
Abstract
Nimodipine has been shown to improve outcomes following aneurysmal subarachnoid hemorrhage. Guidelines recommend that all patients receive a fixed dose of oral nimodipine for 21 days. However, pharmacokinetic studies have suggested variability of nimodipine pharmacokinetics in subarachnoid hemorrhage and in other patient populations. The clinical relevance of such variability is unknown. Therefore, the objective of the present review is, first, to conduct a literature review and summarize nimodipine pharmacokinetic data and sources of variability in various patient groups. Second, to determine if there is any evidence reporting an association between nimodipine exposure and clinical outcomes in patients with subarachnoid hemorrhage. A systematic literature search was performed in MEDLINE and EMBASE. The following keywords were used: (“nimodipine” OR “nymalize” OR “nimotop”) AND (“pharmacokinetic*”, OR “PK”). The search results were limited to English language and human studies. A large interpatient variability in nimodipine pharmacokinetics has been reported. Patient-specific factors that had an influence on pharmacokinetic parameters are age, comorbidities, variabilities in metabolism due to genetic polymorphism and co-administered medications, as well as nimodipine administration technique. The association between nimodipine exposure and clinical outcomes remains unclear and data available are too scarce to reach a firm conclusion. Here, we present a narrative review with a systematic literature search discussing nimodipine pharmacokinetic variability in various patient populations. It is not clear if minimal or lack of systemic exposure to nimodipine denies its benefit and contributes to worsening outcomes in patients with subarachnoid hemorrhage. Further studies are needed to determine if such an association exists.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nimodipine exposure is highly variable among individuals. |
Age, liver disease, genetic make-up, and nimodipine administration techniques influence nimodipine exposure. |
1 Introduction
Nimodipine is a dihydropyridine calcium channel blocker with greater selectivity for cerebral blood vessels than other agents within the same class [1, 2]. As a result, nimodipine has been tested in the setting of aneurysmal subarachnoid hemorrhage (SAH), a life-threatening brain bleed. Neurological and medical complications are common following SAH and contribute significantly to the overall prognosis. Cerebral vasospasm and delayed cerebral ischemia (DCI) are examples of those complications and are considered as significant contributors of disability in patients with SAH who survive the initial bleed [3, 4]. Effective prevention of DCI can significantly improve the functional outcomes of patients. Therefore, a substantial amount of research in this area has been focused on understanding the mechanisms of those complications and exploring potential therapeutic modalities for improving patient outcomes. Several agents have been investigated to target vasospasm and DCI; however, nimodipine was the only pharmacological agent that has been shown to improve neurological outcomes following SAH by several randomized clinical trials [5,6,7,8]. Therefore, the current guidelines for treating SAH recommend that a fixed dose of oral nimodipine to be administered within 96 h from ictus to all patients regardless of their weight, age, disease severity, comorbidities, and other patient-specific characteristics and to be continued for 21 days [9]. Despite the strong evidence for the use of nimodipine in patients with SAH, there have been reports of significant variations in the pharmacokinetics of nimodipine among different patient groups. An individualized approach for the administration of nimodipine remains an area of ongoing research, and the association between variations in pharmacokinetic (PK) parameters and clinical outcomes remains unclear.
The objective of the present review is, therefore, bifaceted. The first objective is to conduct a comprehensive literature review and summarize nimodipine pharmacokinetic data and sources of variability in various patient groups and healthy volunteers. Second, to determine if there is any evidence reporting an association between systemic nimodipine exposure and clinical outcomes in patients with SAH.
2 Evidence Used in this Review
A systematic literature search was performed in MEDLINE (1946 to 30 August, 2019), and EMBASE (1974 to 30 August, 2019). The following keywords were used: (“nimodipine” OR “nymalize” OR “nimotop”) AND (“pharmacokinetic*”, OR “PK”). The search results were limited to English language and human studies. Search strategies were developed in collaboration with a librarian from the University of Alberta Library. In addition, relevant studies were obtained from references of articles identified in the initial search. A total of 808 articles were found. After duplicate removal, title and abstract screening, full-text screening, and the addition of newly published articles since the original search, a total of 87 references were included in this review. Article selection was independently performed by all authors. In case of any discrepancies, further discussion was undertaken to reach a consensus. The included articles studied a wide range of patient populations including, but not limited to, healthy volunteers, patients with SAH, renal or liver disease, and other cerebral disorders.
3 Chemical Properties of Nimodipine
Nimodipine, 3-O-(2-methoxyethyl) 5-O-propan-2-yl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate, has a 1,4-dihydropyridine ring structure (Fig. 1). Nimodipine is a yellow crystalline compound that is insoluble in water but soluble in ethanol, polyethylene glycol 400, and dimethyl sulfoxide [10]. It is sensitive to light with a degradation half-life of 56 h and 16 h when 50 ng/mL nimodipine solution was exposed to daylight and ultraviolet light, respectively [11]. Nimodipine is a chiral compound with an asymmetric carbon at position 4 and it is marketed as a racemic mixture of (+)-R and (−)-S nimodipine. Towart et al. have found that (−)-S nimodipine is approximately twice as potent a vasorelaxant as the racemic mixture; however, the clinical relevance of such differential pharmacology is unclear [12]. Nimodipine is an amphiphilic compound and its selectivity towards cerebral blood vessels has been attributed to the increased lipophilicity of the drug and its ability to cross the blood–brain barrier [13, 14].

Chemical structure of nimodipine. * indicates chiral carbon
4 Nimodipine Pharmacodynamics
Nimodipine inhibits the influx of calcium ions through voltage-gated L-type calcium channels of vascular smooth muscles, thereby, causing vasorelaxation [15, 16]. Nimodipine has been shown to dilate blood vessels and prevent vasoconstriction particularly in small arterioles whose diameters are 70–100 µm [17, 18]. Despite that, nimodipine reported benefits in patients with SAH were not related to its effects on vasospasm, suggesting other potential mechanisms. Furthermore, nimodipine elevates adenosine levels in the central nervous system with subsequent inhibition of the excitatory neurotransmitter glutamate, a potential neuroprotective mechanism [19,20,21,22].
5 Nimodipine Pharmacokinetics
5.1 Absorption
Nimodipine undergoes rapid absorption from the gastrointestinal tract followed by extensive first-pass hepatic metabolism. The oral bioavailability of nimodipine has been reported to range from 3 to 30%. Both the parent drug and the metabolites start to circulate 10–15 min after ingestion with the time to peak concentration (Tmax) from 0.5 to 1 h [23, 24]. Nimodipine follows a linear PK profile where the area under the concentration–time curve (AUC) is proportional to the administered dose [25].
5.2 Distribution
Volume of distribution (Vd) of oral nimodipine ranges from as low as 0.94 L/kg to as high as 2.46 L/kg. Nimodipine is highly bound to plasma proteins (> 95%) particularly, alpha-acid glycoprotein (AAG) [26]. Therefore, the distribution of nimodipine can be affected by the concentration of AAG. To illustrate, Ensom et al. compared AAG levels in patients with SAH with controls before and after surgery [26]. Preoperative values of AAG in patients with SAH were 39% higher than the values of controls. However, AAG levels of patients declined significantly postoperatively and reached control levels 48 h post-surgery. This transient shift in AAG levels had an impact on serum nimodipine total concentration and the unbound fraction. Furthermore, the concentration of nimodipine in cerebrospinal fluid was inversely proportional to the level of AAG [26].
5.3 Metabolism and Excretion
Nimodipine undergoes extensive hepatic metabolism with cytochrome P450 (CYP) 3A4 and 3A5 enzymes [23, 24, 27]. It undergoes multiple metabolic pathways (Fig. 2) and more than 18 metabolites have been reported [28, 29]. Nimodipine plasma concentrations decline rapidly with a half-life of 1–2 h and a reported terminal half-life of 9 h [11, 30, 31]. As nimodipine has a chiral carbon atom, it exists as (+)-R and (−)-S enantiomers. The (−)-S enantiomer is more rapidly eliminated than (+)-R following oral dosing [32,33,34,35]. However, such differential effect was not apparent when nimodipine was administered intravenously, suggesting enantioselective first-pass metabolism. It is excreted in urine and bile mainly as metabolites.

Common biotransformation reactions of nimodipine
6 Nimodipine Pharmacokinetic Variability
A large interpatient variability in nimodipine pharmacokinetics has been reported in different patient populations and healthy volunteers. Variabilities in the bioavailability (F) and clearance (CL) of nimodipine with resultant altered nimodipine systemic concentrations were reported. Figures 3, 4, 5 summarize observed nimodipine concentrations in various pharmacokinetic studies. As seen in Fig. 3, steady-state plasma concentrations following intravenous infusion (Cssivi) had a range from as low as 10.8 up to 73 ng/mL with equivalent daily dosing. Similarly, a wide range of peak plasma concentrations following oral administration (CmaxPO) of a single 60-mg dose (Fig. 4) and at steady state (Fig. 5) was observed. Patient-specific factors that had an influence on PK parameters are age and sex of the participants, comorbidities, and variabilities in metabolism due to genetic polymorphisms. This section provides a summary of the potential covariates contributing to nimodipine PK variability.

Steady-state nimodipine plasma concentrations following intravenous infusion (Cssivi) in healthy individuals, patients with subarachnoid hemorrhage (SAH), skull base lesions, cerebral vasospasm, and subjects who underwent coronary artery bypass grafts (CABG) [35, 38, 56,57,58, 67,68,69,70,71,72,73,74]. In Scheller et al. (n = 22), patients started on 1 mg/h for 2 h, increased to 2 mg/h, then decreased to 1 mg/h due to low blood pressure. The duration that patients were taking 2 mg/h was not reported in the article. The data for this study are grouped under a daily dose of 24 mg [71]


6.1 Effect of Age on Nimodipine Pharmacokinetics
Muck et al. specifically compared the PK parameters of nimodipine in young vs old populations [36]. The young age group included 24 subjects aged 22–40 years and the older age group included 24 subjects aged 59–79 years. The study showed no statistically significant differences in PK parameters between the two groups after a single intravenous infusion of 15 µg/kg infused over 1 h. However, a higher Cmax and AUC was observed in the older age group when both groups were administered nimodipine 30 mg orally as a single dose and as a three times-daily multiple-dose regimen. The reported Cmax and AUC for the older age group were 23.3 ± 1.62 ng/mL and 47.5 ± 1.62 ng·h/mL, respectively; while for the young group the Cmax and AUC were 13.5 ± 2.03 ng/mL and 25.7 ± 1.73 ng·h/mL, respectively [36]. This suggests that older adults have reduced first-pass metabolism of oral nimodipine compared with younger patients. Similar Cmax and AUC values have been reported in a study of 21 elderly patients with various central nervous system disorders [37]. In addition, in a population PK analysis of previous PK studies, the authors have found that nimodipine CL was reduced by 32% in elderly subjects (aged ≥ 65 years). Furthermore, the authors reported a significant inter-individual variability of nimodipine PK parameters: 60% in CL, 59% in apparent volume of distribution of the central compartment, and 95% in apparent volume of distribution of the peripheral compartment [30]. Similar to what was observed in healthy subjects, there was also a negative correlation between nimodipine CL and age seen in 24 patients with SAH treated with surgical clipping (r = − 0.4, p < 0.05) [38]. Taken together, the first-pass metabolism and CL of nimodipine are age dependent.
6.2 Effect of Sex on Nimodipine Pharmacokinetics
Sex differences in nimodipine pharmacokinetics were not as apparent as the effect of age. Results from Muck et al. have reported that nimodipine CL among male individuals (n = 12) is slightly lower than female individuals (n = 11) [36]. However, there was no sex differences (p = 0.573) in the observed nimodipine levels in a study involving patients with SAH [39].
6.3 Effect of Renal Function on Nimodipine Pharmacokinetics
The influence of renal function on oral nimodipine (30 mg administered every 8 h) has been studied in two reports [40, 41]. In both studies, CmaxPO was at least 17 ng/mL higher in subjects with chronic kidney disease subjects compared with those with normal kidney function (Fig. 5). Kirch et al. have concluded that the nimodipine half-life is prolonged in patients with CKD, defined as estimated glomerular filtration rate < 60 mL/min. However, both groups were not age matched, where patients in the CKD group were older (65.3 ± 4.1 vs 25.2 ± 2.4 years), which could have contributed to this observation. Furthermore, Terziivanov et al. have reported increased nimodipine PK variability among patients with creatinine CL 51–80 mL/min. The authors concluded that although nimodipine is mainly metabolized by the liver, renal function could be an additional factor contributing to nimodipine PK variability.
6.4 Effect of Liver Disease on Nimodipine Pharmacokinetics
As hepatic metabolism is the main route of nimodipine elimination, the influence of liver cirrhosis on nimodipine pharmacokinetics has been studied. It has been reported that the apparent oral CL of nimodipine is substantially lower in patients with liver cirrhosis than in healthy subjects. As a result, the observed CmaxPO in patients with liver disease was 1.4- to ninefold higher than CmaxPO observed in those with normal liver function (Fig. 4). Furthermore, there was a large variability in the influence of liver disease on nimodipine oral CL with CL ranges from as low as 60 L/h to as high as 652 L/h. Like other highly protein bound lipophilic compounds that are extensively metabolized, the observed variability may be due to disease-induced changes in protein binding, gastric and enterohepatic circulation, and hepatic blood flow, as well as an intrinsic decrease in the metabolic capacity of the liver [42]. The clinical significance of decreased nimodipine CL in patients with cirrhosis was evidenced by a reduction in main arterial pressure in cirrhotic patients. A concentration-related blood pressure reduction was seen in the individual data of cirrhotic patients, which was not seen in control subjects [42].
6.5 Effect of Genetic Polymorphism on Nimodipine Pharmacokinetics
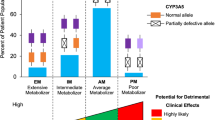
As discussed under “Metabolism”, nimodipine is mainly metabolized by the CYP3A enzyme family. Genetic polymorphism of the enzyme CYP3A5 has been reported to alter the disposition of nimodipine. Depending on their metabolizing enzyme genotype, patients can fall into four categories: extensive, normal, intermediate, and poor metabolizers [43]. In a pharmacogenetic study conducted in healthy Chinese individuals, participants who were carriers of homozygous CYP3A5 (*3/*3) and received oral nimodipine had higher Cmax and AUC compared with those with heterozygous CYP3A5 (*1/*3) or wild-type CYP3A5 (*1/*1). This has been attributed to reduced nimodipine CL in the homozygous CYP3A5 (*3/*3) group (538 ± 153 vs 758 ± 260 L/h, p = 0.03) [44]. Furthermore, a recent case study reported severe bradycardia with junctional atrioventricular heart block and hypotension for an elderly patient treated with nimodipine who had the CYP3A5 genotype (*3/*3) [poor metabolizer] [45]. This is believed to be partially caused by the advanced age of the patient and the altered genotype status for one of the major enzymes responsible to nimodipine metabolism (CYP3A5) [45]. This suggests the potential utility of pharmacogenomic testing in patients with SAH to predict nimodipine tolerability [43]. Further studies are needed.
6.6 Drug Interactions and Nimodipine Pharmacokinetics
Being a substrate of CYP3A enzymes, nimodipine is susceptible to drug–drug interactions with liver microsomal enzymes inducers (such as phenytoin and carbamazepine) and inhibitors (such as cimetidine and grapefruit juice). As patients with SAH often present with seizures, interactions with antiepileptic drugs (AEDs) is of most relevance. Effects of enzyme-inducing AEDs such as phenytoin, carbamazepine, and phenobarbital were investigated in patients with epilepsy. After a single oral dose of 60 mg nimodipine, patients who have been taking enzyme-inducing AEDs for at least 4 months had significantly lower nimodipine plasma concentrations compared with healthy controls (CmaxPO 4.2 vs 39 ng/mL, respectively) [46]. Similarly, in a study involving patients with SAH, concomitant administration of phenytoin in two patients resulted in lower nimodipine concentrations compared with those not taking phenytoin [47]. However, patients with epilepsy taking enzyme-inhibiting AEDs such as valproic acid had slightly higher nimodipine plasma concentrations compared with normal subjects (CmaxPO 48 vs 39 ng/mL, respectively) [46]. It is not clear; however, if altered nimodipine concentration translates into altered drug response. To illustrate, administration of cimetidine and grapefruit juice with nimodipine resulted in 75 and 51% increase in AUC of nimodipine, respectively. However, there were no significant differences in blood pressure despite the increase in AUC [48, 49]. Several studies have also reported no interactions when nimodipine was co-administered with ranitidine, clazosentan, tirilazad, diazepam, propranolol, and indomethacin [48, 50,51,52,53,54].
6.7 Influence of Nimodipine Formulations on Nimodipine Pharmacokinetics
Nimodipine is marketed as soft gelatin capsule, oral tablet, intravenous solution, and oral suspension. Both tablets and capsules have comparable AUCs (91.1 ng·h/mL and 103.5 ng·h/mL, respectively). However, CmaxPO is lower with the tablet formulation compared with the capsule (45.6 ng/mL vs 69.1 ng/mL, respectively) and Tmax is longer with tablets (0.77 h vs 0.59 h) [55]. This suggests that the tablet formulation has slightly delayed absorption compared with the capsule. There was no direct comparison of nimodipine oral liquid to other oral formulations.
6.8 Influence of Administration Technique on Nimodipine Pharmacokinetics
Patients with SAH who are able to swallow will administer the whole capsules or tablets, otherwise, nimodipine liquid needs to be drawn from the capsules, tablets to be crushed or commercially available liquid to be administered through enteral feeding tubes for those who are unable to swallow, such as those with altered mental status or mechanically ventilated. It is not clear, however, whether these techniques of administration are equivalent. Few studies have shown a decreased nimodipine systemic exposure and increased PK variability when it is administered via feeding tubes. To illustrate, Soppi et al. have reported nimodipine concentrations following the standard 60 mg po every 4 h dosing schedule in patients with SAH [56]. Nimodipine maximum concentrations had a range from as low as 1 ng/mL up to 56.7 ng/mL for those receiving tablets and 0.9–1.7 ng/ml for those receiving an extemporaneously prepared oral suspension. Similarly, Abboud et al. compared plasma nimodipine concentrations administered parenterally followed by enteral administration. The AUC of the parenteral route was significantly higher than that of the oral route. Moreover, nimodipine AUC for those who swallowed whole nimodipine tablets was higher than those who received it through enteral feeding tube [median 52 (IQR 26–1411) ng·h/ml vs. 23 (IQR 6–1272 ng·h/ml), respectively, p value 0.006] [57]. In addition, two patients with high grade SAH had undetectable nimodipine concentrations. Similarly, Kumana et al. have reported reduced nimodipine systemic exposure in a patient given crushed tablets through gastric tube [58]. The reason for this reduced exposure is unclear. In vitro experiments indicated that adsorption of nimodipine by the nasogastric tubing was limited (< 20%) and that was unlikely to have been the cause of a low plasma concentration [58]. In a retrospective study in SAH patients, after adjusting for disease severity nimodipine administration technique was associated with vasospasm where patients receiving nimodipine enterally had increased prevalence of vasospasm compared to those administered it as whole tablets [59]. Further studies are needed to examine the clinical relevance and causes of the observed reduced nimodipine exposure. Nutritional intake is another factor that may contribute to nimodipine PK variability. Presence of food in the gut delays the rate of nimodipine absorption as evidenced by lower CmaxPO and prolonged Tmax [55]. In a post hoc analysis study, absorption rate constant (Ka) was shown to be decreased if nimodipine was administered with a meal [30].
6.9 SAH Characteristics and Nimodipine Exposure
The effects of SAH severity and occurrence of DCI on nimodipine pharmacokinetics have been explored in few studies. Hunt and Hess score and World Federation of Neurological Surgeons Grade are used to grade SAH severity on a scale of 1–5, where 5 is the most severe SAH. Four PK studies have reported SAH grades [39, 47, 57, 58]. Following intravenous administration of nimodipine, it appears that there is no correlation between SAH grade and Cssivi. However, following oral administration, poor-grade patients had significantly lower Cmax and AUC values than those observed in good-grade patients. This suggests that the observed differential effects of SAH grade could be attributed to reduced nimodipine bioavailability secondary to administration via feeding tube rather than altered drug clearance. Further studies are needed. With regards to DCI, three patients developed DCI in a study involving 24 SAH patients. All subjects were treated with clipping and were administered IV nimodipine for 7 days. The three patients that developed DCI had plasma nimodipine concentrations similar to patients who did not. Authors concluded that it seems unlikely that the therapeutic failure could be attributed to individual deviations in the pharmacokinetics of the drug [38]. However, the study was underpowered to detect such differences.
7 Nimodipine Exposure and Outcomes Following SAH
Guidelines recommend that all patients presenting with aneurysmal SAH should receive a fixed dose of oral nimodipine 60 mg every 4 h for 21 days from SAH onset regardless of weight, age, disease severity, comorbidities, and other patient-specific characteristics [60]. Pharmacokinetic studies have reported extensive variability of nimodipine concentrations in various populations (Figs. 3, 4, 5) and in the setting of SAH, with some patients had undetectable nimodipine plasma levels [38, 39, 56,57,58]. The observed variability in nimodipine exposure may have been attributed to practice variations in nimodipine administration, disease severity, administration of concomitant interacting drugs and cytochrome P450 polymorphism [35, 43, 46, 61]. While previous randomized controlled trials have found that nimodipine reduces the incidence of poor neurologic outcomes (defined by death, persistent vegetative state, and severe disability) by 40–86%, still up to 22% patients in the nimodipine arm experienced poor outcomes [5, 7, 8]. Therefore, it is not clear if all patients are getting the full benefit of nimodipine using a fixed dose regimen and the evidence supporting a correlation between nimodipine concentrations and patient outcomes is scarce and not clear. Riva et al. have reported an association between nimodipine CSF concentrations and neurological outcomes at 9 months following SAH onset but they were unable to find such correlation with plasma concentrations [62]. It should be noted, however, that all patients were dosed using nimodipine IV infusion and their plasma concentrations ranged from 24.9 to 71.8 ng/mL, concentrations way above what has been reported in some patients given oral dosing [23, 56]. Nevertheless, it is not clear if minimal or lack of systemic exposure to oral nimodipine denies its benefit and contributes to worsening patient outcomes. The intravenous formulation of nimodipine is not available in Canada and United States and was banned due to accidental intravenous administration of the capsule content that has resulted in death or near-death events [63]. Intravenous nimodipine was compared to the oral route in two small randomized trials [64, 65]. Both studies have found no difference in patient outcomes; however, the number of patients with high Hunt and Hess grade (IV and V) was small to draw conclusions on the comparability of both routes in high grade patients. Further research is needed. In a way to potentially minimize the systemic adverse reactions of nimodipine and enhance its cerebrospinal fluid exposure, single dose intraventricular nimodipine administration has been compared to oral nimodipine in a double-blind, double-dummy, randomized controlled trial in SAH patients (NEWTON-2 study) [66]. The intraventricular formulation was composed of sustained-release microparticles avoiding repeated dosing. Intraventricular nimodipine had acceptable safety profile but was not superior to oral nimodipine in achieving favorable outcome defined as extended Glasgow Outcome scale of ≥ 6 at 90 days following SAH. It worth to mention that upon subgroup analysis, intraventricular nimodipine demonstrated a trend towards more favorable outcomes compared to oral nimodipine in those with WFNS grades 3–4 [66]. Further research is needed in poor grade SAH patients.
8 Limitations
This review has limitations. Many of the included studies had small sample sizes. In addition, studies were heterogenous in terms of the included patient population, nimodipine dosage regimen and formulations, making it difficult for direct comparisons or producing a summary estimate. However, taken together, the included studies underlined the wide interpatient variability of nimodipine pharmacokinetics.
9 Conclusion
Nimodipine has been shown to improve outcomes following SAH. Guidelines recommend that all patients receive a fixed dose of oral nimodipine for 21 days. However, pharmacokinetic studies have reported extensive variability of nimodipine concentrations in SAH. The observed variability may have been attributed to practice variations in nimodipine administration, disease severity, administration of concomitant interacting drugs and cytochrome P450 polymorphism. Therefore, it is not clear if all patients are getting the full benefit of nimodipine using a fixed dose regimen and the evidence supporting a correlation between nimodipine concentrations and patient outcomes is scarce. Further studies are needed to determine if such association exists and if there is a need for nimodipine dosage individualization in SAH patients.
References
Lopez-Arrieta JM, Birks J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. 2002;3:Cd000147. https://doi.org/10.1002/14651858.Cd000147.
Wessell A, Kole MJ, Badjatia N, Parikh G, Albrecht JS, Schreibman DL, et al. High compliance with scheduled nimodipine is associated with better outcome in aneurysmal subarachnoid hemorrhage patients cotreated with heparin infusion. Front Neurol. 2017;8:268. https://doi.org/10.3389/fneur.2017.00268.
Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KT. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth. 2012;109(3):315–29. https://doi.org/10.1093/bja/aes264.
Dorsch NW. Therapeutic approaches to vasospasm in subarachnoid hemorrhage. Curr Opin Crit Care. 2002;8(2):128–33. https://doi.org/10.1097/00075198-200204000-00007.
Petruk KC, West M, Mohr G, Weir BK, Benoit BG, Gentili F, et al. Nimodipine treatment in poor-grade aneurysm patients. Results of a multicenter double-blind placebo-controlled trial. J Neurosurg. 1988;68(4):505–17. https://doi.org/10.3171/jns.1988.68.4.0505.
Philippon J, Grob R, Dagreou F, Guggiari M, Rivierez M, Viars P. eCt ahirurgica prevention of vasospasm in subarachnoid haemorrhage. A controlled study with nimodipine. Acta Neurochir (Wien). 1986;82:110–4.
Pickard JD, Murray GD, Illingworth R, Shaw MD, Teasdale GM, Foy PM, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ. 1989;298(6674):636–42. https://doi.org/10.1136/bmj.298.6674.636.
Allen GS, Ahn HS, Preziosi TJ, Battye R, Boone SC, Boone SC, et al. Cerebral arterial spasm–a controlled trial of nimodipine in patients with subarachnoid hemorrhage. N Engl J Med. 1983;308(11):619–24. https://doi.org/10.1056/NEJM198303173081103.
Connolly ES, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American heart association/American stroke association. Stroke. 2012;43(6):1711–37. https://doi.org/10.1161/STR.0b013e3182587839.
Scriabine A, Battye R, Hoffmeister F, Kazda S, Towart R, Garthoff B, Schlüter G, Rämsch KD, Scherling D. Nimodipine. Cardiovasc Drug Rev. 1985;3:197–21818.
Ms L, Sorkin EM. Nimodipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in cerebrovascular disease. Drugs. 1989;37(5):669–99.
Towart R, Wehinger E, Meyer H, Kazda S. The effects of nimodipine, its optical isomers and metabolites on isolated vascular smooth muscle. Arzneimittelforschung. 1982;32(4):338–46.
Herbette LG, Trumbore M, Chester DW, Katz AM. Possible molecular basis for the pharmacokinetics and pharmacodynamics of three membrane-active drugs: propranolol, nimodipine and amiodarone. J Mol Cell Cardiol. 1988;20(5):373–8. https://doi.org/10.1016/s0022-2828(88)80128-7.
Herbette LG, Mason PE, Sweeney KR, Trumbore MW, Mason RP. Favorable amphiphilicity of nimodipine facilitates its interactions with brain membranes. Neuropharmacology. 1994;33(2):241–9. https://doi.org/10.1016/0028-3908(94)90015-9.
Uchida S, Yamada S, Nagai K, Deguchi Y, Kimura R. Brain pharmacokinetics and in vivo receptor binding of 1,4-dihydropyridine calcium channel antagonists. Life Sci. 1997;61(21):2083–90. https://doi.org/10.1016/s0024-3205(97)00881-3.
Scriabine A, van den Kerckhoff W. Pharmacology of nimodipine. A review. Ann N Y Acad Sci. 1988;522:698–706. https://doi.org/10.1111/j.1749-6632.1988.tb33415.x.
Kazda S, Garthoff B, Krause HP, Schlossmann K. Cerebrovascular effects of the calcium antagonistic dihydropyridine derivative nimodipine in animal experiments. Arzneimittelforschung. 1982;32(4):331–8.
Towart R, Meyer H, Kazda S. The effect of nimodipine, its optical isomers and metabolites on isolated vascular smooth muscle. ArzneimForsch/Drug Res. 1982;32(1):338–46.
Li RW, Tse CM, Man RY, Vanhoutte PM, Leung GP. Inhibition of human equilibrative nucleoside transporters by dihydropyridine-type calcium channel antagonists. Eur J Pharmacol. 2007;568(1–3):75–82. https://doi.org/10.1016/j.ejphar.2007.04.033.
Blardi P, Urso R, De Lalla A, Volpi L, Perri TD, Auteri A. Nimodipine: drug pharmacokinetics and plasma adenosine levels in patients affected by cerebral ischemia. Clin Pharmacol Ther. 2002;72(5):556–61. https://doi.org/10.1067/mcp.2002.128127.
Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13(12):439–45. https://doi.org/10.1016/0165-6147(92)90141-r.
Sweeney MI. Neuroprotective effects of adenosine in cerebral ischemia: window of opportunity. Neurosci Biobehav Rev. 1997;21(2):207–17. https://doi.org/10.1016/s0149-7634(96)00011-5.
Langley MS, Sorkin EM. Nimodipine: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in cerebrovascular disease. Drugs. 1989;37(5):669–99.
Muck W, Ahr G, Kuhlmann J. Nimodipine. Potential for drug-drug interactions in the elderly. Drugs Aging. 1995;6(3):229–42.
Ramsch KD, Ahr G, Tettenborn D, Auer LM. Overview on pharmacokinetics of nimodipine in healthy volunteers and in patients with subarachnoid hemorrhage. Neurochirurgia. 1985;28(SUPPL. 1):74–8.
Woodward DK, Hatton J, Ensom MH, Young B, Dempsey R, Clifton GD. Alpha1-acid glycoprotein concentrations and cerebrospinal fluid drug distribution after subarachnoid hemorrhage. Pharmacother. 1998;18(5):1062–8.
Ha HR, Pletscher W, Leuenberger PM, Sticher O, Meier PJ. Measurement of nimodipine metabolism in rat liver microsomes by high-performance liquid chromatography. J Liq Chromatogr. 2007;18(11):2243–55. https://doi.org/10.1080/10826079508010268.
Scherling D, Bühner K, Krause HP, Karl W, Wünsche C. Biotransformation of nimodipine in rat, dog, and monkey. Arzneimittelforschung. 1991;41(4):392–8.
Rämsch KD, Ahr G, Tettenborn D, Auer LM. Overview on pharmacokinetics of nimodipine in healthy volunteers and in patients with subarachnoid hemorrhage. Neurochirurgia. 1985;28(Suppl 1):74–8.
Anonymous. Population pharmacokinetic analysis for nimodipine. Jpn J Clin Pharmacol Ther. 1999;30(1):229–30. https://www.jstage.jst.go.jp/article/jscpt1970/30/1/30_1_229/_pdf/-char/en.
Becker C, Hilgert D, Platt D, Krauss D, Spahn H, Mutschler E. Pharmacokinetics of nimodipine and its major metabolites in elderly patients with cerebral disorders. Eur J Pharmacol. 1990;183(6):2298. https://doi.org/10.1016/0014-2999%2890%2993850-P.
Fischer C, Schonberger F, Muck W, Heuck K, Eichelbaum M. Simultaneous assessment of the intravenous and oral disposition of the enantiomers of racemic nimodipine by chiral stationary-phase high-performance liquid chromatography and gas chromatography/mass spectroscopy combined with a stable isotope technique. J Pharm Sci. 1993;82(3):244–50.
Michelson G, Wärntges S, Leidig S, Lötsch J, Geisslinger G. Nimodipine plasma concentration and retinal blood flow in healthy subjects. Invest Ophthalmol Vis Sci. 2006;47(8):3479–86.
Muck W, Heine PR, Breuel HP, Niklaus H, Horkulak J, Ahr G. The effect of multiple oral dosing of nimodipine on glibenclamide pharmacodynamics and pharmacokinetics in elderly patients with type-2 diabetes mellitus. Int J Clin Pharmacol Ther. 1995;33(2):89–94.
Wanner-Olsen H, Gaarskaer FB, Mikkelsen EO, Jakobsen P, Voldby B. Studies on concentration-time profiles of nimodipine enantiomers following intravenous and oral administration of nimodipine in patients with subarachnoid hemorrhage. Chirality. 2000;12(9):660–4. https://doi.org/10.1002/1520-636X%282000%2912:9%3C660:AID-CHIR3%3E3.0.CO;2-1.
Muck W, Kuhlmann J. The influence of age on pharmacokinetics of nimodipine*. Int J Clin Pharmacol Ther. 1996;34(7):293–8.
Eicher H, Hilgert D, Zeeh J, Platt D, Becker C, Mutschler E. Pharmacokinetics of nimodipine in multimorbid elderly patients with chronic brain failure. Arch Gerontol Geriatr. 1992;14(3):309–19. https://doi.org/10.1016/0167-4943%2892%2990030-8.
Vinge E, Andersson KE, Brandt L, Ljunggren B, Nilsson LG, Rosendal-Helgesen S. Pharmacokinetics of nimodipine in patients with aneurysmal subarachnoid haemorrhage. Eur J Clin Pharmacol. 1986;30(4):421–5.
Albanna W, Weiss M, Conzen C, Clusmann H, Schneider T, Reinsch M, et al. Systemic and cerebral concentration of nimodipine during established and experimental vasospasm treatment. World Neurosurg. 2017. https://doi.org/10.1016/j.wneu.2017.03.062.
Kirch W, Rämsch KD, Dührsen U, Ohnhaus EE. Clinical pharmacokinetics of nimodipine in normal and impaired renal function. Int J Clin Pharmacol Res. 1984;4(5):381–4.
Terziivanov D, Atanasova I, Dimitrova V, Robeva R, Unger S, Mück W. Pharmacokinetic variability of nimodipine disposition after single and multiple oral dosing to hypertensive renal failure patients: parametric and nonparametric population analysis. Int J Clin Pharmacol Ther. 1999;37(8):404–12.
Gengo FM, Fagan SC, Krol G, Bernhard H. Nimodipine disposition and haemodynamic effects in patients with cirrhosis and age-matched controls. Br J Clin Pharmacol. 1987;23(1):47–53.
Peacock SH, James C, Turnbull MT, Cowart JB, Reid JM, Freeman WD. Pharmacogenomics of cytochrome P450 of nimodipine metabolism after aneurysmal subarachnoid hemorrhage. J Neurosci Nurs. 2019;51(5):238–42. https://doi.org/10.1097/JNN.0000000000000464.
Zhao Y, Zhai D, He H, Li T, Chen X, Ji H. Effects of CYP3A5, MDR1 and CACNA1C polymorphisms on the oral disposition and response of nimodipine in a Chinese cohort. Eur J Clin Pharmacol. 2009;65(6):579–84. https://doi.org/10.1007/s00228-009-0619-6.
James CL, Turnbull MT, Freeman WD. Nimodipine-induced junctional bradycardia in an elderly patient with subarachnoid hemorrhage. Pharmacogenomics. 2020;21(6):387–92. https://doi.org/10.2217/pgs-2019-0136.
Tartara A, Galimberti CA, Manni R, Parietti L, Zucca C, Baasch H, et al. Differential effects of valproic acid and enzyme-inducing anticonvulsants on nimodipine pharmacokinetics in epileptic patients. Br J Clin Pharmacol. 1991;32(3):335–40.
Soppi V, Kokki H, Koivisto T, Lehtonen M, Helin-Tanninen M, Lehtola S, et al. Early-phase pharmacokinetics of enteral and parenteral nimodipine in patients with acute subarachnoid haemorrhage—a pilot study. Eur J Clin Pharmacol. 2007;63(4):355–61.
Mück W, Wingender W, Seiberling M, Woelke E, Rämsch KD, Kuhlmann J. Influence of the H2-receptor antagonists cimetidine and ranitidine on the pharmacokinetics of nimodipine in healthy volunteers. Eur J Clin Pharmacol. 1992;42(3):325–8.
Fuhr U, Maier-Brüggemann A, Blume H, Mück W, Unger S, Kuhlmann J, et al. Grapefruit juice increases oral nimodipine bioavailability. Int J Clin Pharmacol Ther. 1998;36(3):126–32.
Fleishaker JC, Hulst LK, Peters GR. Lack of a pharmacokinetic/pharmacodynamic interaction between nimodipine and tirilazad mesylate in healthy volunteers. J Clin Pharmacol. 1994;34(8):837–41.
Heine PR, Weyer G, Breuel HP, Muck W, Schmage N, Kuhlmann J. Lack of interaction between diazepam and nimodipine during chronic oral administration to healthy elderly subjects. Br J Clin Pharmacol. 1994;38(1):39–433.
Breuel HP, Heine PR, Mück W, Niklaus H, Schmage N, Kuhlmann J. Chronic administration of nimodipine and propranolol in elderly normotensive subjects–an interaction study. Int J Clin Pharmacol Ther. 1995;33(2):103–8.
Bruderer S, Zisowsky J, Fuseau E, Gutierez M, Dingemanse J. Investigation of pharmacokinetic and pharmacodynamic interactions between clazosentan and nimodipine in asah patients: a population analysis. Basic Clin Pharmacol Toxicol. 2011;109:150. https://doi.org/10.1111/j.1742-7843.2011.00722.x.
Mück W, Heine PR, Schmage N, Niklaus H, Horkulak J, Breuel HP. Steady-state pharmacokinetics of nimodipine during chronic administration of indometacin in elderly healthy volunteers. Arzneimittelforschung. 1995;45(4):460–2.
NIMOTOP®PRODUCT MONOGRAPH M, Ontario, 2014, Bayer Inc.
Soppi V, Kokki H, Koivisto T, Lehtonen M, Helin-Tanninen M, Lehtola S, et al. Early-phase pharmacokinetics of enteral and parenteral nimodipine in patients with acute subarachnoid haemorrhage—a pilot study. Eur J Clin Pharmacol. 2007;63:355–61. https://doi.org/10.1007/s00228-007-0267-7.
Abboud T, Andresen H, Koeppen J, Czorlich P, Duehrsen L, Stenzig J, et al. Serum levels of nimodipine in enteral and parenteral administration in patients with aneurysmal subarachnoid hemorrhage. Acta Neurochir (Wien). 2015;157(5):763–7. https://doi.org/10.1007/s00701-015-2369-9.
Kumana CR, Kou M, Yu YL, Fong KY, Fung CF, Chang CM, et al. Investigation of nimodipine pharmacokinetics in Chinese patients with acute subarachnoid haemorrhage. Eur J Clin Pharmacol. 1993;45(4):363–6.
Isse FA, Abdallah YEH, Mahmoud SH. The impact of nimodipine administration through feeding tube on outcomes in patients with aneurysmal subarachnoid hemorrhage. J Pharm Pharm Sci. 2020;23(1):100–8. https://doi.org/10.18433/jpps30960.
Diringer MN, Bleck TP, Claude Hemphill J 3rd, Menon D, Shutter L, Vespa P, et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society's Multidisciplinary Consensus Conference. Neurocrit Care. 2011;15(2):211–40. https://doi.org/10.1007/s12028-011-9605-9.
Schmith VD, Foss JF. Effects of inflammation on pharmacokinetics/pharmacodynamics: increasing recognition of its contribution to variability in response. Clin Pharmacol Ther. 2008;83(6):809–11. https://doi.org/10.1038/clpt.2008.62.
Riva R, Pegoli M, Contin M, Perrone A, Mohamed S, Zanello M. Cerebrospinal fluid concentrations of nimodipine correlate with long-term outcome in aneurysmal subarachnoid hemorrhage: pilot study. Clin Neuropharmacol. 2019. https://doi.org/10.1097/WNF.0000000000000356.
Aschenbrenner DS. Oral nimodipine given intravenously can be fatal. AJN Am J Nurs. 2010;110(12):27. https://doi.org/10.1097/01.NAJ.0000391235.60640.da.
Soppi V, Karamanakos PN, Koivisto T, Kurki MI, Vanninen R, Jaaskelainen JE, et al. A randomized outcome study of enteral versus intravenous nimodipine in 171 patients after acute aneurysmal subarachnoid hemorrhage. World Neurosurg. 2012;78(1–2):101–9. https://doi.org/10.1016/j.wneu.2011.09.030.
Kronvall E, Undren P, Romner B, Saveland H, Cronqvist M, Nilsson OG. Nimodipine in aneurysmal subarachnoid hemorrhage: a randomized study of intravenous or peroral administration. J Neurosurg. 2009;110(1):58–63. https://doi.org/10.3171/2008.7.JNS08178.
Carlson AP, Hanggi D, Wong GK, Etminan N, Mayer SA, Aldrich F, et al. Single-dose intraventricular nimodipine microparticles versus oral nimodipine for aneurysmal subarachnoid hemorrhage. Stroke. 2020;51(4):1142–9. https://doi.org/10.1161/STROKEAHA.119.027396.
Carcas AJ, Abad-Santos F, De Rosendo JM, Frias J. Nimodipine transfer into human breast milk and cerebrospinal fluid. Ann Pharmacother. 1996;30(2):148–50.
Hynynen M, Siltanen T, Sahlman A, Pohjasvaara T, Muck W, Kaste M. Continuous infusion of nimodipine during coronary artery surgery: haemodynamic and pharmacokinetic study. Br J Anaesth. 1995;74(5):526–33.
Jakobsen P, Mikkelsen EO, Laursen J, Jensen F. Determination of nimodipine by gas chromatography using electron-capture detection; External factors influencing nimodipine concentrations during intravenous administration. J Chromatogr Biomed Appl. 1986;374(2):383–7.
Ramsch KD, Graefe KH, Scherling D. Pharmacokinetics and metabolism of calcium-blocking agents nifedipine, nitrendipine, and nimodipine. Am J Nephrol. 1986;6(SUPPL. 1):73–80.
Scheller C, Vogel AS, Simmermacher S, Rachinger JC, Prell J, Strauss C, et al. Prophylactic intravenous nimodipine treatment in skull base surgery: pharmacokinetic aspects. J Neurol Surg Part A Central Eur Neurosurg. 2012;73(3):153–9. https://doi.org/10.1055/s-0032-1313724.
Scheller C, Wienke A, Wurm F, Vogel AS, Simmermacher S, Prell J, et al. Enteral or parenteral nimodipine treatment: a comparative pharmacokinetic study. J Neurol Surg Part A Central Eur Neurosurg. 2014;75(2):84–90. https://doi.org/10.1055/s-0033-1337608.
Steudel WI, Carvi M, Lorenz R. Prognostic significance of the concentration of nimodipine in the CSF and plasma in acute subarachnoid hemorrhage. New Trends Clin Neuropharmacol. 1989;3(2):100.
Sramek JJ, Heller AH, Sundaresan PR, Lettieri J, Sawin S, Cutler NR. Safety and tolerance of intravenous nimodipine. Ann Pharmacother. 1994;28(10):1143–8. https://doi.org/10.1177/106002809402801001.
Gualano V, Ntsikoussalabongui B, Mignot A, Duvauchelle T, Felices M, Guillaume M, et al. Comparative bioavailability of two oral nimodipine formulations after administration to 24 healthy volunteers. Clin Drug Investig. 1999;17(6):475–82. https://doi.org/10.2165/00044011-199917060-00008.
He Z, Zhong D, Chen X, Liu X, Tang X, Zhao L. Development of a dissolution medium for nimodipine tablets based on bioavailability evaluation. Eur J Pharm Sci. 2004;21(4):487–91.
Liu XD, Xie L, Xu GQ, Wang J, Zhou YC, Liu GQ. Variability of nimodipine pharmacokinetics in healthy Chinese males. Chin J Pharmacol Toxicol. 1996;10(1):25–7.
Muck W, Tanaka T, Ahr G, Kuhlmann J. No interethnic differences in stereoselective disposition of oral nimodipine between Caucasian and Japanese subjects. Int J Clin Pharmacol Ther. 1996;34(4):163–71.
Qin F, Ma Y, Wang Y, Chen L, Wang D, Li F. Determination of nimodipine in human plasma by ultra performance liquid chromatography-tandem mass spectrometry and pharmacokinetic application. J Pharm Biomed Anal. 2008;46(3):557–62. https://doi.org/10.1016/j.jpba.2007.10.035.
Qiu F, Chen X, Li X, Zhong D. Determination of nimodipine in human plasma by a sensitive and selective liquid chromatography-tandem mass spectrometry method. J Chromatogr B Anal Technol Biomed Life Sci. 2004;802(2):291–7. https://doi.org/10.1016/j.jchromb.2003.12.001.
Zhao Y, Zhai D, Chen X, Yu Q, He H, Sun Y, et al. Determination of nimodipine in human plasma by HPLC-ESI-MS and its application to a bioequivalence study. J Chromatogr Sci. 2010;48(2):81–5.
Breuel HP, Muck W, Heine PR, Schmage N, Niklaus H, Kuhlmann J. The influence of nimodipine on hemodynamic parameters and peak and trough plasma concentrations of nifedipine chronically administered to elderly hypertensive patients. Int J Clin Pharmacol Ther. 1995;33(2):109–13.
Hernández-Hernández R, Coll T, Rachitzky P, Armas-Hernández MJ, Armas-Padilla MC, Velasco M, et al. Comparison of two nimodipine formulations in healthy volunteers. J Hum Hypertens. 2002;16(Suppl 1):S142–S144144.
Kirch W, Ramsch KD, Duhrsen U, Ohnhaus EE. Clinical pharmacokinetics of nimodipine in normal and impaired renal function. Int J Clin Pharmacol Res. 1984;4(5):381–4.
Muck W, Breuel HP, Kuhlmann J. The influence of age on the pharmacokinetics of nimodipine. Int J Clin Pharmacol Ther. 1996;34(7):293–8.
Muck W, Heine PR, Schmage N, Niklaus H, Horkulak J, Breuel HP. Steady-state pharmacokinetics of nimodipine during chronic administration of indometacin in elderly healthy volunteers. Arzneimittel-Forschung/Drug Res. 1995;45(4):460–2.
Terziivanov D, Atanasova I, Dimitrova V, Robeva R, Unger S, Muck W. Pharmacokinetic variability of nimodipine disposition after single and multiple oral dosing to hypertensive renal failure patients: parametric and nonparametric population analysis. Int J Clin Pharmacol Ther. 1999;37(8):404–12.
Acknowledgements
The authors acknowledge Janice Kung (librarian) for her guidance in the literature search process for this review.
Author information
Authors and Affiliations
Contributions
SHM had the idea of the article. FAI and XJ performed the literature search and data abstraction and summarization. All authors confirmed data collected and search and wrote the manuscript.
Corresponding author
Ethics declarations
Funding
There is no funding associated with this work.
Conflict of interest
All authors declare that they have no conflict of interest.
Ethics approval
No ethics approval required for this work.
Consent to participate
N/A.
Consent to publish
N/A.
Availability of data and material
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Code availability
N/A.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mahmoud, S.H., Ji, X. & Isse, F.A. Nimodipine Pharmacokinetic Variability in Various Patient Populations. Drugs R D 20, 307–318 (2020). https://doi.org/10.1007/s40268-020-00322-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00322-3