
Abstract
Intranasal drug administration is a commonly used route for therapeutic formulations, but there may be challenges associated with a lack of absorption and bioavailability, as well as damage to mucosal tissue. To address these issues, potential absorption enhancers that are generally nonirritating to nasal mucosal tissue have been investigated as excipients in intranasal formulations. Among those studied are alkylsaccharides, which are composed of sugars covalently coupled to at least one alkyl chain. Alkylsaccharides have been shown to be nontoxic and have been used in food products as emulsifiers. In clinical trials, alkylsaccharide excipients have demonstrated substantially increased absorption of therapeutic agents across mucosal membranes and have been shown to be applicable to a wide range of types of molecules and molecular weights. Because they are water and oil soluble, alkylsaccharide excipients can be used in formulations with both hydrophilic and hydrophobic drugs. They are also effective in safely stabilizing protein therapeutics. An example of an alkylsaccharide excipient is dodecyl maltoside (Intravail®; 511 Da, stable long term when stored cold), which provides absorption enhancement by paracellular and transcellular routes. Dodecyl maltoside has been shown to be generally nonirritating to the nose and to promote systemic bioavailability. Dodecyl maltoside is used in US Food and Drug Administration-approved intranasal formulations of sumatriptan for migraine headaches and diazepam nasal spray for patients with epilepsy with acute seizure clusters.
Plain Language Summary
Nasal sprays can offer an easy and efficient route to deliver drugs. The nose can absorb drugs into the bloodstream quickly, and sprays require less patient training than injection or rectal treatments. However, the nasal cavity is small, and the spray must be concentrated and easily absorbed. Alkylsaccharides may enhance absorption in the nose. They have a good safety profile and are used in foods. Intravail® alkylsaccharides include dodecyl maltoside and tetradecyl maltoside. Nasal medications with dodecyl maltoside had drug concentrations and safety similar to injectable forms. Dodecyl maltoside is used in US Food and Drug Administration-approved nasal sprays for migraine (sumatriptan [Tosymra®]) and seizure clusters (diazepam [Valtoco®]). Dodecyl maltoside is also being tested in nasal sprays with other drugs that are absorbed too poorly or slowly on their own. With these new formulations, doctors and patients have more treatment choices and can select the best option.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Intranasal drug administration is an attractive route for therapeutic formulations, but there may be challenges associated with a lack of absorption and bioavailability. |
Dodecyl maltoside is a proprietary alkylsaccharide that can be used with both hydrophilic and hydrophobic drugs and has demonstrated improved delivery of small molecules, peptides, and proteins and improved bioavailability of drugs for nasal administration, addressing the challenges of bioavailability of intranasal formulations. |
Two intranasal formulations using dodecyl maltoside have been approved by the US Food and Drug Administration (sumatriptan nasal spray for migraine and diazepam nasal spray for epilepsy seizure clusters), and dodecyl maltoside is recognized as generally safe for oral administration and may have potential in oral and other pharmaceutical applications. |
1 Introduction
The nasal cavity can be an attractive target for systemic drug delivery because of the potential ease of administration through nasal spray devices [1], as shown through experience with such drugs as intranasal (IN) corticosteroids for allergic rhinitis [2]. Although most of the current drugs for IN administration are topical therapies, this route of delivery can provide systemic exposure, via highly vascularized and relatively permeable mucosa, while reducing gastrointestinal side effects and eliminating first-pass metabolism, thus providing an alternative to oral or parenteral administration [3,4,5,6].
Intranasal formulations have the potential for rapid absorption and onset of action [4, 7], which is ideal for medical conditions requiring rapid-acting drugs, such as migraines, other types of pain, seizure emergencies, panic attacks, cardiovascular events, and narcotic overdose [8, 9]. Rapid initiation also makes IN formulations useful alternatives to intravenous drugs in emergency departments [10] and for administration outside a hospital setting. Additional advantages of IN administration include the potential for direct delivery to the brain, bypassing the blood–brain barrier [11,12,13]. Formulation is a key determinant of the performance of a drug and therapeutic effectiveness, and water-insoluble agents, as well as hydrophilic substances with low permeability, present challenges that may necessitate tailored strategies [6, 14]. In particular, absorption-enhancing excipients may be important for achieving therapeutic drug concentrations; however, substantial nasal mucosal toxicity has been shown to occur with many potential agents [15, 16]. The objective of this review is to summarize the data from studies examining the use of the excipient dodecyl maltoside (DDM; Intravail®) to improve the rapid absorption and delivery of acute-care medications for the treatment of central nervous system disorders including cluster seizures or migraine headaches.
2 Nasal Anatomy
The physical anatomy of the nose can be exceptionally conducive to drug delivery because there is sufficient surface area within the nose, approximately 160 cm2, with approximately 15 mL of total cavity volume [17]. Nasal cavity microvilli protrusions of the cellular membrane increase the surface area of cells and facilitate the absorption of drugs [17]. There is potential for nose-to-brain delivery owing to the presence of the first (olfactory) and fifth (trigeminal) cranial nerves via intracellular, transcellular, and paracellular transport [17, 18]. Although olfactory filaments reach through the nasal mucosal surface, those of the trigeminal nerve are beneath the line of tight junctions [18, 19].
2.1 Obstacles to IN Administration
The nasal anatomy may present obstacles that can limit IN drug administration. The volume of a drug formulation that can be administered in one dose is limited to 100–150 μL because of the size and anatomy of the nasal cavity [4]. Intranasal administration also may be limited by the function of ciliated cells that rapidly sweep the mucus coat to the throat in a process called ciliary clearance, providing a short window for absorption of nasal formulations (15–20 min) [17, 20]. Additionally, molecule size has a role in absorption. In the natural state, nasal mucosal absorption decreases as molecular weight increases, with a cut-off point of 1000 Da, effectively excluding larger molecules [21, 22]. Additionally, lipophilicity is an important factor controlling diffusion, especially for smaller molecules [4]. Generally, drugs of greater lipophilicity will diffuse across biological membranes faster than drugs of lower lipophilicity [15].
2.2 The Role of Absorption Enhancers
Administration of IN formulations that include absorption enhancers in the form of excipients may increase permeation and allow for larger sizes of absorbable molecules up to 30,000 Da [22, 23]. However, it has been a challenge to identify nonirritating excipients with adequate solubility. Hundreds of nasal absorption enhancers have been tested, but almost all of them have been shown to cause substantial or serious damage to the nasal mucosa [19, 23,24,25].
3 Alkylsaccharide Excipients
Alkylsaccharides are compounds that consist of sugars covalently coupled to at least one alkyl chain [23]. Alkylsaccharides are safe, nontoxic, odorless, tasteless, nonmutagenic, and nonsensitizing in the Draize test at concentrations of up to 25%; they are commonly used as food emulsifiers [23]. Some alkylsaccharides have shown promise as absorption enhancers [23].
Dodecyl maltoside (511 Da) and tetradecyl maltoside (TDM; 539 Da) are closely related longer-chain, efficient alkylsaccharide absorption enhancers (collectively designated as Intravail®; Neurelis, Inc.; San Diego, CA, USA) [23, 26]. Dodecyl maltoside and TDM are proprietary transmucosal-absorption enhancers. Maltose is a diglucose that metabolizes to glucose, and DDM also includes dodecanol, a fatty alcohol that is an organic compound manufactured from palm or coconut oil, which metabolizes to the fatty acid lauric acid, a common component of triglycerides [23]. The C12 and C14 alkyl chains of DDM and TDM, respectively, provide maximal and nearly identical absorption enhancement effects [24]. In contrast, longer and shorter alkyl chains are substantially ineffective [24, 27]. In addition to its use in nasal sprays, DDM has applications in oral routes of administration [23]. Although some preclinical in vitro toxicity experiments of DDM have produced equivocal results, clinical studies of drug formulations with DDM have detected no remarkable mucosal toxicity [24, 28,29,30,31,32]. One in vitro study making quantitative comparisons relative to Triton X-100, which served as a cytotoxic control, detected lactate dehydrogenase release, a measure of toxicity (20% considered nontoxic [24]), of 24–28% for 0.1% concentrations for 1 h (approximately four-fold longer than the time of mucociliary clearance) outside the nasal environment [23, 24]. In an in vitro study, DDM enhanced the permeability of insulin in human adenocarcinoma cells (T-84 and Caco-2) without causing damage to the cells [32].
In vitro studies may provide useful comparative data in analyzing multiple absorption enhancement candidates. However, in addition to longer than normal exposure times, in vitro studies often rely on static contact between test cells and the aqueous media containing the test substance, typically in microtiter wells, which does not reflect the natural dynamic biological environment found in healthy airway surfaces. These surfaces are lined with continuously beating ciliated epithelial cells and covered with a two-component surface layer comprising a mucus layer that entraps inhaled particles and foreign pathogens and a low-viscosity periciliary layer that lubricates and continuously cleans the airway surface [33]. Dodecyl maltoside is generally recognized as safe for oral administration [23].
Another relevant property of alkylsaccharide excipients is their ability to stabilize protein therapeutics and reduce aggregation owing to low critical micelle concentrations [34]. By binding to hydrophobic regions on the protein surface, nonionic surfactants like DDM/TDM ultimately reduce intermolecular protein interactions to confer surface-induced anti-aggregation activities [35]. Dodecyl maltoside has been shown to inhibit the aggregation of peptides [34, 35] and proteins [36], while also increasing the stability and solubilization of lyophilized peptides [36]. Other nonionic surfactants, including the polysorbate PS-20, have been considered in protein pharmaceutical formulations as IN surfactants; however, recombinant human interferon β-1b has been shown to be less stable and more prone to aggregation by light with PS-20 compared with DDM [35]. Polysorbates, which contain ether linkages that rapidly auto-oxidize into peroxides, epoxy acids, and reactive aldehydes, may induce immunogenicity [37]. Dodecyl maltoside and other absorption enhancers were evaluated as surfactants to overcome the challenges of solubility, bioavailability, and tolerability of IN formulations [16]. Anesthetized male Sprague-Dawley rats were given 36 mg/mL of Homo sapiens-derived antibody Fc fragment at pH 7.4 as a component of the formulations of the surfactants. The liquid formulations were nasally administered over 1 min as a 50-mL bolus via microcannula inserted into the nostril. After 20 min, cardiac puncture was performed, and brain and other tissues were collected and analyzed for the Homo sapiens-derived antibody Fc fragment content. Dodecyl maltoside had the highest increase in nose-to-brain concentration with a small range of variability and minimal or mild nasal toxicity (Table 1) [16].
4 Alkylsaccharide/DDM Mechanism of Action
Because DDM is soluble both in water and oils, it can be used in the formulations of hydrophilic and hydrophobic drugs [38]. Two mechanisms of action for absorption enhancement with DDM have been suggested, through paracellular and transcellular routes [38]. First, as with many alkylsaccharides, it promotes transient loosening of tight cell junctions that allows permeation of the mucosal barrier [23], as shown in Caco-2 intestinal cells [39], human bronchial epithelial cells [40], T-84 and Caco-2 human carcinoma cells [32], and epithelial tissue [24]. This loosening of junctions is transient, and as the tight junctions close, they block progressively smaller molecules (Fig. 1) [23]. The alkylsaccharide TDM has been shown to work long enough to facilitate drug absorption, after which the normal protective epithelial layer is restored [41]. Second, transcellular transport via vesicle carriers also has been demonstrated with TDM in rats (Fig. 2) [23] and with DDM in a porcine jejunal mucosal explant system [38, 42]. Dodecyl maltoside enables the noninvasive delivery of a broad range of small-molecule drugs, peptides, and therapeutic proteins, through permeation of the nasal mucosal barrier that is controlled and transient without irritation [38].

Loosening of tight mucosal junctions is reversed within 2 h after application of calcitonin with tetradecyl maltoside and somatropin with tetradecyl maltoside. The top line shows the drugs and tetradecyl maltoside administered together; other lines show tetradecyl maltoside administered alone first and drugs 60 and 120 min thereafter [41]. For calcitonin, the smaller of the two drugs (4 kDa), absorption is reduced after 60 and 120 min, indicating that the tight junctions are still partially open. In the case of somatotropin (22 kDa), the junctions are sufficiently closed at 60 min to prevent drug absorption. Reprinted from Journal of Pharmaceutical Sciences, 99 (4), John J. Arnold, Michael D. Fyrberg, Elias Meezan, Dennis J. Pillion, Reestablishment of the Nasal Permeability Barrier to Several Peptides Following Exposure to the Absorption Enhancer Tetradecyl-β-D-Maltoside, pages 1912-1920, Copyright (2010), with permission from Elsevier

Adapted from Journal of Pharmaceutical Sciences, 99 (4), John J. Arnold, Michael D. Fyrberg, Elias Meezan, Dennis J. Pillion, Reestablishment of the Nasal Permeability Barrier to Several Peptides Following Exposure to the Absorption Enhancer Tetradecyl-β-D-Maltoside, pages 1912-1920, Copyright (2010), with permission from Elsevier
Electron microscopy showing rat mucosa before and 10 min after administration of tetradecyl maltoside 0.125% showing vesicle formation [41]. cc ciliated cell, gc goblet cell, n nucleus, v vesicle.
5 Preclinical Studies with IN Formulations
In animal studies, TDM was found to increase the IN absorption of calcitonin [25] and of the low-molecular-weight heparins enoxaparin and dalteparin [43, 44]. In the calcitonin study, levels were measured in rats at various times after nasal administration of the formulation with TDM or the controls with saline or 0.125% octylmaltoside. Alkyl chain length is associated with a balance between hydrophilic and lipophilic characteristics as well as critical micelle concentrations. Octylmaltoside, as a medium-chain alkylsaccharide, exhibits lower lipophilicity compared with the more lipophilic long-chain alkylsaccharide TDM [25]. The controls provided little or no absorption, whereas calcitonin levels were increased with 0.125% or 0.25% TDM formulations [25]. One low-molecular-weight heparin study compared rats administered enoxaparin or dalteparin in formulations with or without TDM (0.25%). The formulations with TDM showed a significant increase in area under the curve and peak plasma concentration for anti-Factor Xa activity, higher bioavailability, and rapid reversibility compared with saline alone [43]. In a subsequent study, rats were administered nasal formulations with different combinations of enoxaparin and four alkylmaltosides, including DDM and TDM. Alkylmaltosides increased absorption in a dose-dependent manner, with rapid reversibility [44].
6 Clinical Trials of IN Formulations with DDM
6.1 Drugs Currently Approved by the US FDA
6.1.1 Sumatriptan Nasal Spray with DDM (Tosymra ® )
Dodecyl maltoside is used as an excipient in US Food and Drug Administration (FDA)-approved medications. Sumatriptan nasal spray with DDM is approved for the treatment of acute migraine headaches with or without aura in adults [45]. Based on its components, and that it breaks down to glucose and a natural fatty acid, DDM might be predicted to have a benign safety profile, and this has been demonstrated in clinical trials in which DDM has been combined with therapeutic agents. In a double-blind, placebo-controlled, two-period study of sumatriptan 10 mg with 0.20% DDM in 107 patients with migraine [46], patients administered IN sumatriptan with a single-use device delivering a 100-µL spray and recorded outcomes in an eDiary. Approximately 2% of patients in the first period and 2.7% in the second period experienced application-site pain with active medication vs none for both periods with placebo [46]. In an open-label single-dose study of diazepam nasal spray with DDM in 57 patients experiencing seizures under seizure and nonseizure conditions, 3.5% of patients reported nasal discomfort [29]. In a 6-month safety study of IN sumatriptan 10 mg with DDM, 51 of 167 (30.5%) patients experienced nasal burning or stinging at least once, usually mild. Application-site reaction and irritation were modest: 5.4% and 4.2%, respectively [30]. In an interim analysis of a long-term (12-month) phase III safety study of diazepam nasal spray with DDM, mild and transient nasal discomfort was reported in 5.3% of patients [47]. Scores for nasal irritation, rated by a trained observer, were similarly mild and transient, and there were no clinically relevant olfactory changes [47]. These studies did not compare the active drug containing DDM with DDM alone; thus, whether any of these adverse events were related to DDM was not determined.
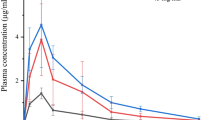
In a randomized, preapproval three-way crossover study (n = 18) comparing sumatriptan 10 mg with 0.20% DDM vs IN sumatriptan 20 mg without DDM, absorption was much more rapid with the DDM formulation (time to maximum plasma concentration [tmax] 15 min for the DDM formulation monodose, 10.2 min for the DDM formulation multidose, and 2 h for the IN sumatriptan 20-mg dose without DDM [with an earlier peak reflecting nasal absorption and a second peak suggesting absorption of a swallowed portion of the dose across intestinal mucosa]; Fig. 3) [31]. In a phase I, open-label, randomized, single-dose, three-way crossover study of relative bioavailability, pharmacokinetic outcomes were compared for IN sumatriptan with DDM (n = 73) and 4 and 6 mg of subcutaneous sumatriptan without DDM (n = 75 each) in healthy subjects [31]. The median tmax was significantly faster with IN sumatriptan with DDM (10 min [range 5–23 min]) than with subcutaneous sumatriptan 4 mg (15 min [range 5–30 min]) or sumatriptan 6 mg (15 min [range 5–26 min]; p < 0.0001 for IN sumatriptan vs both) [31].

Change in sumatriptan plasma concentration over time after administration of intranasal sumatriptan with dodecyl maltoside (DDM) or intranasal sumatriptan without DDM [31]. Cp plasma concentration. Reprinted from Headache: The Journal of Head and Face Pain, 56(9), Sagar Munjal, Anirudh Gautam, Elliot Offman, et al., A Randomized Trial Comparing the Pharmacokinetics, Safety, and Tolerability of DFN‐02, an Intranasal Sumatriptan Spray Containing a Permeation Enhancer, With Intranasal and Subcutaneous Sumatriptan in Healthy Adults, Copyright (2016), with permission from John Wiley and Sons
6.1.2 Diazepam Nasal Spray with DDM (Valtoco ® )
Diazepam nasal spray with DDM is approved for the acute treatment of intermittent stereotypic seizure clusters in patients with epilepsy 6 years of age and older [48]. In a preapproval, open-label crossover pharmacokinetic study of 24 healthy volunteers comparing diazepam nasal spray with diazepam IN suspension and intravenous diazepam, diazepam 10-mg nasal spray with DDM had an absolute bioavailability of 97% compared with 67% for the 10-mg IN suspension and similar variability in area under the curve to that of intravenous diazepam (Fig. 4) [49]. In an open-label randomized crossover study in 48 healthy subjects, overall variability in area under the curve from time 0 to infinity and peak plasma concentration were lower with diazepam nasal spray than with diazepam rectal gel [28]. Measurable plasma concentrations of diazepam were seen for diazepam nasal spray with DDM by the first measurement point (10 min) [28]. In 57 patients with epilepsy with seizure clusters, the pharmacokinetics of diazepam nasal spray with DDM was found to be similar in the seizure and nonseizure state [29].

Concentration–time profiles (mean ± standard deviation; only positive error bars are shown for visual clarity) after administration of diazepam nasal solution, suspension, and intravenous diazepam [49]. Reprinted from Epilepsy Research, 105(3), Suresh K. Agarwal, Robert L. Kriel, Richard C. Brundage, Vijay D. Ivaturi, James C. Cloyd, A Pilot Study Assessing the Bioavailability and Pharmacokinetics of Diazepam After Intranasal and Intravenous Administration in Healthy Volunteers, 362–367, Copyright (2013), with permission from Elsevier
6.2 Drugs in Development
Nalmefene nasal spray with DDM is in development for the treatment of synthetic opioid overdose. The pharmacokinetics of IN administration of the opiate antagonist nalmefene 3 mg with or without DDM and intramuscular nalmefene 1.5 mg was compared in healthy volunteers (n = 14) [50]. The tmax of IN nalmefene with DDM was 15 min compared with 2 h for IN nalmefene alone and 20 min for intramuscular nalmefene. Peak plasma concentration was increased 2.2-fold with IN nalmefene with DDM (4.45 ng/mL) vs IN nalmefene without DDM (1.99 ng/mL). Area under the curve from time 0 to the last quantifiable concentration was increased to 15.2 ng·h/mL for IN nalmefene with DDM vs 12.7 ng·h/mL for IN nalmefene without DDM. Adverse events were mild, with no changes in sense of smell noted [50].
Naltrexone nasal spray with DDM is in development to treat alcohol use disorder. In a study of healthy volunteers (n = 14), the addition of DDM to IN naltrexone decreased tmax from 30 to 10 min and increased peak plasma concentration approximately three-fold (Fig. 5). Adverse events were generally mild, and nasal irritation scores were clinically normal throughout the study [9].

Plasma concentrations of naltrexone following intranasal administration with and without dodecyl maltoside (DDM) [9]. Reprinted from The Journal of Clinical Pharmacology, 59(7), Philip Krieter, Shwe Gyaw, C. Nora Chiang, et al., Enhanced Intranasal Absorption of Naltrexone by Dodecyl Maltopyranoside: Implications for the Treatment of Opioid Overdose, Copyright (2019), with permission from John Wiley and Sons
7 Discussion
Intranasal administration is an appealing option that provides ease of administration for drugs that require a rapid onset of action as well as for biologics that otherwise must be administered by injection. A potential challenge to IN drug administration is the ability to increase absorption through the nasal mucosa, particularly with large-molecule and lipid-soluble drugs, without causing nasal toxicity, and alkylsaccharides may address this need.
Alkylsaccharide excipients such as DDM are soluble in water and oils and are generally nonirritating and nondamaging to mucosal tissue [38]. These excipients markedly enhance absorption to address suboptimal bioavailability or route-of-administration issues with minimal administration-site adverse effects. Dodecyl maltoside has been shown to be a functional absorption-enhancing excipient that transiently and reversibly loosens tight junctions between cells as well as functioning by transcellular routes [23]. Studies of drug formulations with low DDM concentrations (< 0.5%) have demonstrated improved bioavailability following nasal administration of a wide range of molecules up to 30,000 Da, including therapeutic proteins, peptides, and small-molecular-weight molecules, through increased absorption across mucosal membranes [22, 23]. Alkylsaccharide excipients also help stabilize protein therapeutics by safely inhibiting protein aggregation, [34,35,36] potentially decreasing the formation of neutralizing proteins [34, 36].
Two IN formulations using DDM have been approved by the FDA: sumatriptan nasal spray for migraine and diazepam nasal spray for epilepsy seizure clusters. Sumatriptan with DDM has been shown to be more rapidly absorbed than without DDM [31]. Absolute bioavailability of the IN diazepam formulation in healthy volunteers was 97%, and the drug was generally well tolerated with less intrapatient variability than rectal diazepam [28, 49]. Additional IN formulations with DDM are currently being investigated.
8 Conclusions
The nasal route of administration can provide important advantages for access and speed of drug delivery in acute situations and emergencies; however, rapid absorption and optimized bioavailability present a challenge to IN drug formulations. The potent and proprietary alkylsaccharide absorption enhancer DDM has demonstrated improved delivery of small molecules, peptides, and proteins, as well as bioavailability of drugs for nasal administration, addressing the challenges of bioavailability of IN formulations. Because DDM is soluble both in water and oils, it can be used in the formulations of hydrophilic and hydrophobic drugs. Furthermore, it inhibits the aggregation of proteins and peptides, with implications for drug stability and reduced immunogenicity, and avoids the auto-oxidization of ether linkages in polysorbate surfactants that contribute to formulation-associated anaphylaxis for many protein therapeutics. This is particularly relevant today considering the rapidly growing role of protein therapeutics, in particular, monoclonal antibodies, in treating a broad spectrum of cancers and immune system-related diseases. The accepted safety of DDM, despite its recent introduction, is confirmed by its status as being generally recognized as safe for oral administration and by the FDA approval of the nasally administered drugs sumatriptan and diazepam. Additional nasal formulations are in development for emergency and non-emergency use in the community setting, and DDM may have potential in oral, buccal, dermal, and other pharmaceutical applications as well.
References
Djupesland PG. Nasal drug delivery devices: characteristics and performance in a clinical perspective: a review. Drug Deliv Transl Res. 2013;3(1):42–62.
Fromer LM, Ortiz GR, Dowdee AM. Assessment of patient attitudes about mometasone furoate nasal spray: the ease-of-use patient survey. World Allergy Organ J. 2008;1(9):156–9.
Bitter C, Suter-Zimmermann K, Surber C. Nasal drug delivery in humans. Curr Probl Dermatol. 2011;40:20–35.
Costantino HR, et al. Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm. 2007;337(1–2):1–24.
Leonard AK, et al. In vitro formulation optimization of intranasal galantamine leading to enhanced bioavailability and reduced emetic response in vivo. Int J Pharm. 2007;335(1–2):138–46.
Forbes B, et al. A consensus research agenda for optimising nasal drug delivery. Expert Opin Drug Deliv. 2020;17(2):127–32.
Li L, et al. Rapid-onset intranasal delivery of anticonvulsants: pharmacokinetic and pharmacodynamic evaluation in rabbits. Int J Pharm. 2000;199(1):65–76.
Touitou E, Illum L. Nasal drug delivery. Drug Deliv Transl Res. 2013;3(1):1–3.
Krieter P, et al. Enhanced intranasal absorption of naltrexone by dodecyl maltopyranoside: implications for the treatment of opioid overdose. J Clin Pharmacol. 2019;59(7):947–57.
Rech MA, et al. When to pick the nose: out-of-hospital and emergency department intranasal administration of medications. Ann Emerg Med. 2017;70(2):203–11.
Meredith ME, Salameh TS, Banks WA. Intranasal delivery of proteins and peptides in the treatment of neurodegenerative diseases. AAPS J. 2015;17(4):780–7.
Kozlovskaya L, Abou-Kaoud M, Stepensky D. Quantitative analysis of drug delivery to the brain via nasal route. J Control Release. 2014;189:133–40.
Born J, et al. Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci. 2002;5(6):514–6.
Ghadiri M, Young PM, Traini D. Strategies to enhance drug absorption via nasal and pulmonary routes. Pharmaceutics. 2019;11(3):113.
Cloyd J, et al. Overcoming the challenges of developing an intranasal diazepam rescue therapy for the treatment of seizure clusters. Epilepsia. 2021;62(4):846–56.
Maggio ET, Inventor; Aegis Therapeutics, LLC, assignee. Intranasal administration of active agents to the central nervous system. US patent US 8,883,728 B2, 2014.
Gizurarson S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr Drug Deliv. 2012;9(6):566–82.
Djupesland PG, Messina JC, Mahmoud RA. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv. 2014;5(6):709–33.
Bruinsmann FA, et al. Nasal drug delivery of anticancer drugs for the treatment of glioblastoma: preclinical and clinical trials. Molecules. 2019;24(23):4312.
Pires A, et al. Intranasal drug delivery: how, why and what for? J Pharm Pharm Sci. 2009;12(3):288–311.
McMartin C, et al. Analysis of structural requirements for the absorption of drugs and macromolecules from the nasal cavity. J Pharm Sci. 1987;76(7):535–40.
Huang Y, Donovan MD. Large molecule and particulate uptake in the nasal cavity: the effect of size on nasal absorption. Adv Drug Deliv Rev. 1998;29(1–2):147–55.
Maggio ET, Pillion DJ. High efficiency intranasal drug delivery using Intravail® alkylsaccharide absorption enhancers. Drug Deliv Transl Res. 2013;3(1):16–25.
Chen-Quay SC, et al. Identification of tight junction modulating lipids. J Pharm Sci. 2009;98(2):606–19.
Ahsan F, et al. Enhanced bioavailability of calcitonin formulated with alkylglycosides following nasal and ocular administration in rats. Pharm Res. 2001;18(12):1742–6.
Pillion DJ, et al. Insulin delivery in nosedrops: new formulations containing alkylglycosides. Endocrinology. 1994;135(6):2386–91.
Pillion DJ, et al. Synthetic long-chain alkyl maltosides and alkyl sucrose esters as enhancers of nasal insulin absorption. J Pharm Sci. 2002;91(6):1456–62.
Hogan RE, et al. Bioavailability and safety of diazepam intranasal solution compared to oral and rectal diazepam in healthy volunteers. Epilepsia. 2020;61(3):455–64.
Hogan RE, et al. Pharmacokinetics and safety of VALTOCO (NRL-1; diazepam nasal spray) in patients with epilepsy during seizure (ictal/peri-ictal) and nonseizure (interictal) conditions: a phase 1, open-label study. Epilepsia. 2020;61(5):935–43.
Munjal S, et al. A multicenter, open-label, long-term safety and tolerability study of DFN-02, an intranasal spray of sumatriptan 10 mg plus permeation enhancer DDM, for the acute treatment of episodic migraine. J Headache Pain. 2017;18(1):31.
Munjal S, et al. A randomized trial comparing the pharmacokinetics, safety, and tolerability of DFN-02, an intranasal sumatriptan spray containing a permeation enhancer, with intranasal and subcutaneous sumatriptan in healthy adults. Headache. 2016;56(9):1455–65.
Tirumalasetty PP, Eley JG. Evaluation of dodecylmaltoside as a permeability enhancer for insulin using human carcinoma cells. J Pharm Sci. 2005;94(2):246–55.
Bustamante-Marin XM, Ostrowski LE. Cilia and mucociliary clearance. Cold Spring Harb Perspect Biol. 2017;9(4):a028241.
Maggio ET. Use of excipients to control aggregation in peptide and protein formulations. J Excip Food Chem. 2010;1(2):40–9.
Mahjoubi N, et al. Effect of nonionic surfactants (dodecyl maltoside and polysorbate 20) on prevention of aggregation and conformational changes of recombinant human IFNbeta_1b induced by light. Iran J Pharm Res. 2017;16(1):103–11.
Rifkin RA, et al. n-Dodecyl-beta-D-maltoside inhibits aggregation of human interferon-beta-1b and reduces its immunogenicity. J Neuroimmune Pharmacol. 2011;6(1):158–62.
Maggio ET. Alkylsaccharides: circumventing oxidative damage to biotherapeutics caused by polyoxyethylene-based surfactants. Ther Deliv. 2013;4(5):567–72.
Maggio ET. Absorption enhancing excipients in systemic nasal drug delivery. J Excip Food Chem. 2014;5(2):100–12.
Gradauer K, et al. Dodecylmaltoside modulates bicellular tight junction contacts to promote enhanced permeability. Mol Pharm. 2017;14(12):4734–40.
Ahsan F, et al. Effects of the permeability enhancers, tetradecylmaltoside and dimethyl-beta-cyclodextrin, on insulin movement across human bronchial epithelial cells (16HBE14o-). Eur J Pharm Sci. 2003;20(1):27–34.
Arnold JJ, et al. Reestablishment of the nasal permeability barrier to several peptides following exposure to the absorption enhancer tetradecyl-beta-d-maltoside. J Pharm Sci. 2010;99(4):1912–20.
Danielsen EM, Hansen GH. Probing the action of permeation enhancers sodium cholate and N-dodecyl-β-D-maltoside in a porcine jejunal mucosal explant system. Pharmaceutics. 2018;10(4):172.
Arnold J, et al. Nasal administration of low molecular weight heparin. J Pharm Sci. 2002;91(7):1707–14.
Mustafa F, et al. Chain length-dependent effects of alkylmaltosides on nasal absorption of enoxaparin. J Pharm Sci. 2004;93(3):675–83.
Upsher-Smith Laboratories, LLC. Tosymra (sumatriptan). Full Prescribing Information. Maple Grove, MN: Upsher-Smith Laboratories, LLC; 2019.
Lipton RB, et al. DFN-02 (sumatriptan 10 mg with a permeation enhancer) nasal spray vs placebo in the acute treatment of migraine: a double-blind, placebo-controlled study. Headache. 2018;58(5):676–87.
Wheless JW, et al. Safety of Valtoco (NRL-1; diazepam nasal spray) in patients with epilepsy: interim results from a phase 3, open-label, 12-month repeat dose study. Presented at: 73rd American Epilepsy Society Annual Meeting, December 6-10, 2019; Baltimore, MD.
Neurelis, Inc. Valtoco (diazepam nasal spray). Full Prescribing Information. San Diego, CA: Neurelis, Inc.; 2021.
Agarwal SK, et al. A pilot study assessing the bioavailability and pharmacokinetics of diazepam after intranasal and intravenous administration in healthy volunteers. Epilepsy Res. 2013;105(3):362–7.
Krieter P, et al. Fighting fire with fire: development of intranasal nalmefene to treat synthetic opioid overdose. J Pharmacol Exp Ther. 2019;371(2):409–15.
Acknowledgements
Medical writing support was provided at the direction of the authors by Laura J. Herold of The Curry Rockefeller Group, LLC, which also provided additional editorial assistance including formatting and proofreading. Development of this manuscript was supported by Neurelis, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Development of this manuscript was supported by Neurelis, Inc., which also sponsored open access.
Conflict of Interest
Adrian L. Rabinowicz is an employee of and has received stock options from Neurelis, Inc. Enrique Carrazana is an employee of and has received stock and stock options from Neurelis, Inc. Edward T. Maggio is a consultant to Neurelis, Inc.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Data sharing is not applicable to this article; no datasets were generated or analyzed during the current study.
Code Availability
Not applicable.
Author Contributions
ETM provided initial interpretation of the studies. All authors provided additional interpretation, developed the initial outline, and contributed to the manuscript. All authors meet International Committee of Medical Journal Editors criteria for authorship. All authors approved the final version of this manuscript and agreed to submit to Drugs in R&D.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Rabinowicz, A.L., Carrazana, E. & Maggio, E.T. Improvement of Intranasal Drug Delivery with Intravail® Alkylsaccharide Excipient as a Mucosal Absorption Enhancer Aiding in the Treatment of Conditions of the Central Nervous System. Drugs R D 21, 361–369 (2021). https://doi.org/10.1007/s40268-021-00360-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-021-00360-5