
Abstract
Genetic complexity and DNA damage repair defects are common in different cancer types and can induce tumor-specific vulnerabilities. Poly(ADP-ribose) polymerase (PARP) inhibitors exploit defects in the DNA repair pathway through synthetic lethality and have emerged as promising anticancer therapies, especially in tumors harboring deleterious germline or somatic breast cancer susceptibility gene (BRCA) mutations. However, the utility of PARP inhibitors could be expanded beyond germline BRCA1/2 mutated cancers by causing DNA damage with cytotoxic agents in the presence of a DNA repair inhibitor. US Food and Drug Administration (FDA)-approved PARP inhibitors include olaparib, rucaparib, and niraparib, while veliparib is in the late stage of clinical development. Talazoparib inhibits PARP catalytic activity, trapping PARP1/2 on damaged DNA, and it has been approved by the US FDA for the treatment of metastatic germline BRCA1/2 mutated breast cancers in October 2018. The talazoparib side effect profile more closely resembles traditional chemotherapeutics rather than other clinically approved PARP inhibitors. In this review, we discuss the scientific evidence that has emerged from both experimental and clinical studies in the development of talazoparib. Future directions will include optimizing combination therapy with chemotherapy, immunotherapies and targeted therapies, and in developing and validating biomarkers for patient selection and stratification, particularly in malignancies with ‘BRCAness’.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
PARP inhibitors are a family of enzymes that play a role in DNA repair. |
Tumors carrying mutations in BRCA1/2 and other genes implicated in homologous repair deficiency are particularly sensitive to PARP inhibition. |
Talazoparib has greater stereospecific PARP-DNA trapping ability than other PARP inhibitors. |
Evidence supporting the use of talazoparib in the treatment of ovarian cancer is limited in comparison with other PARP inhibitors. |
Talazoparib has mostly been investigated in breast cancer. |
1 Introduction
Ovarian cancer is one of the most common malignancies of the female genital tract, ranking third after cervical and uterine cancer. In 2017, there were 22,440 estimated new diagnoses of ovarian cancer and 14,080 deaths from the disease in the United States; deaths were higher than from cancer of the corpus uteri but lower than from cervical cancer [1]. Usually, patients with epithelial ovarian cancer (EOC) respond well to the initial standard treatment, which includes cytoreductive surgery followed by adjuvant platinum-based chemotherapy. Furthermore, it has been supported that neoadjuvant treatment is non-inferior to the standard primary debulking strategy in management of those who were fit for either procedure [2]. However, up to 80% of patients relapse and the estimated median progression-free survival (PFS) is approximately 12–18 months [3].
Recent advances in next-generation sequencing (NGS) have shown that the development of EOC is a complex multi-step process. Diverse genetic and epigenetic alterations play a fundamental role in tumorigenesis, progression, and development of drug resistance during the treatment course [4, 5]. Furthermore, two-thirds of patients are initially diagnosed with advanced or metastatic disease [6]. Together, chemoresistance and late diagnosis make EOC an incurable disease with an overall 5-year survival rate of < 30% [6].
Inhibition of poly(ADP-ribose) polymerase (PARP) in tumor cells in which repair of DNA is already impaired can lead to tumor cell death by increasing genomic instability [7]. The antitumor activity of PARP inhibition was first demonstrated in ovarian cancer cells [8]. Talazoparib’s mechanism of action includes inhibition of PARP1/2 enzymes, which play an instrumental role in detection and repair of single-strand DNA damage; subsequent PARP trapping, in which PARP proteins remain bound to a PARP inhibitor and with DNA, prevents DNA repair, replication, and transcription, ultimately leading to cell death. Cells with mutations in breast cancer susceptibility genes 1 or 2 (BRCA1/2) have an impaired double-strand DNA break (DSB) repair mechanism and are highly dependent on the single-strand repair pathway, regulated by PARP [9, 10]. In the case of genomic loss of DNA repair function or other impairment of homologous recombination (HR), synthetic lethality induced by PARP inhibition occurs and can be used to target tumor tissue selectively.
From the original in vitro observation to the clinical evidence of anticancer activity in BRCA-related recurrent EOC, PARP inhibitors have changed the therapeutic approach to patients with BRCA-mutated ovarian cancer. Currently, there are multiple PARP inhibitors approved and in clinical development for EOC therapy. Olaparib, rucaparib, and niraparib have all obtained US Food and Drug Administration (FDA) and/or European Medicines Agency (EMA) approval in ovarian cancer in different settings while veliparib is being investigated mainly in combination with chemotherapy or targeted agents and it is not currently approved in USA or Europe [11]. Talazoparib is the most potent of the PARP inhibitors, based on its enhanced capability to trap PARP on the DNA and its subsequent higher cytotoxicity [12]. However, this enhanced potency is accompanied by a toxicity profile more similar to that observed with chemotherapeutics, as compared with other clinically approved PARP inhibitors [12, 13]. Interestingly, combinations of PARP inhibitors with drugs that inhibit HR may sensitize EOC with primary or secondary HR proficiency to PARP inhibitors [11]. This observation suggests that PARP inhibitor therapeutic utility could be expanded beyond HR-deficient ovarian cancers.
In this review, we provide an updated overview of talazoparib clinical development, we discuss available results from recent and ongoing clinical trials and future challenges including identification of a wider population that can benefit from this class of agents.
2 Mechanisms of DNA Repair
DNA damage is a frequent event during cell life. It can be spontaneous or caused by cell metabolism or by environmental agents and can result in single-strand DNA breaks (SSBs) or DSBs [14]. In HR-deficient cells, DNA repair is impaired, leading to cell cycle arrest and/or cell death, whereas, in HR-proficient cells, DSBs are repaired, maintaining cell viability [15]. HR and non-homologous end joining (NHEJ) recombination are the two major pathways responsible for repairing DNA DSBs [16]. HR is a high fidelity repair mechanism, active during phase G2-S of the cell cycle [17], whereas NHEJ is faster but error prone [18]. Additional DNA damage repair mechanisms in mammalian cells include base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), and translesional synthesis, which are the primary mechanisms to resolve DNA SSBs [19]. In the case of functional defects of both HR and classical NHEJ, inhibition of PARP1 avoids activation of alternative NHEJ causing the death of HR-deficient cells [20].
Activation of PARP1 appears to affect DNA methylation, thus suggesting the existence of a close connection between DNA methylation and PARylation. If PARylation is inhibited and the BER pathway is impaired, SSBs persist and cause stalled replication forks and DSBs. This could lead to loss of genomic instability, cell death, and even carcinogenesis if not correctly repaired [21]. PARP1 also contributes to HR system functioning by either recruiting critical DNA repair factors such as NBS1 and MRE11 to sites of DSBs or preventing the Ku70/80 proteins’ interaction with free DNA ends [22]. Ku proteins are essential components of the NHEJ pathway, and PARP1 exerts an active role in their inhibition [22].
PARP1 accounts for approximately 90% of the total PARP activity and consists of three main domains; an NH2-terminal DNA binding domain, an automodification domain, and a C-terminal catalytic domain [7, 23]. The involvement of PARP2 and PARP3 in the DNA damage response has not been equally investigated. Nevertheless, PARP2 is known to be involved in BER/SSBs repair pathways, while PARP3 participates in DSBs repair through NHEJ [24].
PARP inhibitors may also function by trapping PARP1 and PARP2 at DNA lesions, thus abolishing PARylation-mediated biological processes such as DNA damage repair. PARP–DNA complexes have the ability to interfere with DNA replication, and recent studies have indicated that PARP trapping is important for the cytotoxicity of PARP inhibitors [25, 26]. This explains the different magnitude of cytotoxicity exerted by different PARP inhibitors. Among PARP inhibitors that have already been evaluated, olaparib, niraparib, and rucaparib trap PARP approximately 100-fold more efficiently than veliparib, while talazoparib appears to be the most potent PARP trapper investigated so far [25].
3 BRCA Genes and Cancer Susceptibility
Identification of BRCA genes as risk factors for cancer development and the availability of effective cancer treatments for patients with these mutations has promoted BRCA mutational analysis, genetic counseling, and risk assessment and treatment and has led to the framework of the management of breast and ovarian cancers [27]. The BRCA1 gene was identified in 1990 [28], whilst simultaneously, Stratton and Wooster working at the Institute of Cancer Research, London, UK discovered the BRCA2 gene [29].
The BRCA1 gene is located on the long arm of chromosome 17, consisting of 24 exons. A large number of deletions, insertions, or duplications have been reported in its sequence. BRCA1 takes part in response signaling of the DNA DSB damage, and the following repair depending on HR repair. It also participates in transcription regulating and cell-cycle checkpoint controlling. The BRCA2 gene plays a more direct repair role in HR repair relying on the regulation of RAD51, and it is located on the long arm of chromosome 13. It is larger than BRCA1 and consists of 27 exons. Approximately 2000 different mutations have been identified in both genes; nevertheless, they are not all risk-associated.
In terms of the risk for specific cancers in BRCA1 or BRCA2 mutation carriers, a prospective study reported cumulative risks of breast and ovarian cancer of 72% and 44%, respectively, for germline BRCA1, and 69% and 17% for germline BRCA2 mutations [30]. Of note, in the general population, the cumulative breast and ovarian cancer risk is 12% and 1.3%, respectively [31]. Germline BRCA1/2 accounts for 22.6% of mutations in high-grade serous EOC, usually accompanied by the loss of heterozygosis (LOH) [32]. On the other hand, somatic mutations are present in 6–7% [33], and BRCA1 hypermethylation occurs in around 10% of high-grade serous EOC [34]. Although BRCA-mutated ovarian cancers are frequently diagnosed at an advanced stage, and in young women, their prognosis remains better than that of BRCA wild-type EOC [32, 35].
4 Synthetic Lethality and BRCAness
Two preclinical studies published in 2005 demonstrated the exquisite in vitro sensitivity of BRCA-mutated cells to treatment with a selective inhibitor of PARP, providing strong support for their clinical development [8, 36]. The model proposed was based on the concept of synthetic lethality, which describes the combined lethal effect of two genetic variations that are otherwise non-lethal when occurring in isolation [37]. Based on this phenomenon known to geneticists since the last century, PARP inhibition has been evaluated in BRCA1/2-deficient tumor cells, with up to a 1000-fold increased sensitivity relative to BRCA wild-type tumor cells [15]. In addition to mutations in BRCA1/2 genes, genomic alterations involving other genes in HR pathways have been recognized [34], resulting in a BRCA-like phenotype, and conferring sensitivity to PARP inhibition [38, 39]. Indeed, the concept of ‘BRCAness’ has been introduced to describe the shared phenotype between sporadic cancers and familial cancers with BRCA1/2 mutations. This BRCAness phenotype may be attributed in part to defective HR secondary to several mechanisms, including hypermethylation of the BRCA1 promoter, somatic mutations of BRCA1/2, or EMSY amplification. Furthermore, several somatic mutations in genes beyond BRCA have been recognized so far, which are also involved in HR repair, such as ATM, ATR, BAP1, CDK12, CHEK2, FANCA, FANCC, FANCD2, FANCE, FANCF, PALB2, NBS1, WRN, RAD51C, RAD51D, MRE11A, CHEK1, BLM, and RAD51B. This is therapeutically important as the activity of PARP inhibitors has now been demonstrated in trials to extend beyond germline BRCA mutation-associated ovarian cancer [40]. The first demonstration of a clinically meaningful activity of olaparib in BRCA-proficient ovarian tumors was provided by Gelmon and colleagues in a phase II study (NCT00679783) [41]. Moreover, therapeutic interventions in BRCA proficient tumors and in tumors without BRCAness that could result in HR impairment with subsequent sensitization to PARP inhibitors is an approach that merits further investigation.
Ongoing efforts are directed towards the clinical application of synthetic lethality and the development of novel inhibitors that target the various components of the DNA repair mechanisms. Indeed, blocking of the BER pathway with PARP inhibitors significantly improves survival of cancer patients. Within this context, it has been demonstrated that Αtaxia telangiectasia and Rad3-related protein (ATR) inhibitor NU6027 inhibits RAD51 foci formation and exhibits a synthetic lethal relationship with BER inactivation in ovarian cancer cell line [42]. Similarly, targeted modulation of HR and NHEJ factors may develop novel cancer inhibitors. DNA-dependent protein kinase (DNA-PKcs) involved in the NHEJ process has been suggested as a therapeutic target [43].
5 Clinical Development of Poly(ADP-ribose) Polymerase (PARP) Inhibitors
PARP inhibitors were originally developed for cancer treatment as radio- and chemosensitizing drugs. Their activity is based on the concept of synthetic lethality. Olaparib, rucaparib, niraparib, veliparib, and talazoparib have been developed to a different extent either as a single agent or in combination therapy for the management of EOC.
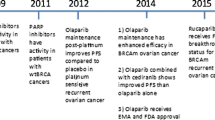
Historically, the EMA approved in 2014 a capsule formulation of olaparib in a maintenance setting for BRCA carriers with recurrent high-grade serous EOC, fallopian tube, or primary peritoneal carcinoma [40]. The maximum tolerated dose (MTD) of 400 mg twice daily was established based on the dose-limiting toxicities of fatigue, somnolence, and thrombocytopenia. A few months later, the FDA granted approval to olaparib for germline BRCA-mutated advanced EOC, fallopian tube, or primary peritoneal carcinoma, in patients previously treated with three or more lines of chemotherapy [44]. The tablet formulation of olaparib has been approved by both agencies for the maintenance therapy of platinum-sensitive recurrent ovarian cancer regardless of BRCA mutational status [40, 45]. Due to the enhanced bioavailability, the approved dose of the tablet is 300 mg twice daily [46]. The FDA approved olaparib maintenance treatment on December 19, 2018, based on the results of the SOLO-1 trial (NCT01844986), examined the efficacy of olaparib versus placebo in subjects with BRCA-mutated advanced EOC, fallopian tube, or primary peritoneal carcinoma, who responded to first-line platinum-based chemotherapy [47]. The estimated median PFS was not met in the olaparib arm and was 13.8 months in the placebo arm [hazard ratio (HazR) 0.30, 95% confidence interval (CI) 0.23–0.41; p < 0.001]. Overall survival (OS) data were not yet realized in the study. Very recently, the phase III PAOLA-1/ENGOT-ov25 trial (NCT02477644) assessed the addition of olaparib to bevacizumab maintenance therapy in unselected patients with advanced EOC, fallopian tube, or primary peritoneal carcinoma, who achieved complete response (CR) or partial response (PR), following first-line platinum-based therapy accompanied by bevacizumab [48]. As presented at the 2019 European Society for Medical Oncology (ESMO) congress, overall median PFS was significantly longer with the doublet compared with bevacizumab monotherapy (22.1 vs 16.6 months, respectively; HazR 0.59, 95% CI 0.49–0.72; p < 0.001). The cohorts with a BRCA mutation or HR deficiency achieved even longer PFS.
Following approval of olaparib, rucaparib and niraparib received authorization for the treatment of relapsed BRCA-mutated ovarian cancer. Rucaparib was approved by the FDA in December 2016 and by the EMA in May 2018 for the treatment of high-grade serous EOC, fallopian tube, or primary peritoneal carcinoma patients with germline or somatic BRCA mutations, relapsed after at least two chemotherapy lines. The efficacy was demonstrated by a pooled analysis of two multicenter, single-arm clinical trials; study 10 and ARIEL 2 study [49,50,51]. The established MTD in study 10 was 600 mg twice daily, whereas rucaparib treatment showed an ORR of 59.5%, with a median duration of response of 7.8 months in 42 pre-treated, platinum-sensitive, high-grade serous EOC patients carrying a germline BRCA mutation [49]. ARIEL 2 enrolled 192 platinum-sensitive EOC patients, stratified into three subgroups according HR deficiency status: BRCA1/2 mutated, BRCA wild-type with LOH high, and BRCA wild-type with LOH low, respectively [50]. The median PFS was significantly longer in the BRCA mutated subgroup (12.8 months; HazR 0.27; p < 0.0001) and in the BRCA wild-type/LOH high (5.7 months; HazR 0.62; p = 0.011), as compared with BRCA wild-type/LOH low subgroup (5.2 months). Similarly, the objective response rate (ORR) was higher in the BRCA1/2 mutated and BRCA wild-type/LOH high, than the BRCA wild-type/LOH low subgroup (80, 29, and 10%, respectively).
In March 2017, the FDA approved niraparib at a dosage of 300 mg daily as maintenance treatment of recurrent EOC, fallopian tube, or primary peritoneal carcinoma in patients obtaining CR or PR to platinum-based chemotherapy. The EMA’s approval for the same indication was established in November 2017. In October 2019, the FDA approved niraparib for patients with advanced HR-deficient EOC, or primary peritoneal cancer treated with at least three prior chemotherapy regimens, based on the results of the Quadra trial (NCT02354586) [52]. Efficacy was investigated in 98 patients with either tumor BRCA-mutated (n = 63) and/or a genomic instability score ≥ 42 (n = 35). The ORR was 24% (95% CI 16–34), whereas the estimated median duration of response reached 8.3 months (95% CI 6.5–not estimable). In the subset of BRCA-mutated EOC, the ORR was 39% (95% CI 17–64) in patients with platinum-sensitive disease, compared with 29% (95% CI 11–52) and 19% (95% CI 4–46) in those with platinum-resistant and platinum-refractory disease, respectively. Adverse events led to dose reduction or interruption in 73% of patients treated with niraparib in Quadra. Furthermore, the phase III PRIMA study (NCT02655016) compared niraparib with placebo in newly diagnosed, advanced, platinum-sensitive EOC regardless of BRCA mutation status. Niraparib extended PFS significantly more than placebo in both the overall population and the subset of patients with HR deficiency [53].
As far as veliparib is concerned, the phase III VELIA trial (NCT02470585) presented at the 2019 ESMO congress demonstrated that patients with high-grade serous EOC, fallopian tube, or primary peritoneal carcinoma experienced a 32% reduction in the risk of progression or death with frontline combination veliparib plus carboplatin and paclitaxel followed by veliparib maintenance treatment [54]. The VELIA study randomized patients in three arms. The control arm consisted of carboplatin and paclitaxel with placebo followed by placebo as maintenance (n = 375). In the second arm, veliparib was added at 150 mg twice daily to carboplatin and paclitaxel as induction therapy followed by placebo maintenance (n = 383). In the third arm, the maintenance treatment consisted of veliparib 400 mg twice daily (n = 382). The overall median PFS for the induction and maintenance phases combined in the veliparib arm was 23.5 months compared with 17.3 months in the placebo arm (HazR 0.68, 95% CI 0.56–0.83; p < 0.001). The benefit was more pronounced in BRCA mutants as the median PFS was 34.7 months compared with 22.0 months for veliparib and placebo, respectively (HazR 0.44, 95% CI 0.28–0.68; p < 0.001). A response to chemotherapy was not needed for inclusion in this trial, and PFS was measured from randomization (start of chemotherapy), in contrast to the trials of PARP inhibitor used only as maintenance treatment (SOLO-1, PAOLA-1/ENGOT-ov25, and PRIMA).
Finally, in 2018, both olaparib and talazoparib were FDA approved for BRCA carriers diagnosed with epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer, with disease recurrence following chemotherapy regardless of treatment setting [55, 56].
Talazoparib is a potent PARP1/2 inhibitor (PARP1 IC50: 0.57 nM), effective against BRCA1/2 and PTEN mutants in preclinical models. Its molecular formula is C19H14F2N6O and has a molecular weight equal to 380.359 g/mol. The chemical structure of talazoparib is shown in Fig. 1. It is well distributed into tissues, and its half-life observed at the MTD of 1.0 mg once daily is nearly 48 h. Talazoparib is characterized by greater stereospecific PARP-DNA trapping ability than other PARP inhibitors, and also potentiates cytotoxic effects of temozolomide, carboplatin, and the active metabolite of irinotecan SN-38 [12, 26].

Talazoparib compound summary
PARP inhibitors differ in their potency for catalytic inhibition and ability to trap PARP, which are related to the size, structure and allosteric differences in the NAD+ binding site of the relevant molecule. In this regard, talazoparib has the greatest in vitro cytotoxicity and PARP-trapping [26], followed by niraparib, whereas veliparib is the weakest one [25, 57]. This results in significant differences in doses among PARP inhibitors. As the most potent PARP trapping agent, talazoparib is recommended at a dose of 1 mg daily, as compared with 300 mg or greater for the remaining PARP inhibitors [58].
In terms of cost effectiveness, olaparib, niraparib, and rucaparib extend PFS in platinum-sensitive recurrent EOC, but are not currently priced in alignment with this benefit, with the exception of olaparib in recurrent, BRCA-mutated ovarian cancer [59]. It is essential that comparisons between trials require robust statistical methods, carefully selected data, and proper adjustments to ensure the control of inter-trial variability. Interestingly enough, a recent study compared olaparib in the upfront (SOLO-1) versus recurrent maintenance setting (SOLO-2). Although the higher cost associated with olaparib in SOLO-1 reflects the longer time patients stay on drug due to extended PFS, the incremental cost-effectiveness ratio recommends early initiation of first-line maintenance therapy among women with germline BRCA-mutated advanced EOC [60]. Overall, mature OS data are required to confirm whether olaparib, niraparib, and rucaparib are cost effective at the current high prices. It seems that the high costs of the drugs rather than associated adverse events may limit their cost effectiveness. Nevertheless, they may have only a modest budget impact from the public healthcare systems perspective.
6 Developing Predictive Biomarkers for PARP Inhibitors
As interest has evolved beyond germline BRCA1/2 mutations, multiplexed NGS assays of both germline and somatic DNA have accelerated the identification of cancers susceptible to PARP inhibitor therapy [61, 62]. Genomic signatures or scars could be used as an alternative to single gene mutation, deletion, or methylation analyses [63]. However, HR deficiency genomic signatures can be the result of so-called mutational genomic scars, which persist while the cancer cells reactivate HR.
Single nucleotide polymorphism array-based signatures of chromosome instability have been evaluated to measure ‘genomic scarring’. There are several important ‘scars’ used in HR deficiency tests. Numeric chromosomal instability represents the amplification or deletion of large fragments of chromosomes [64, 65]. Additional assays illustrating the burden of genomic instability include telomeric allelic imbalance (TAI), large-scale transition (LST) and LOH. They have been tested as predictive biomarkers of sensitivity to PARP inhibitors [66]. Among them, two have been approved by the FDA as companion diagnostics for PARP inhibitors in ovarian cancer. ‘FoundationFocus CDx BRCA LOH’ evaluates the frequency of LOH events throughout the genome, whereas ‘myChoice HRD’ (Myriad) is a composite signature of LOH, TAI, and LST events [67]. The ARIEL2 trial (NCT01891344) suggests that an assay using LOH to identify genomic scarring may be useful to predict PARP inhibitor response in ovarian cancer without BRCA1/2 mutations [50]. The study demonstrated significant difference in PFS between the rucaparib-treated high genomic LOH and biomarker-negative groups (HazR 0.67, 95% CI 0.45–0.99; p = 0.04), accompanied by more than doubling of the ORR (BRCA-like 36% versus biomarker-negative 16%).
Functional assays may directly inform on the capacity of the tumor cell to repair the damage, translating upstream DNA/RNA alterations. Accumulation of RAD51 at the DNA lesion is a recognized marker of HR proficiency; its absence following DNA damage represents a functional biomarker of HR dysfunction [68]. As such, γH2AX-RAD51 recently showed promising results in breast cancer biopsies, predicting PARP inhibitor sensitivity but also capturing the emergence of secondary resistance [69]. Indeed, the demethylation of RAD51C is associated with mRNA re-expression and development of resistance [70].
Finally, the role of Schlafen 11 (SLFN11) as a predictive biomarker for PARP inhibitor sensitivity was supported by the reported bimodal distribution in SLFN11 expression in treatment-naive patient tumor samples [71].
7 Development of Talazoparib in Ovarian Cancer
Evidence supporting the use of talazoparib specifically in the treatment of EOC is limited in comparison with other PARP inhibitors and it is still at an early stage of clinical development. However, some studies have evaluated the activity of talazoparib in several solid tumors. Chronologically, talazoparib therapy was evaluated in a two-part, phase I, first-in-human trial in 2017, in patients with germline BRCA1/2 mutated advanced or recurrent solid tumors, or selected sporadic cancers (NCT01286987) [13]. Thirty-four ovarian cancer patients were enrolled in nine cohorts and received doses of 25–1100 μg daily. A sustained PARP inhibition was observed at doses ≥ 60 μg daily. The recommended MTD was 1 mg daily. Seventeen patients with BRCA1/2-mutant high-grade serous EOC were treated at doses of at least 0.1 mg daily, and radiological, biochemical, and clinical benefit responses were achieved by 44, 70, and 82%, respectively. The median PFS reached was 36.4 weeks. In BRCA1/2 patients, the ORR to talazoparib was 50% (7/14). Tolerance over a treatment period exceeding 2 years was reasonable. Fatigue, nausea, and alopecia were observed in 30% of patients, followed by myelosuppression-related dose reductions (15%) and grade 3/4 anemia (13%), thrombocytopenia (14%), and neutropenia (6%). No patients withdrew permanently from treatment because of toxicity.
In 2017, Dhawan et al. published the results of a phase I/II trial on the safety and efficacy of talazoparib in combination with carboplatin in patients with solid tumors regardless of germline mutational status [72]. Two out of 24 enrolled patients (8%) were diagnosed with EOC, and 20% had a BRCA1/2 mutation. Grade 3/4 toxicities included fatigue (13%), neutropenia (63%), thrombocytopenia (29%), and anemia (38%). Neutropenia was more prominent in germline BRCA carriers. Three patients (14%) showed a PR or CR among BRCA-mutated patients. Among 21 evaluable patients, 11 (52%) experienced disease stabilization (range 7–22 weeks; median 10.5 weeks).
Furthermore, POSITION is a phase I study evaluating the effects of talazoparib on DNA copy number, RNA expression and protein levels in patients with stage IIIA–IV ovarian cancer (NCT02316834) [73]. Additionally, two trials have already been withdrawn. NCT02326844 was a phase II single-arm study of talazoparib in women with recurrent BRCA1/2-mutated ovarian cancer who have progressed on prior PARP inhibitor therapy after attaining a response [74]. The rationale was to establish whether re-challenge with an alternative PARP inhibitor can induce a further clinical response. Finally, a withdrawn phase II randomized study, NCT02836028, had been planned to compare the efficacy and safety of talazoparib monotherapy versus talazoparib with temozolomide in patients with BRCA-mutated or HR-deficient relapsed ovarian cancer [75]. Clinical trials of talazoparib for treatment of ovarian cancer are summarized in Table 1.
8 Development of Talazoparib in Breast Cancer
Among solid tumors, talazoparib has mostly been investigated in breast cancer. At the 2017 San Antonio Breast Cancer Symposium, the results of the EMBRACA study (NCT01945775) were presented. This open-label, randomized, two-arm, phase III trial compared the efficacy and safety of talazoparib with the physician’s choice of chemotherapy, which included eribulin, vinorelbine, capecitabine, or gemcitabine [56]. Patients with advanced and unresectable or metastatic HER2-negative breast cancer and a germline BRCA1/2 mutation were randomly assigned in a 2:1 ratio to talazoparib 1 mg once daily (n = 287) or chemotherapy (n = 144). The primary endpoint was PFS, whilst secondary endpoints included safety, OS, ORR, clinical benefit rate (CBR), at 24 weeks, and quality-of-life measurements. Within a follow-up of 14.5 months, median PFS was 8.6 and 5.6 months in the talazoparib and chemotherapy arms, respectively (HazR 0.54, 95% CI 0.41–0.71; p < 0.0001). This represents a 46% reduction in the risk of disease progression, whereas all key secondary efficacy endpoints demonstrated benefit with talazoparib treatment. ORR in the talazoparib cohort was more than double compared with the control arm (62.6 vs 27.2% for chemotherapy; p < 0.0001). This benefit was consistent and unrelated to hormone receptor expression, BRCA mutational status, previous systemic treatment, and history of intracranial disease. Talazoparib was generally well tolerated; nevertheless, grade 3/4 myelosuppression was higher with talazoparib than chemotherapy (55 vs 39%), but still manageable by dose modifications or treatment delays. This grade 3/4 hematological toxicity is more prominent in EMBRACA as compared with the previously reported NCT01286987 dose-escalation study of talazoparib. Nevertheless, the combination of talazoparib with carboplatin in the study of Dhawan et al. [72] was associated with higher rates of grade 3/4 neutropenia (63%). Non-hematologic toxicity was minimal and patients experienced fewer grade 3/4 gastrointestinal side effects with talazoparib as compared with chemotherapy (5.6 vs 11.9%, respectively). The FDA granted priority review designation for talazoparib based on the results of the EMBRACA study.
Efficacy of talazoparib in the EMBRACA study was comparable to that observed in a similar phase III, OlympiAD trial (NCT02000622), which compared olaparib versus single-agent chemotherapy in metastatic breast cancer [55]. The primary endpoint of median PFS in the OlympiAD trial was 7.0 months in the olaparib group versus 4.2 months in the standard therapy group (HazR 0.58, 95% CI 0.43–0.80; p < 0.001); nevertheless, there was no difference in OS between the two arms (19.3 vs 19.6 months, respectively; HazR 0.90, 95% CI 0.63–1.29; p = 0.57), likely due to a high degree of cross over. Patients previously treated with platinum chemotherapy were less likely to benefit from olaparib (HazR 0.67, 95% CI 0.48–1.14), suggesting overlap in the mechanisms of resistance to olaparib and platinum agents.
Encouraging signs of efficacy of talazoparib came also from the phase II ABRAZO trial (NCT02034916), conducted in patients with germline BRCA1/2 mutated, locally advanced or metastatic breast cancers with or without prior exposure to platinum agents [76]. Those who were enrolled on the platinum-exposed arm were further required to have a platinum-sensitive disease with a documented PR or CR to platinum agents and no history of disease progression on a platinum agent. Patients who had not been exposed to platinum were required to have had two or more non-platinum regimens in the metastatic setting. The primary outcome was ORR, whereas CBR, PFS, and OS were among the secondary outcome measures. Response rates to talazoparib were higher in platinum-naive patients [37.1% (95% CI 21.49–55.08)] than those with prior platinum exposure [0.8% (95% CI 10.47–34.99)], suggesting some degree of cross-resistance between platinum agents and the PARP inhibitor. This has also been demonstrated in women with high-grade serous EOC. Median PFS was 4.0 months (95% CI 2.8–5.4) with a median OS of 11.8 months (95% CI 8.8–15.0) in patients with prior platinum exposure (n = 49), as compared with 5.6 months (95% CI 5.5–7.8) and 16.5 months in those who were platinum-naive (n = 35), respectively. The reported ORR in patients with triple-negative breast cancer and hormone receptor-positive disease was 26% and 29%, respectively, whilst for BRCA1 and BRCA2 mutations carriers, it was 24% and 34%, respectively. As with other PARP inhibitors, myelosuppression was the predominant toxicity [anemia (52%), thrombocytopenia (33%), and neutropenia (27%)], in addition to nausea (42%), and diarrhea (33%).
Currently, there is a trend for investigation of PARP inhibitors in earlier stages of the disease, given that fewer resistance mechanisms will be present in this setting. Single-agent talazoparib has been assessed preoperatively in BRCA-mutated metastatic breast cancer in a small phase II trial (NCT03499353) [77]. The treatment produced significant pathologic CR with manageable toxicity. Clinical trials exploring talazoparib in ovarian cancer are listed in Table 2.
9 Expanding the Indications for Talazoparib to Other Tumor Types with Homologous Recombination (HR) Defects
It has been identified that patients with germline BRCA1/2 mutations beyond ovarian or breast cancer could respond to olaparib [78]. Based on that, PARP inhibitors can be expanded into additional HR-defective tumors.
Genomic studies have demonstrated that 8–12% of metastatic prostate cancers are characterized by BRCA2 mutations and homozygous deletions [79]. Incorporation of mutations in other genes involved in the DSB DNA repair, such as ATM, RAD51, PALB2, FANC, and PTEN, leads to elevation up to 20–25% of metastatic prostate cancers that harbor gene defects in different DNA-damage response pathways [80]. PARP1 represents a novel therapeutic target in prostate cancer based on the cross-talk between the androgen receptor and DNA-damage response pathways [81]. In the previously reported study by de Bono et al. [13], 4 out of 13 patients with pancreatic cancer treated with talazoparib at 1.0 mg once daily experienced CBR of 31% for at least 16 weeks. Among those responders, one harbored a PALB2 mutation, which is suggestive of potentially expanding applications for talazoparib therapy.
Beyond that, Table 3 depicts two additional trials of single-agent talazoparib that are currently recruiting patients with solid tumors. Phase II study NCT02286687 will explore talazoparib activity in advanced neoplasms with BRCA1/2 germline or somatic alterations, PTEN mutations or PTEN loss and HR deficiency defect [82], whilst the phase I/II NCT01989546 study will evaluate talazoparib efficacy in platinum-sensitive BRCA1/2-mutant tumors [83]. In this study, tumor biopsies for DNA damage response markers are taken prior to treatment, during cycle 1, and if the disease progresses. On the other hand, a phase I study (NCT02567396) designed to assess the utility and tolerability of talazoparib in the treatment of advanced or metastatic non-resectable solid tumors with liver or kidney dysfunction has been withdrawn [84].
10 PARP Inhibitors and Antiangiogenic Agents
The rationale for the combination of PARP inhibitors with anti-angiogenic agents is based on the fact that PARP inhibition decreases angiogenesis, whereas hypoxic state and vascular endothelial growth factor receptor 3 (VEGFR3) inhibitors induce down-regulation of HR repair proteins, such as BRCA1/2 and RAD51. However, hypoxia is also associated with hypoxia inducible factor 1 alpha (HIF1α) up-regulation. Due to PARP1 involvement in HIF1α stabilization, inhibition of PARP may prevent HIF1α accumulation that leads to targeted hypoxic-induced apoptosis.
The combination of the antiangiogenic agent cediranib and olaparib versus olaparib monotherapy has been evaluated in a randomized, open label, phase II study (NCT01116648) [85]. Interim analysis revealed a significantly longer PFS of 17.7 months in the experimental arm compared with 9.0 months for patients treated with single-agent olaparib (HazR 0.42; p = 0.005). The purpose of the GY004 trial (NCT02446600) was to compare olaparib monotherapy versus doublet therapy of olaparib and cediranib versus standard platinum-based chemotherapy in patients with platinum-sensitive recurrent ovarian cancer [86]. Similarly, the ICON 9 trial (NCT03278717) is examining maintenance therapy with the doublet of cediranib and olaparib, versus single agent olaparib [87]. With regards to platinum-resistant disease, three phase II/III trials are currently in progress [88,89,90].
As previously mentioned, PAOLA-1/ENGOT-ov25 (NCT02477644) is the first phase III trial that evaluated efficacy and safety of a PARP inhibitor plus bevacizumab as first-line maintenance therapy in advanced EOC [48]. Patients were randomized 2:1 to receive oral olaparib at 300 mg twice daily or placebo for up to 24 months. All subjects received standard maintenance bevacizumab (15 mg/kg every 3 weeks) for up to 15 months, and were stratified by first-line treatment outcome and tumor BRCA mutation. The reported median PFS was 22.1 months on the olaparib arm, compared with 16.6 months on the placebo (HazR 0.59, 95% CI 0.49–0.72; p < 0.0001). In the subset with BRCA mutation, median PFS with olaparib treatment was 37.2 months, compared with 21.7 months with placebo (HazR 0.31, 95% CI 0.20–0.47). BRCA wild-type patients demonstrated median PFS of 18.9 months with olaparib versus 16.0 months with placebo (HazR 0.71, 95% CI 0.58–0.88). Among tumors with HR deficiency, median PFS with olaparib was 37.2 months, compared with 17.7 months with placebo (HazR 0.33, 95% CI 0.25–0.45). HR-deficient BRCA wild-type patients treated with olaparib reached a median PFS of 28.1 months versus 16.6 months with placebo (HazR 0.43, 95% CI 0.28–0.66). However, the benefit of olaparib in negative or unknown HR deficiency status was limited; median PFS was 16.9 months versus 16.0 months for olaparib and placebo, respectively (HazR 0.92, 95% CI 0.72–1.17). Overall, PAOLA-1/ENGOT-ov25 met its primary objective, demonstrating a statistically significant prolongation of PFS in the intent-to-treat population. Patients with BRCA mutation and those with HR deficiency are most likely to benefit from this maintenance therapeutic strategy.
Similarly, in the phase II OVARIO study (NCT03326193), niraparib plus bevacizumab is evaluated as a maintenance treatment in patients with advanced EOC who have recovered from primary debulking surgery and have responded to frontline platinum-based chemotherapy with bevacizumab [91]. The starting dose of niraparib was based on baseline body weight and/or platelet count, whereas bevacizumab dosage was 15 mg/kg on a 3-weekly schedule up to 15 months, until disease progression or unacceptable toxicity.
Finally, AVANOVA (NCT02354131) is a phase II superiority trial, comparing single-agent niraparib 300 mg with combination niraparib-bevacizumab (15 mg/kg once every 3 weeks), until disease progression [92]. Ninety-seven enrolled women with high-grade serous or endometrioid platinum-sensitive EOC were assessed based on myChoice HR-deficient scores. Niraparib plus bevacizumab significantly improved PFS compared with niraparib alone [median PFS 11.9 months (95% CI 8.5–16.7) vs 5.5 months (95% CI 3.8–6.3), respectively; HazR 0.35, 95% CI 0.21–0.57; p < 0.0001].
11 PARP Inhibitors and Immune Checkpoint Inhibitors
The therapeutic strategy of combining PARP inhibitors with immunotherapies is based on the hypothesis that BRCA1/2 and wild-type BRCA1/2 HR deficiency ovarian tumors display a higher neo-antigen load than HR-proficient cancers [93]. The targeted blockade of the PD-L1 pathway can restore antitumor immunity and potentiate the antitumor activity of PARP inhibitors. Currently, there are available data from olaparib-durvalumab, and niraparib-pembrolizumab combinations [94, 95]. The reported toxicities are compatible with those that were observed for the relevant agents in monotherapy settings.
The phase I/II basket MEDIOLA trial (NCT02734004), evaluated the combination of olaparib and durvalumab in selected advanced solid cancers [94]. The phase I trial in patients with triple-negative breast and gynecological cancers demonstrated early evidence of efficacy, without overlapping toxicities [95]. In the phase II study among 32 patients with germline BRCA1/2 mutant platinum-sensitive ovarian cancer, disease control rate at 12 weeks and ORR were 81% and 63%, respectively. The most common reported adverse events of grade 3 or more were anemia (12%) and increased lipase (9%), along with any-grade hypothyroidism (15%) and rash (12%) [94].
The phase I/II TOPACIO trial (NCT02657889) enrolled heavily pretreated patients with platinum-resistant, or secondarily platinum-refractory EOC [96]. Based on dose finding in phase I, the recommended phase II dose of niraparib and pembrolizumab was 200 mg orally once daily and 200 mg intravenously 3-weekly, respectively. Among 60 patients evaluable for initial response assessment, 64% had platinum-resistant, 19% had platinum-refractory, and 17% had platinum-sensitive ovarian cancer, respectively [97]. The overall ORR and disease control rate were 25% and 68%, respectively, whilst in the BRCA1/2 mutant cohort they were higher (45% and 73%, respectively). Interestingly enough, the ORR in BRCA wild-type and HR deficiency negative subgroups was 24% and 27%, respectively. This is suggestive of treatment efficacy even in populations not typically responsive to single-agent PARP inhibitors. Preliminary data revealed adverse events compatible with treatment with single agents. Grade 3 or more toxicities included anemia (17%), fatigue (6%), and thrombocytopenia (3%) [98].
Currently, there are five ongoing phase III studies in the maintenance setting. FIRST (NCT03602859) was designed to assess platinum and the PD-L1 inhibitor TSR-042, followed by niraparib and TSR-042 maintenance therapy, versus standard platinum-based treatment followed by maintenance niraparib or placebo, as first-line treatment of advanced EOC [99]. The ENGOT-ov46/AGO/DUO-O study (NCT03737643) evaluates the efficacy and safety of the standard platinum-based chemotherapy and bevacizumab followed by maintenance bevacizumab as monotherapy, or in combination with durvalumab, either alone or combined with olaparib [100]. ATHENA is a four-arm study (NCT03522246) that is currently evaluating the combination of rucaparib with nivolumab for those responded to front-line platinum-based chemotherapy [101]. Pembrolizumab in combination with chemotherapy followed by olaparib maintenance therapy is being investigated in the phase III, randomized, double‐blind ENGOT‐ov43/KEYLYNK‐001 study (NCT03740165) in first‐line treatment of patients with BRCA1/2 wild-type advanced EOC [102]. Randomization is in a 1:1:1 ratio to pembrolizumab + chemotherapy followed by olaparib maintenance; pembrolizumab + chemotherapy followed by placebo; or placebo + chemotherapy followed by placebo. Bevacizumab use is determined at the investigator’s discretion before randomization. Finally, JAVELIN Ovarian PARP 100 (B9991030) is an open-label, international, multi-center, randomized study designed to evaluate the efficacy and safety of the combination of avelumab and chemotherapy followed by avelumab and talazoparib maintenance compared with chemotherapy and bevacizumab followed by bevacizumab maintenance in treatment-naïve patients with locally advanced or metastatic EOC (NCT03642132) [103]. Approximately 720 patients will be randomized in a 2.5:1:2.5 ratio to receive paclitaxel + carboplatin + avelumab, followed by avelumab + talazoparib maintenance for up to 24 months (Arm A); paclitaxel + carboplatin, followed by talazoparib maintenance for up to 24 months (Arm B); or paclitaxel + carboplatin + bevacizumab, followed by bevacizumab maintenance for up to 21 doses or 22 per local approval (Arm C). Patients are stratified by germline BRCA1/2 mutation.
12 Talazoparib Combination Therapies
PARP inhibitors have been combined with chemotherapy based on the fact that they block BER, and consequently may potentiate the efficacy of cytotoxins [104]. A major concern for this therapeutic strategy is the high risk of overlapping myelotoxicity; for this reason, initiation of standard chemotherapy doses has been accompanied by dose escalation of the PARP inhibitor. Furthermore, intermittent dosing schedules and supportive treatment with granulocyte colony-stimulating factors are also reasonable strategies to improve the tolerability of chemotherapy in combination with PARP inhibitors.
A phase III neoadjuvant BrighTNess trial (NCT02032277) in patients with stage II–III, operable triple-negative breast cancer is a placebo-controlled study, where patients were randomized 2:1:1 to veliparib 50 mg twice daily in combination with the standard chemotherapy doublet of carboplatin and paclitaxel, versus paclitaxel plus carboplatin plus veliparib placebo (twice daily), versus paclitaxel plus carboplatin placebo plus veliparib placebo [105]. Pathologic CR for the experimental arm was not significantly better than standard chemotherapy. As such, subtherapeutic tolerated doses of PARP inhibitors with chemotherapy may not potentiate therapeutic efficacy.
Among studies that investigate the utility of a combination of immune checkpoint blockade with PARP inhibitors, a phase Ib/II JAVELIN PARP MEDLEY (NCT03330405) trial is currently recruiting patients with germline BRCA1/2 mutations or ATM-deficient advanced solid tumors including breast and ovarian cancers, for evaluation of talazoparib in combination with PD-L1 inhibitor avelumab [106]. Given the non-overlapping toxicities of immune checkpoint therapies and PARP inhibitors, the combination is expected to be well tolerated.
Α study demonstrated that combination treatment of talazoparib and the next-generation DNA methyltransferase inhibitor guadecitabine optimized PARP inhibitors’ response in breast and high-grade serous EOC cell lines, both in vitro and in vivo, regardless of BRCA status [107]. Indeed, the combination of guadecitabine and talazoparib in vitro decreased clonogenic survival, cell proliferation, and increased caspase 3/7 cleavage, whereas, in vivo, it reduced tumor burden in BRCA-proficient ovarian and breast cancer xenograft models, and prolonged survival. Furthermore, enhanced PARP activation by guadecitabine was in part dependent on reactive oxygen species and protein kinase A (PKA) activation, which represents the major regulator of PARP activation, and which may have led to PARP inhibitor response. Tolerance in mice was not problematic and this treatment doublet should be further explored for patients with either intrinsic or treatment-induced resistance to the PARP inhibitors. Table 4 depicts the ongoing studies of talazoparib in combination with immunotherapy and cytotoxic chemotherapy.
13 PARP Inhibitors Resistance
PARP inhibitor research has led to important new understandings about PARP inhibitor resistance mechanisms. Each PARP inhibitor has a separate chemical structure with diverse off-target effects [108]. This indicates that the utilization of a secondary PARP inhibitor could be therapeutically beneficial in a resistant tumor. Among the resistance mechanisms identified to date, restoration of homology-directed DNA repair is frequently observed in vitro and in vivo. Indeed, the restoration of BRCA activity starts from BRCA-deficient and chemo-sensitive cells as a result of several mutations that are induced by platinum agents. This initial restored clone expands in the setting of treatment-specific selective pressure [109].
Compensatory deleterious mutations have also been detected to confer PARP inhibitor resistance. TP53 binding protein 1 (53BP1) maintains the balance between HR and NHEJ [110]. Loss of 53BP1 function by either mutation or downregulation accelerates the BRCA1-independent end-resection and provides PARP inhibitor resistance [111]. It has been demonstrated that the inactivation of downstream factors of 53BP1-mediated repair, typically RIF1 and REV7, also leads to the restoration of DNA end resection, and consequently promotes homology-mediated repair [112]. Loss of the 53BP1-RIF1-REV7 pathway in PARP inhibitor-resistant BRCA1-deficient cells results in hypersensitivity to ionizing radiation.
The knockdown of cyclin-dependent kinase 12 (CDK12) is associated with concomitant downregulation of DNA repair proteins. In vitro, pharmacological inhibition of CDK12 with dinaciclib reverses acquired PARP inhibitor resistance [113]. Furthermore, it has been shown that the inhibition of cell cycle regulator WEE1 leads cells to enter the S-phase of the cell cycle. This enforces accumulation of DNA DSBs in the context of HR deficiency and PARP inhibition [114]. Overall, a combined inhibition of CDK12 or WEE1 could be therapeutically indicated for overcoming HR-restored PARP inhibitor resistance.
Beyond mechanisms of HR restoration in BRCA1-deficient cells, it has been recently observed in vitro that loss of PAR glycohydrolase (PARG), responsible for degrading nuclear PAR, is involved in a major resistance mechanism [112]. Endogenous PARG activity is crucial for the success of PARP inhibition and PARG suppression restores PARP1 signaling upon PARP inhibitor treatment. Based on that, PARG activity may serve as a predictive marker for PARP inhibitor therapy. Furthermore, there is a correlation between increased radiosensitivity of PARP inhibitor-resistant tumors and lost PARG [112]. As such, radiotherapy could be a therapeutic option for PARP inhibitor-resistant tumors in patients without BRCA1/2 reversion mutations. However, radiotherapy is not typically included in the treatment of EOC. Consequently, alternating treatment cycles of PARP inhibitor and radiomimetic drugs would be more effective than the PARP inhibitor maintenance treatment in platinum-sensitive EOC.
Another mechanism of inherent or acquired resistance is the upregulation of genes encoding p-glycoprotein efflux pumps, related to decreased intracellular drug levels. This resistance can be reverted by the ABCB1 inhibitors verapamil, elacridar, and tariquidar [115].
Overall, there are several ongoing combination clinical trials seeking to abrogate PARP inhibitor resistance up front, and we eagerly anticipate results.
14 Conclusions and Future Perspectives
More research is required to improve our understanding of DNA repair mechanisms. With a multitude of ongoing studies in different patient populations and clinical settings, it is likely that the indications for PARP inhibitors in ovarian cancer will continue to expand. The analytical validation and clinical qualification of biomarkers enabling patient stratification are of critical importance to deliver precision treatment. In this regard, several assays have already been developed to test tumor tissue for a BRCA-like phenotype, loosely defined as HR repair deficiency. Indeed, molecular signatures for the prediction of response to PARP inhibitors include DNA repair and replication defects, expression of SLFN11, and hyperactive drug efflux pumps. Furthermore, widespread somatic BRCA testing should be incorporated in routine clinical practice, given that somatic BRCA1/2 mutants are potentially PARP inhibitor candidates. An open question is whether HER2-negative breast cancers harboring somatic mutations may also benefit from this novel agent.
Resistance to PARP inhibitors also needs to be further explored. The most common acquired resistance mechanism to PARP inhibitors consists of secondary mutations restoring the BRCA1/2 protein functionality. Recently, secondary mutations that restored the open reading frame of BRCA or HR-related genes have been detected by NGS in ovarian cancer patients at disease progression during treatment with PARP inhibitors. Therapeutically, combinations of PARP inhibitors with drugs that inhibit HR might be an effective approach to sensitize ovarian cancers with de novo or acquired HR proficiency to PARP inhibitors. The discovery and characterization of talazoparib as a potent, selective, orally bioavailable PARP1/2 inhibitor provides an important addition to the field of PARP inhibitors. Its potency in PARP trapping is the early evidence that talazoparib could potentially lead to improved clinical outcomes in BRCA mutant malignancies.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Kehoe S, Hook J, Nankivell M, Jayson GC, Kitchener H, Lopes T, et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet. 2015;386:249–57.
Papa A, Caruso D, Strudel M, Tomao S, Tomao F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J Transl Med. 2016;14:267.
Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res. 2013;19:961–8.
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34.
Bowtell DD, Böhm S, Ahmed AA, Aspuria PJ, Bast RC Jr, Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668–79.
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5:387–93.
Javle M, Curtin NJ. The potential for poly (ADP-ribose) polymerase inhibitors in cancer therapy. Ther Adv Med Oncol. 2011;3:257–67.
Boussios S, Karihtala P, Moschetta M, Abson C, Karathanasi A, Zakynthinakis-Kyriakou N, et al. Veliparib in ovarian cancer: a new synthetically lethal therapeutic approach. Investig New Drugs. 2020;38:181–93.
Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19:5003–15.
de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7:620–9.
Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15.
Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–90.
Drew Y. The development of PARP inhibitors in ovarian cancer: from bench to bedside. Br J Cancer. 2015;113:S3–9.
Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:366–74.
Radhakrishnan SK, Jette N, Lees-Miller SP. Non-homologous end joining: emerging themes and unanswered questions. DNA Repair (Amst). 2014;17:2–8.
Plummer R. Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res. 2010;16:4527–31.
Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hoser G, Maifrede S, et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Investig. 2017;127:2392–406.
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74.
Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA. 2011;108:3406–11.
Ohmoto A, Yachida S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 2017;10:5195–208.
Langelier MF, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–75.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–99.
Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433–43.
Audeh MW. Novel treatment strategies in triple-negative breast cancer: specific role of poly(adenosine diphosphate-ribose) polymerase inhibition. Pharmgenom Pers Med. 2014;7:307–16.
Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–9.
Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265:2088–90.
Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317:2402–16.
Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, et al (eds). SEER Cancer Statistics Review, 1975–2014, National Cancer Institute. Bethesda, MD. https://seer.cancer.gov/csr/1975_2014/.
Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30:2654–63.
Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28:3570–6.
Bell D, Berchuck A, Birrer M, Chien J, Cramer D, Dao F, Cancer Genome Atlas Research Network, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, Cancer Genome Atlas Research Network, et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. 2012;307:382–90.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18:613–23.
Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–97.
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852–61.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–61.
Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011;105:372–81.
Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, et al. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103:4659–65.
Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–50.
Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1274–84.
Mateo J, Moreno V, Gupta A, Kaye SB, Dean E, Middleton MR, et al. An adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparib. Target Oncol. 2016;11:401–15.
Olaparib Maintenance Monotherapy in Patients With BRCA Mutated Ovarian Cancer Following First Line Platinum Based Chemotherapy. (SOLO-1). https://clinicaltrials.gov/ct2/show/NCT01844986. Accessed 29 Nov 2019.
Ray-Coquard IL, Pautier P, Pignata S, Pérol D, González-Martín A, Sevelda P, et al. Phase III PAOLA-1/ENGOT-ov25 trial: olaparib plus bevacizumab (bev) as maintenance therapy in patients (pts) with newly diagnosed, advanced ovarian cancer (OC) treated with platinum-based chemotherapy (PCh) plus bev. Ann Oncol. 2019;30:v853–934.
Kristeleit R, Shapiro GI, Burris HA, Oza AM, LoRusso P, Patel MR, et al. A phase I–II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res. 2017;23:4095–106.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87.
Oza AM, Tinker AV, Oaknin A, Shapira-Frommer R, McNeish IA, Swisher EM, et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from study 10 and ARIEL2. Gynecol Oncol. 2017;147:267–75.
Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20:636–48.
González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381:2391–402.
Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381:2403–15.
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–33.
Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753–63.
Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer. 2016;115:1157–73.
Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8:362ps17.
Liu AY, Cohen JG, Walsh CS, Holschneider CH, Sinno AK. A cost-effectiveness analysis of three PARP inhibitors for maintenance therapy in platinum-sensitive recurrent ovarian cancer. Gynecol Oncol. 2017;147:196.
Wolford JE, Tewari KS, Liang SY, Bai J, Mann AK, Moore KN, et al. SOLO1 versus SOLO2: cost-effectiveness of olaparib as maintenance therapy for newly diagnosed and platinum-sensitive recurrent ovarian carcinoma among women with germline BRCA mutations (gBRCAmut). J Clin Oncol. 2019;37:5545.
Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32:2001–9.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–6.
Vanderstichele A, Busschaert P, Olbrecht S, Lambrechts D, Vergote I. Genomic signatures as predictive biomarkers of homologous recombination deficiency in ovarian cancer. Eur J Cancer. 2017;86:5–14.
Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014;16:211.
Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–25.
Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2014;33:3894–907.
Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol. 2018;29:1203–10.
Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun. 2018;9:3970.
Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res. 2017;23:523–35.
Dhawan MS, Bartelink IH, Aggarwal RR, Leng J, Zhang JZ, Pawlowska N, et al. Differential toxicity in patients with and without DNA repair mutations: phase I study of carboplatin and talazoparib in advanced solid tumors. Clin Cancer Res. 2017;23:6400–10.
Talazoparib in Determining Genetic Effects on Disease Response in Patients With Advanced Ovarian, Fallopian Tube, or Primary Peritoneal Cancer (POSITION). https://clinicaltrials.gov/ct2/show/NCT02316834. Accessed 29 Nov 2019.
BMN 673 (Talazoparib), an Oral PARP Inhibitor, in People With Deleterious BRCA1/2 Mutation-Associated Ovarian Cancer Who Have Had Prior PARP Inhibitor Treatment. https://clinicaltrials.gov/ct2/show/NCT02326844. Accessed 29 Nov 2019.
A Study Evaluating Talazoparib in Relapsed Ovarian, Fallopian Tube, and Peritoneal Cancer. https://clinicaltrials.gov/ct2/show/NCT02836028. Accessed 29 Nov 2019.
Turner NC, Telli ML, Rugo HS, Mailliez A, Ettl J, Grischke EM, et al. A phase II study of talazoparib after platinum or cytotoxic nonplatinum regimens in patients with advanced breast cancer and germline BRCA1/2 mutations (ABRAZO). Clin Cancer Res. 2019;25:2717–24.
Litton JK, Scoggins ME, Hess KR, Adrada BE, Murthy RK, Damodaran S, et al. Neoadjuvant talazoparib for patients with operable breast cancer with a germline BRCA pathogenic variant. J Clin Oncol. 2020;38:388–94.
Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol. 2016;27:1013–9.
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–53.
Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50:645–51.
Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19:975–86.
Talazoparib in Treating Patients With Recurrent, Refractory, Advanced, or Metastatic Cancers and Alterations in the BRCA Genes. https://clinicaltrials.gov/ct2/show/NCT02286687. Accessed 29 Nov 2019.
Pilot Trial of BMN 673, an Oral PARP Inhibitor, in Patients With Advanced Solid Tumors and Deleterious BRCA Mutations. https://clinicaltrials.gov/ct2/show/NCT01989546. Accessed 29 Nov 2019.
Talazoparib in Treating Patients With Advanced or Metastatic Solid Tumors That Cannot Be Removed by Surgery and Liver or Kidney Dysfunction. https://clinicaltrials.gov/ct2/show/NCT02567396. Accessed 29 Nov 2019.
Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15:1207–14.
Olaparib or Cediranib Maleate and Olaparib Compared with Standard Platinum-Based Chemotherapy in Treating Patients with Recurrent Platinum-Sensitive Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. https://clinicaltrials.gov/ct2/show/NCT02446600. Accessed 15 Feb 2020.
Study Evaluating the Efficacy of Maintenance Olaparib and Cediranib or Olaparib Alone in Ovarian Cancer Patients (ICON9). https://clinicaltrials.gov/ct2/show/NCT03278717. Accessed 15 Feb 2020.
Cediranib Maleate and Olaparib or Standard Chemotherapy in Treating Patients with Recurrent Platinum-Resistant or -Refractory Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. https://clinicaltrials.gov/ct2/show/NCT02502266. Accessed 15 Feb 2020.
Olaparib +/- Cediranib or Chemotherapy in Patients With Platinum-resistant Ovarian Cancer (OCTOVA). https://clinicaltrials.gov/ct2/show/NCT03117933. Accessed 15 Feb 2020.
Efficacy and Safety Study of Cediranib in Combination with Olaparib in Patients with Recurrent Platinum-Resistant Ovarian Cancer (CONCERTO). https://clinicaltrials.gov/ct2/show/NCT02889900. Accessed 15 Feb 2020.
Hardesty M, Hope JM, Krivak T, Chen J, Wainszelbaum M, Gupta D, et al. OVARIO: a single-arm, open-label phase 2 study of maintenance therapy with niraparib + bevacizumab (bev) in patients (pts) with advanced ovarian cancer (OC) after response to frontline platinum-based chemotherapy (chemo). Ann Oncol. 2018;29:v332–58.
Mirza MR, Åvall Lundqvist E, Birrer MJ, dePont Christensen R, Nyvang GB, Malander S, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol. 2019;20:1409–19.
Boussios S, Karihtala P, Moschetta M, Karathanasi A, Sadauskaite A, Rassy E, et al. Combined strategies with poly(ADP-Ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer: a literature review. Diagnostics (Basel). 2019;9:E87.
Drew Y, de Jonge M, Hong SH, Park YH, Wolfer A, Brown J, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol. 2018;149:246–7.
Lee JM, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, Annunziata CM, et al. Safety and clinical activity of the programmed death-ligand 1 inhibitor durvalumab in combination with poly(ADP-Ribose) polymerase inhibitor olaparib or vascular endothelial growth factor receptor 1–3 inhibitor cediranib in women’s cancers: a dose-escalation. Phase I study. J Clin Oncol. 2017;35:2193–202.
Niraparib in Combination with Pembrolizumab in Patients with Triple-negative Breast Cancer or Ovarian Cancer (TOPACIO). https://clinicaltrials.gov/ct2/show/NCT02657889. Accessed 15 Feb 2020.
Konstantinopoulos PA, Waggoner SE, Vidal GA, Mita MM, Fleming GF, Holloway RW, et al. TOPACIO/Keynote-162 (NCT02657889): a phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—results from ROC cohort. J Clin Oncol. 2018;36:106.
Konstantinopoulos PA, Munster P, Forero-Torez A, Holloway RW, Schwartzberg L, Matulonis UA, et al. Topacio: preliminary activity and safety in patients (pts) with platinum-resistant ovarian cancer (PROC) in a phase 1/2 study of niraparib in combination with pembrolizumab. Gynecol Oncol. 2018;149:246.
A Phase 3 Comparison of Platinum-Based Therapy With TSR-042 and Niraparib Versus Standard of Care Platinum-Based Therapy as First-Line Treatment of Stage III or IV Nonmucinous Epithelial Ovarian Cancer (FIRST). https://clinicaltrials.gov/ct2/show/NCT03602859. Accessed 15 Feb 2020.
Durvalumab Treatment in Combination with Chemotherapy and Bevacizumab, Followed by Maintenance Durvalumab, Bevacizumab and Olaparib Treatment in Advanced Ovarian Cancer Patients. (DUO-O). https://clinicaltrials.gov/ct2/show/NCT03737643. Accessed 15 Feb 2020.
A Study in Ovarian Cancer Patients Evaluating Rucaparib and Nivolumab as Maintenance Treatment Following Response to Front-Line Platinum-Based Chemotherapy (ATHENA). https://clinicaltrials.gov/ct2/show/NCT03522246. Accessed 15 Feb 2020.
Fujiwara K, Vergote IB, Sehouli J, Salutari V, Zola P, Madry R, et al. 264TiP—ENGOT-ov43/KEYLYNK-001: a phase III trial of pembrolizumab plus chemotherapy with olaparib maintenance for first-line treatment of BRCA-nonmutated advanced epithelial ovarian cancer. Ann Oncol. 2019;30:ix89.
Eskander RN, Ledermann JA, Birrer MJ, Fujiwara K, Gaillard S, Richardson GE, et al. JAVELIN ovarian PARP 100 study design: phase III trial of avelumab + chemotherapy followed by avelumab + talazoparib maintenance in previously untreated epithelial ovarian cancer. J Clin Oncol. 2019;37:TPS9.
Boussios S, Karathanasi A, Cooke D, Neille C, Sadauskaite A, Moschetta M, et al. PARP inhibitors in ovarian cancer: the route to “Ithaca”. Diagnostics (Basel). 2019;9:E55.
Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19:497–509.
Javelin Parp Medley: Avelumab Plus Talazoparib In Locally Advanced Or Metastatic Solid Tumors. https://clinicaltrials.gov/ct2/show/NCT03330405. Accessed 29 Nov 2019.
Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H, et al. An effective epigenetic-PARP inhibitor combination therapy for breast and ovarian cancers independent of BRCA mutations. Clin Cancer Res. 2018;24:3163–75.
Antolín AA, Mestres J. Linking off-target kinase pharmacology to the differential cellular effects observed among PARP inhibitors. Oncotarget. 2014;5:3023–8.
Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell. 2018;33:1078–93.
Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54.
Makvandi M, Xu K, Lieberman BP, Anderson RC, Effron SS, Winters HD, et al. A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP inhibitor therapy. Cancer Res. 2016;76:4516–24.
Francica P, Rottenberg S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018;10:101.
Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, et al. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016;17:2367–81.
Garcia TB, Snedeker JC, Baturin D, Gardner L, Fosmire SP, Zhou C, et al. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017;16:2058–68.
Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer. 2016;115:431–41.
Phase II Talazoparib in BRCA1 +BRCA2 Wild-Type & Triple-Neg /HER2-Negative Breast Cancer /Solid Tumors. https://clinicaltrials.gov/ct2/show/NCT02401347. Accessed 29 Nov 2019.
I-SPY 2 TRIAL: Neoadjuvant and Personalized Adaptive Novel Agents to Treat Breast Cancer (I-SPY 2). https://clinicaltrials.gov/ct2/show/NCT01042379. Accessed 29 Nov 2019.
Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–54.
Study of BMN-673 With Carboplatin and Paclitaxel in Patients With Advanced BRCA-mutated Solid Tumor or Triple Negative Metastatic Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT02358200. Accessed 29 Nov 2019.
PARP Inhibitor BMN-673 and Temozolomide or Irinotecan Hydrochloride in Treating Patients With Locally Advanced or Metastatic Solid Tumors. https://clinicaltrials.gov/ct2/show/NCT02049593. Accessed 29 Nov 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Stergios Boussios declares that he has no conflict of interest. Charlotte Abson declares that she has no conflict of interest. Michele Moschetta declares that he has no conflict of interest. Elie Rassy declares that he has no conflict of interest. Afroditi Karathanasi declares that she has no conflict of interest. Tahir Bhat declares that he has no conflict of interest. Faisal Ghumman declares that he has no conflict of interest. Matin Sheriff declares that he has no conflict of interest. Nicholas Pavlidis declares that he has no conflict of interest.
Funding
The work was supported by the Medway NHS Foundation Trust, Gillingham, Kent, UK.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Boussios, S., Abson, C., Moschetta, M. et al. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 20, 55–73 (2020). https://doi.org/10.1007/s40268-020-00301-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00301-8