (1) healthy male volunteers ≥18 years of age; (2) weight not less than 50 kg and body mass index between 19 and 28 kg/m2; (3) no unhealthy habits, such as smoking or alcohol consumption, and no history of drug abuse; and (4) participants who volunteered to participate in the clinical trial, understood the study procedures and signed written informed consent and were able to complete the study according to the test protocol.
The exclusion criteria were (1) abnormal results of vital sign assessments, physical examination, electrocardiogram and other laboratory examinations were clinically significant; (2) diseases of the central nervous system, the cardiovascular system, liver and kidney function, the digestive system (including acute gastroenteritis and diarrhea 2 weeks prior to drug delivery) and the respiratory system (not including upper respiratory infection 2 weeks prior to drug delivery), metabolic or skeletal system diseases, or any other diseases or physiological conditions that may have affected the results of the study; (3) a history of hospitalization or surgery within 3 months prior to the trial; (4) known active hepatitis B, human immunodeficiency virus (HIV), hepatitis C virus (HCV) or Treponema pallidum infection; (5) a history of specific allergic reactions (such as atopic dermatitis, and asthma), a history of allergy to drugs or biological agents, or a history of known allergy to the ingredients of this drug; (6) positive tests for alcohol during urine drug screening; (7) regular drinkers who consumed more than 14 units of alcohol per week (1 unit is 360 mL beer or 45 mL liquor of 40% alcohol, or 150 mL wine) during the 3 months prior to the trial; (8) smoking more than five cigarettes per day within 3 months before the trial; (9) participation as a subject in any drug clinical trial within 3 months prior to the trial; (10) people who had donated blood or lost more than 400 mL of blood during the 3 months before the experiment; (11) a history of prescription drug use during the 14 days prior to the trial; (12) a history of over-the-counter medicine use or consumption of any functional vitamins or herbal products within 48 h prior to the trial; (13) consumption of any xanthine-rich beverage or food, or grapefruit or products containing grapefruit, within 48 h prior to the test; (14) consumption of any food or drink containing caffeine (such as tea or coffee) and any alcoholic products within 48 h before the trial; (15) special requirements on diet and failure to follow the diet and corresponding regulations provided; and (16) volunteers who were considered by the investigator to exhibit poor compliance or any factors making them unsuitable for participating in the study.
2.4 Study Design
A single-center, open, randomized, single-dose, three-period, three-sequence, half-repeat (only repeated reference formulation), and reference formulation-corrected fasting reference-scaled average bioequivalence (RSABE) trial was conducted in the phase I Clinical Research Center of the Affiliated Hospital of Qingdao University. Thirty-seven healthy volunteers were enrolled in this study (one subject was replaced before initial administration). SAS v9.3 software was used to generate a random table, and the volunteers were randomly divided into three groups according to a 1:1:1 ratio. Subjects were randomly assigned to one of the three groups according to the random table (Table 1) generated before the subjects were enrolled. There was a minimum 7-day washout interval between dosing, and subjects took the study drug, according to a randomized schedule, on days 1, 8, and 15 of the trial. Qualified subjects were screened to enter the phase I ward 1 day before the first periods of administration, to participate in the baseline examination. Qualified subjects were admitted to the phase I ward and fasted overnight. Subjects were forbidden to drink water from 1 h before to 1 h after taking the medicine, and could drink water freely at other times. They fasted within 4 h after taking the medicine.
2.5 Blood Sampling and Analysis
During the three-period test, 4 mL of whole venous blood was collected at 0 h (within 1 h before administration) and 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 12, 24 and 48 h after administration and placed into an EDTA-K2 anticoagulant tube. The plasma concentration of abiraterone acetate tablets in plasma was determined by centrifugation. Blood samples were immediately placed in crushed ice or other refrigeration equipment after collection, placed in a precooled low temperature (2–8℃) centrifuge within 60 min, and centrifuged at 3500 rpm for 10 min. The time between the blood sample being collected and the plasma sample being placed in the refrigerator was < 120 min. Plasma samples from intensive blood collection sites were temporarily stored in a refrigerator at − 20 °C and transferred to a refrigerator at − 80 °C within 24 h. Plasma samples from non-intensive blood collection sites were stored directly in a − 80 °C refrigerator until being transported to the sample testing laboratory. Plasma samples were analyzed using the proven high-performance liquid chromatography-tandem mass spectrometry (LC‒MS/MS) method of Shanghai WuXi AppTec New Drug Development Co., Ltd. The LC‒MS/MS system consisted mainly of Shimadzu’s Shimadzu LC-30AD system and Applied Biosystems/SCIEX API 6500. Mobile phase A consisted of 0.1% FA aqueous solution containing 2 mM ammonium acetate, and mobile phase B consisted of 95% acetonitrile solution containing 2 mM ammonium acetate and 0.1% FA. The flow rate was set at 0.500 mL/min. The maximum intra- and interbatch accuracies of abiraterone detection were 4.7% and 4.3%, respectively, and the intrabatch accuracies ranged from 3.2 to 6.6%. The linear range of abiraterone concentration was 0.500–200 ng/mL and the lower limit of quantification was 0.500 ng/mL.
2.6 Safety Assessment
During the three periods, the vital signs of the enrolled subjects were measured (body temperature, pulse and blood pressure) before and 2, 8, 24 and 48 h after administration, as were the levels of electrolytes (potassium, sodium and phosphorus) 24 and 48 h after administration. Subjects were also asked about their subjective feelings to evaluate whether there were adverse reactions. Safety tests, including assessments of vital signs, physical examination, 12-lead electrocardiogram, routine blood tests, routine urine tests, and blood biochemical tests, were performed before subjects withdrew from the study. The clinical characteristics and the occurrence time, duration, treatment measures and severity of adverse reactions of subjects during the trial were recorded in detail.
2.7 Pharmacokinetic Analysis
Pharmacokinetic metrics of abiraterone were estimated using non-compartmental methods based on plasma concentrations using SAS v9.3. The pharmacokinetic metrics estimated included time to reach Cmax (Tmax), maximum plasma concentration (Cmax), area under the concentration-time curve from time zero to time t (AUCt) or AUC from time zero to infinity (AUC∞), and elimination half-life (T½), etc.
2.8 Statistical Analysis
SAS v9.3 software was used for statistical analysis. After logarithmic transformation of the major pharmacokinetic metrics (Cmax, AUCt and AUC∞), bidirectional and unidirectional T tests were performed to calculate the geometric mean ratios (GMRs) and 90% confidence intervals (CIs) of these major pharmacokinetic metrics. When the individual coefficient of variation (CVWR) of the pharmacokinetic metrics (AUCt, AUC∞ and Cmax) of reference formulations was < 30%, two formulations were considered bioequivalent if the 90% CIs of the GMRs of their main pharmacokinetic parameters were within a predefined acceptable range of 80–125%. When the CVWR of the pharmacokinetic metrics (AUCt, AUC∞ and Cmax) of the reference formulation was ≥ 30%, the reference formulation was used to correct for RSABE: if the Critbound of the pharmacokinetic metrics (AUCt, AUC∞ and Cmax) of the test and reference formulations was < 0 and the geometric average ratio was not beyond the range of 0.8000–1.2500, this method can be used to determine whether the test and reference formulations have bioequivalence.
3 Results
3.1 Characteristics of the Subjects
Figure 1 shows the features of the study subjects. A total of 79 subjects were screened for the study and a total of 37 healthy volunteers were enrolled in this clinical study, of whom 35 volunteers completed the whole study. One subject withdrew from the study and was replaced by another subject due to difficulties in blood collection before the first administration, and one subject withdrew voluntarily. All volunteers in this study were male. The patient demographic characteristics are shown in Table 2.

Study design and subject disposition, T test formulation, R reference formulation, PK pharmacokinetic, BE bioequivalence
3.2 Pharmacokinetics
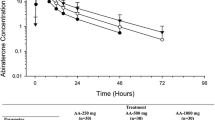
Tables 3 and 4 show the specific pharmacokinetic metrics or variance analysis results of the test and reference formulations of abiraterone acetate tablet under a fasting state. The mean concentration-time curves of plasma abiraterone acetate tablets after one fasting oral administration of the test formulation and two fasting oral reference formulations are shown in Fig. 2. Individual plasma concentration-time data on abiraterone acetate tablets are presented in the electronic supplementary file. The 90% CIs of the GMR of AUCt to AUC∞ of the test and reference formulations were in the range of 0.8000–1.2500; the CVWR of Cmax for the reference formulation was > 30%, the Critbound result was − 0.0522, and the GMR was between 0.8000 and 1.2500 (Table 5).

Average concentration-time curve under fasting conditions. T test formulation, abiraterone acetate tablet, 250 mg/tablet, 1 tablet; R reference formulation, Zecke® (abiraterone acetate tablet), 250 mg/tablet, 1 tablet
3.3 Safety Evaluation
A total of 18 adverse events (AEs) occurred in 11 subjects in this study. Of the 36 subjects enrolled in the safety analysis dataset, 17 AEs occurred in 10 subjects, all of which were mild AEs. After analysis, nine AEs in seven volunteers were found to be ‘probably related’ or ‘very likely related’ to the test drug and were considered adverse reactions. Eight AEs in six subjects were found to be ‘definitely not’ or ‘probably not’ related to the test drug (see Table 6 for details).
4 Discussion
Starting in 2006, the US FDA has proposed an average bioequivalence method for the reference formulation [10]. For bioequivalence studies, using the bioequivalence approach, the investigator may use either a partial copy (three-way cross, RRT or TRR) or a complete copy (four-way cross, RTRT or TRTR) design [11], and in this study a partial copy design was used (only the reference formulation was repeated). Because the abiraterone acetate tablet is an inhibitor of androgen synthesis, only male volunteers were selected for this study.
According to the National Medical Products Administration’s (NMPA) bioequivalence guidelines [12], “Postprandial bioequivalence studies may not be performed if the reference drug specification clearly states that the drug should be taken on an empty stomach only (1 hour before a meal or 2 hours after a meal to take)”, and the instructions for the use and dosage of the original drug used in this experiment indicate that “This product must be taken on an empty stomach, and taking it with food will increase the systemic exposure of this product at least 2 hours before and at least 1 hour after the administration of this product”. Therefore, only a fasting test was conducted in China in accordance with NMPA guidelines.
Studies have shown that the Cmax and AUCt values of abiraterone acetate tablets have high variability, i.e. 46.58% and 34.16%, respectively [13]. Assuming that the GMR of the test and reference formulations are 0.90 or 0.95, the degree of certainty is 0.80, α is 0.05, and the CVWR is 30% or 45%, and the maximum estimated sample size is 33 cases. Thirty-six healthy male volunteers participated in the study, and the possibility that the volunteers could drop out was considered, among other factors. Studies have shown that the plasma elimination half-life of abiraterone acetate tablets in healthy volunteers is 12 ± 5 h [13], therefore the washout period was designed to be 7 days per adjacent 2-week period to ensure that the drug was completely eluted before the second and third periods of administration.
No serious AEs were observed during the study period for either the test or reference formulations. All reported AEs were mild, therefore the test abiraterone acetate tablet (250 mg) and the reference formulation ‘Zecke®’ (abiraterone acetate tablets, 250 mg) are safer. Second, the results showed that the Tmax of abiraterone in the plasma of 35 volunteers was basically the same after oral administration of the test or reference formulations on an empty stomach, and the absorption rate and degree were similar. The 90% CIs of the GMR of AUCt and AUC∞ were in the range of 0.8000–1.2500, which was consistent with average bioequivalence (ABE) evaluation criteria. The CVWR of Cmax for the reference formulation exceeded 30%, therefore using the evaluation standard of RSABE, the Critbound result was − 0.0522 and the GMR was between 0.8000 and 1.2500. Therefore, the abiraterone acetate tablets (250 mg) and reference formulation ‘Zecke®’ (abiraterone acetate tablets, 250 mg) were judged to be bioequivalent formulations. The availability of generic formulations may help increase access to abiraterone acetate for prostate cancer patients and reduce the burden on the Chinese health care system. As this study was designed as a bioequivalence trial, pharmacokinetic parameters were obtained from individuals who were characterized as healthy adults. Therefore, further studies are needed to obtain more detailed pharmacokinetic parameters.
5 Conclusions
The results of this study showed that the 90% CIs of the GMR of AUCt to AUC∞ of the test and reference formulations were within the range of ABE evaluation criteria, and Cmax was consistent with the evaluation criteria of RSABE, indicating that test and reference formulations of abiraterone acetate tablets were bioequivalent under fasting conditions. In addition, both abiraterone acetate tablets had good safety and tolerability, and the absorption rate and degree of the two drugs were similar.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Pernar CH, Ebot EM, Wilson KM, Mucci LA. The epidemiology of prostate cancer. Cold Spring Harb Perspect Med. 2018;8(12): a030361.
Rawla P. Epidemiology of prostate cancer. World J Oncol. 2019;10(2):63–89.
Lowrance WT, Roth BJ, Kirkby E, Murad MH, Cookson MS. Castration-resistant prostate cancer: AUA Guideline Amendment 2015. J Urol. 2016;195(5):1444–52.
Swami U, McFarland TR, Nussenzveig R, Agarwal N. Advanced prostate cancer: treatment advances and future directions. Trends Cancer. 2020;6(8):702–15.
Rehman Y, Rosenberg JE. Abiraterone acetate: oral androgen biosynthesis inhibitor for treatment of castration-resistant prostate cancer. Drug Des Devel Ther. 2012;6:13–8.
Scott LJ. Abiraterone acetate: a review in metastatic castration-resistant prostrate cancer. Drugs. 2017;77(14):1565–76.
Takezawa M. Good clinical practice (GCP) in clinical trials [in Japanese]. Nihon Yakurigaku Zasshi. 2011;138(5):205–8.
General Assembly of the World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human volunteers. J Am Coll Dent. 2014;81(3):14–8.
Davit BM, Chen ML, Conner DP, Haidar SH, Kim S, Lee CH, et al. Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J. 2012;14(4):915–24.
Haidar SH, Davit B, Chen ML, Conner D, Lee L, Li QH, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res. 2008;25(1):237–41.
National Medical Products Administration (NMPA). Technical guidelines for the study of human bioequivalence of generic chemical drugs with pharmacokinetic metrics as the end point evaluation index. https://www.nmpa.gov.cn/zhuanti/ypqxgg/ggzhcfg/20160318210001633.html. Accessed 18 Mar 2016.
Wang C, Hu C, Gao D, Zhao Z, Chen X, Hu X, et al. Pharmacokinetics and bioequivalence of generic and branded abiraterone acetate tablet: a single-dose, open-label, and replicate designed study in healthy Chinese male volunteers. Cancer Chemother Pharmacol. 2019;83(3):509–17.
Acknowledgments
The authors are grateful to Qilu Pharmaceutical Co., Ltd, Shandong, China, for their support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study passed the Medical Ethics Committee of the Affiliated Hospital of Qingdao University review on 13 September 2017, and approval was obtained (no. QYFYEC 2017–057-01). Written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Availability of data and materials
The data supporting the conclusions of this article are included within the article and its additional file.
CONSORT guidelines
This study adheres to the Consolidated Standards of Reporting Trials (CONSORT) guidelines and includes a completed CONSORT checklist as an additional file.
Competing interests
Zhao-xin Wu, Chen-jing Wang, Ping Shi, Yan-ping Liu, Ting Li, Fei-fei Sun, Yao Fu, Xiao-meng Gao, Ya-ping Ma, Yu Cao declare no conflicts of interest.
Funding
Test execution and data collection in this work were supported by grants from the National Major Science and Technology Projects of China Special Project for ‘Significant New Drugs Development’ (2020ZX09201-018, 2020ZX09201-025, 2017ZX09304-024).
Authors’ contributions
This study was designed by YC. CJW and ZXW contributed equally to this work. PS, YPL,TL, FFS, YF, YPM and XMG performed the research, and all authors read and approved the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wu, Zx., Wang, Cj., Shi, P. et al. Pharmacokinetics and Bioequivalence of Abiraterone Acetate Tablets in Healthy Chinese Volunteers: An Open, Randomized, Single-Dose, Three-Period, Three-Sequence Crossover Study. Drugs R D 23, 121–127 (2023). https://doi.org/10.1007/s40268-023-00418-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-023-00418-6