
Abstract
Hyperphosphatemia in chronic kidney disease (CKD) has been associated with elevated cardiovascular morbidity and mortality. Serum phosphate control remains a cornerstone of the clinical management of patients with CKD, in order to both attenuate the progression of secondary hyperparathyroidism or bone disease and (possibly) reduce the risk of vascular calcification. Despite technical improvements in dialysis and the use of dietary restrictions, drug therapy is often required to control phosphate levels in patients with end-stage renal disease (ESRD). Currently available medications for hyperphosphatemia in ESRD are very expensive and not always well tolerated. The discovery and development of new drugs in this indication is therefore a priority for both medical and health-economic reasons. Nicotinamide (an amide derivative of the water-soluble vitamin B3) is a potentially interesting alternative to phosphate binders. In vitro and in vivo data show that nicotinamide reduces hyperphosphatemia by inhibiting sodium-dependent phosphate co-transport in the renal proximal tubule and in the intestine. Accordingly, targeting the sodium-dependent phosphate co-transporter 2b by using nicotinamide as an alternative or adjunct to classical phosphate binders may be a therapeutic option for modulating serum phosphate in CKD. Several recent clinical studies have explored the potential value of nicotinamide in phosphate control (as well as its effects on lipid levels) in dialysis patients. However, we consider that more data on pharmacodynamics, pharmacokinetics and safety are needed before this compound can be recommended as a treatment for hyperphosphatemia in ESRD patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Hyperphosphatemia is a common complication of chronic kidney disease (CKD) and particularly affects dialysis patients. A decline in renal function leads to phosphate retention, elevated parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23) levels, and low 1,25-dihydroxy vitamin D levels [1]. In patients with end-stage renal disease (ESRD), phosphate intake in the diet exceeds phosphate excretion by the kidneys; hence, serum phosphate levels rise progressively. Indeed, in patients with advanced CKD, hyperphosphatemia is a serious clinical problem and leads to a variety of complications, such as secondary hyperparathyroidism, vascular disease and increased vascular calcification [2]. Epidemiological studies have demonstrated a significant association between hyperphosphatemia and increased mortality in ESRD patients [3, 4] and between hyperphosphatemia and increased cardiovascular mortality and hospitalization in dialysis patients [5].
In subjects with unimpaired renal function, the normal range for serum phosphorus is 2.7–4.6 mg/dL (0.9–1.5 mmol/L). The ‘Kidney Disease: Improving Global Outcomes’ (KDIGO) guidelines state that (1) phosphorus concentrations in CKD patients should be lowered toward the normal range; and (2) phosphate binders (whether calcium-based or not) can be used as part of an individualized therapeutic approach [6]. The guidelines therefore recommend correction of phosphate levels in ESRD patients for prevention of hyperparathyroidism, renal osteodystrophy, vascular calcification, and cardiovascular complications [6].
Hyperphosphatemia is a modifiable risk factor. Restriction of the dietary phosphorus intake to 800–1,200 mg/day is the cornerstone of serum phosphorus control. Continuing patient education with a knowledgeable dietitian is the best method for establishing and maintaining adequate dietary habits in CKD patients in general and dialysis patients in particular.
Phosphorus restriction may be instrumental in countering progressive renal failure and soft-tissue calcification [7, 8]. However, dietary restriction is of limited efficacy in ESRD, where a net positive phosphorus balance is inevitable [9, 10]. The current clinical strategy in ESRD involves (1) attempts to restrict dietary phosphorus intake; (2) removal of phosphate with three-times-weekly dialysis or (even better when possible) by daily or more prolonged dialysis sessions; and (3) reduction of intestinal phosphate absorption by the use of binders.
All currently available, orally administered phosphate binders (summarized in Table 1) have broadly the same efficacy in reducing serum phosphate levels (for reviews, see [11–14]). Recently, Block et al. [15] compared the respective effects of three phosphate binders (lanthanum carbonate, sevelamer carbonate, and calcium acetate) in moderate CKD. The researchers found that use of these binders in CKD stage four patients reduced urinary phosphorus excretion and attenuated the progression of secondary hyperparathyroidism but did not prevent the progression of vascular calcification—particularly in patients treated with the combination of calcium acetate and activated vitamin D, as is typically administered in the USA [15]. However, a recent pilot study in 212 non-dialysis CKD patients revealed that calcium-containing and calcium-free phosphate binders differed in their impacts on coronary artery calcification and on survival [16].
Currently available binders also differ in terms of their formulation, taste, tablet burden, and gastric intolerance. These factors clearly influence patient choice and should be carefully considered before prescription, in order to target an efficacious, well-tolerated, and cost-effective solution. Although a number of phosphate binders are in clinical development, none appears to constitute a significant step forward in phosphate control. However, nicotinamide (NAM, also known as niacinamide) may be a useful pharmacological alternative to binder-based approaches. Here, we review the data on NAM as a potentially valuable therapy for hyperphosphatemia. To this end, we searched the PubMed database with the keywords ‘nicotinamide’, ‘niacin’, ‘hyperphosphatemia’, ‘chronic kidney disease’, and ‘phosphate binder’, with a focus on the efficacy of NAM in dialysis patients.
1.1 Nicotinamide as an Alternative to Phosphate Binders
Nicotinamide is a water-soluble, amide derivative of nicotinic acid (niacin; vitamin B3). It is an old drug, with many indications and therapeutic applications. Since the identification of niacin in the 1930s as the pellagra-preventing factor, NAM has been used clinically (1) to treat schizophrenia and psoriasis; (2) to prevent type I diabetes mellitus; and (3) as a potent radiosensitizer [17–21].
Nicotinic acid is also an old medication and was mainly used to treat hyperlipidemia. Nicotinic acid inhibits adipocyte lipolysis via specific nicotinic acid receptors; it lowers low-density lipoprotein (LDL) and very-LDL cholesterol levels, and it increases high-density lipoprotein (HDL) cholesterol levels [22, 23]. Nicotinic acid and NAM have slightly different mechanisms of action. Nicotinic acid alone causes flushing (i.e. prominent cutaneous vasodilatation, particularly in the face) due to its stimulation of prostaglandin D2 and E2 secretion by subcutaneous Langerhans cells via the G-protein-coupled receptor (GPCR) 109A niacin receptor [24]. It was recently reported that both nicotinic acid and NAM showed efficacy in the treatment of hyperphosphatemia [25]. This review focuses on NAM’s pharmacokinetics, pharmacodynamics, efficacy, and safety.
1.2 Pharmacodynamic Properties
The directly absorbed dietary forms of niacin include NAM (the main source, obtained from animal-based foods) and nicotinic acid (obtained from plants). Dietary nicotinic acid is first converted into nicotinamide adenine dinucleotide (NAD) in the intestine and liver and is then cleaved to release NAM into the bloodstream for uptake by extrahepatic tissues [26]. However, the human body is not completely dependent on direct dietary sources of niacin, since NAM can also be synthesized from the tryptophan amino acid present in most proteins. Furthermore, NAM is produced by the catabolism of pyridine nucleotides.
Nicotinamide’s mechanism of action is not completely understood. In contrast to nicotinic acid, NAM is not a vasodilator, does not bind to GPCR 109A and 109B [27], and thus does not produce flushing.
Following filtration in the kidneys, most of the phosphate in the serum is reabsorbed across the proximal tubule epithelium. Indeed, it has been suggested that the sodium-dependent phosphate cotransport protein 2a (NaPi2a), the cotransporter NaPi2c, and the sodium-dependent phosphate transporter 2 mediate phosphate transport across the apical brush border of proximal tubule cells.
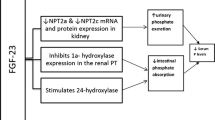
In vitro studies have shown that NAM decreases phosphate uptake by inhibiting the cotransporter NaPi2a in the renal proximal tubule and cotransporter NaPi2b in the intestine [28–31] (Fig. 1). Moreover, NAM reduced intestinal phosphate absorption in a rat model of chronic renal failure by inhibiting expression of NaPi2b [30]. The latter transporter’s major role in phosphate regulation in the intestine was recently confirmed by a study of NaPi2b knockout (−/−) mice in which phosphate absorption was half that seen in wild-type animals [32]. Moreover, an in vitro analysis of active phosphate transport in ilial segments from wild-type and NaPi2b knockout mice demonstrated that the transporter is responsible for over 90 % of total active phosphate absorption. Overall, NaPi2b is largely responsible for intestinal phosphate absorption and contributes to the maintenance of systemic phosphate homeostasis [32]. In a recent study, Schiavi et al. [33] found that uremic NaPi2b knockout mice had significantly lower serum phosphate levels and a significant attenuation of elevation of FGF23 levels (relative to uremic wild-type mice). Treating the NaPi2b knockout mice with the phosphate binder sevelamer carbonate further reduced serum phosphate levels. These data suggest that in addition to using dietary phosphorus binders, targeting NaPi2b could also be of value in the modulation of serum phosphate in CKD [33].

Nicotinamide’s mechanism of action at the brush border membrane of the enterocyte in the intestine. ADP adenosine diphosphate, ATP adenosine triphosphate
Thus, NAM decreases circulating phosphate levels in a different way to currently marketed orally administered compounds, which bind phosphate in the gastrointestinal tract by forming an insoluble complex or by binding the ion into a resin. Hence, less phosphate is available for absorption by the gastrointestinal tract and more is excreted in the feces. The NAM-mediated modulation of renal and/or intestinal phosphate transport processes constitutes a new approach for controlling serum phosphate levels.
1.3 Pharmacokinetic Properties
In a clinical study, twice-daily oral administration of NAM (total daily dose 25 mg/kg) was associated with a plasma half-life of 3.5 h and a mean peak plasma concentration of 42.1 μg/mL (0.3 mM) [34].
In pharmacokinetic studies in healthy volunteers, orally ingested NAM doses of 1–6 g were associated with dose-dependent peak plasma concentrations and showed a relative lack of toxicity [35, 36].
1.3.1 Administration
Dietary NAM is readily absorbed by the stomach and small intestine. The serum NAM concentration peaks 1 h after oral ingestion of a standard preparation [34]. The administration route determines how NAM is metabolized. When NAM is taken orally, it is metabolized by the small intestine and liver before being diluted in the systemic circulation.
1.3.2 Metabolism
As the main precursor for the formation and maintenance of a cellular pool of NAD, NAM is metabolized in the liver by cytochrome P450 to form nicotinamide-N-oxide (via an oxidative reaction), 6-hydroxy-nicotinamide (via a hydroxylation reaction), and N-methyl-nicotinamide (MNA, through catalysis by nicotinamide-N-methyltransferase). In mammals, MNA is further metabolized to N-methyl-2-pyridone-5-carboxamide (2PY) or N-methyl-4-pyridone-5-carboxamide (4PY) by aldehyde oxidase (Fig. 2). The 2PY/4PY ratio differs as a function of species and gender. In the context of uremia, studies in mice have evidenced the accumulation of plasma 4PY [37]. Although 4PY can be detected in the plasma in humans, the main metabolic product of MNA is 2PY [38]. Rutkowski et al. [37] have shown that the blood 2PY concentration increases as renal function deteriorates. Furthermore, 2PY may be a novel uremic toxin, since it significant inhibits poly(ADP-ribose) polymerase 1 [39] (PARP-1, an abundant nuclear enzyme that may be involved in the cellular response to DNA injury). Figure 3 shows PARP-1’s various roles. Slominska et al. [40] have suggested that NAM, 2PY, and 4PY accumulate in the plasma of children with chronic renal failure and that the combined effect of these three compounds could lead to inhibition of PARP-1 activity. The same researchers hypothesized that 4PY is a toxic compound that is actively absorbed by erythrocytes and is metabolized to 4-pyridone-3-carboxamide-1-β-ribonucleoside-triphosphate and 4-pyridone-3-carboxamide-1-β-ribonucleoside-monophosphate—both of which may interfere with cell function and survival [41]. The potential cellular toxicity of NAM metabolites needs to be confirmed in clinical studies.

Schematic description of nicotinamide metabolism. In summary, nicotinamide is metabolized to N-methylnicotinamide (MNA) by nicotinamide-N-methyltransferase, and MNA is further metabolized to N-methyl-2-pyridone-5-carboxamide (2PY) or N-methyl-4-pyridone-5-carboxamide (4PY) by aldehyde oxidase (for more details, please refer to the body of the text). 6HN 6 hydroxynicotinamide, ADP adenosine diphosphate, NA nicotinic acid, NAAD nicotinic acid adenine dinucleotide, NAD nicotinamide adenine dinucleotide, NAMN nicotinamide acid mononucleotide, NMN nicotinamide mononucleotide, NNO nicotinamide N oxide. Enzymes: 1 nicotinamide-N-methyltransferase, 2 aldehyde oxidase, 3–5 nicotinamide deamidase, 6 nicotinamide phosphoribosyltransferase, 7 NAMN adenylyltransferase, 8 nicotinamide synthetase, 9 poly(ADP-ribose) synthetase, 10 nicotinamide glycohydrolase, 11 nicotinamide phosphoribosyltransferase

Nicotinamide metabolites as inhibitors of poly(ADP-ribose) [pADPr] polymerase 1 (PARP-1). Nicotinamide derivatives such as N-methyl-2-pyridone-5-carboxamide (2PY) and N-methyl-4-pyridone-5-carboxamide (4PY) may disturb cellular repair processes via inhibition of PARP-1 activity. PARP-1 catalyzes the formation of adenosine diphosphate (ADP)-ribose polymers on a variety of protein acceptors in a nicotinamide adenine dinucleotide (NAD+-dependent manner. The enzyme plays a key role in DNA damage repair in general and base excision repair in particular. Over-activation of PARP1 leads to a depletion of NAD+/adenosine triphosphate (ATP) energy stores and, ultimately, to necrotic cell death
1.3.3 Distribution
As mentioned above, NAM is a circulating form of nicotinic acid. NAM disappears rapidly from the circulation and distributes into all tissues. Rutkowski et al. have shown that in rats, NAM is present in the plasma, erythrocytes, lungs, liver, heart, and brain but only weakly in fat tissue. Accumulation of NAM end products was observed in the liver, lungs, and skeletal muscles but not in fatty tissue or the brain [37]. Nicotinamide has a high hepatic extraction ratio, and plasma clearance is often abnormally low in patients with liver failure [42].
1.3.4 Elimination
Nicotinamide end products are excreted by the kidney, whereas NAM itself is reabsorbed by renal tubules [43]. This is why only small amounts of the unmodified NAM appear in the urine—even after administration of pharmacological (high) doses of the compound.
1.4 Therapeutic Efficacy
A number of clinical studies have explored the potential value of niacin and its analogs in phosphate control in dialysis patients [25]. Some have shown that nicotinic acid is effective in the treatment of hyperphosphatemia [44–47] as well as hyperlipidemia (historical use). In vivo conversion of nicotinic acid to NAM is required for this action. We focus on NAM in this respect.
Table 2 summarizes the results of clinical studies of NAM in dialysis patients.
The first study to show that NAM decreased serum phosphorus (from 6.9 to 5.4 mg/dL) and iPTH (without increasing serum calcium levels) was published by Takahashi et al. [48]. This open-label study was carried out in 65 hyperphosphatemic dialysis patients receiving NAM in divided doses (mean daily dose 1,080 mg) for 12 weeks. Furthermore, NAM treatment significantly increased serum HDL cholesterol levels and decreased LDL cholesterol levels over the course of the study. Other authors have since reported significant reductions in phosphatemia in NAM-treated dialysis patients [49–52].
Cheng et al. [49] were the first to perform a double-blind, placebo-controlled, randomized clinical trial of NAM (300–1,800 mg) in the treatment of hyperphosphatemia in 33 dialysis patients. After 8 weeks of treatment, the mean serum phosphate level had fallen significantly in the NAM group (from 6.26 to 5.47 mg/dL) but not in the placebo group (with a rise from 5.85 to 5.98 mg/dL, in fact). Moreover, mean serum HDL levels rose in the NAM group (from 50 to 61 mg/dL) but not in the placebo group. Nicotinamide had no effect on serum calcium levels in the study population [49].
In another prospective, randomized, double blind, placebo-controlled trial of NAM in 15 dialysis patients, it was found that an initial daily dose of 750 mg of NAM resulted in a slight but significant decrease in plasma phosphorus levels (from 5.9 to 5.2 mg/dL) in the active treatment group (but not in the placebo group) at 8 weeks [50].
Similarly, Shahbazian et al. [51] performed a placebo-controlled clinical study of the efficacy and safety of a 4-week course of NAM in 48 dialysis patients. The researchers found that administration of NAM 500 mg/day was associated with a decrease in serum phosphate levels (from 5.9 to 4.77 mg/dL). Moreover, NAM was associated with clinically important differences, such as higher HDL levels and fasting glycemia and lower LDL and triglyceride levels vs. placebo. However, the authors observed that NAM was associated with a significantly low platelet count and emphasized the need to monitor for thrombocytopenia when the compound is used therapeutically [51].
Recently, Vasantha et al. [52] reported an open-label study in which 30 dialysis patients receiving a mean dose of NAM 750 mg per day experienced a mean 2.3 mg/dL decrease in serum phosphorus levels after 8 weeks of treatment. A decrease in alkaline phosphatase levels was also observed [52].
However, none of these studies included large numbers of dialysis patients, and the follow-up periods were short. Furthermore, NAM was used as an adjunct to phosphate binders in some studies [49, 51, 53] but was studied alone in others [48, 52]. We consider that it will be essential to perform large-scale clinical studies of the efficacy and especially the safety of long-term NAM use as an alternative therapy in CKD patients.
1.5 Tolerability
A considerable body of literature data shows that NAM in adults is safe at doses of below 3 g/day [42].
Nicotinamide’s long-term safety in patients with normal renal function was examined in the European Nicotinamide Diabetes Intervention Trial [18]. Although the researchers could not demonstrate a preventive effect of NAM on type 1 diabetes, they did conclude that tolerance was good. The main side effects at therapeutic doses are gastrointestinal symptoms (mainly diarrhea) that generally resolve on treatment withdrawal. Delanaye et al. reported that five of six patients included in an open-label study developed diarrhea; the symptoms emerged at a mean ± SD dose of 1,050 ± 447 mg/day and resolved after withdrawal of the drug. The researchers pointed out that all of the patients were also taking calcium binders and/or sevelamer, which may have facilitated the emergence of these adverse events [54].
There is also a case report of severe hepatotoxicity in a patient who was taking NAM 9 g/day. Again, the event resolved upon discontinuation of treatment [55].
Rottembourg et al. [56] reported that six dialysis patients being treated with NAM 1,000 mg/day developed significant thrombocytopenia within 3 months of treatment initiation. These results were confirmed by Shahbazian et al. Although the mechanism of this side effect has not yet been clearly elucidated, it is possible that thrombocytopenia results from the low levels of thyroxin-binding globulin induced by NAM and its derivatives [51].
Nicotinamide’s long-term safety in ESRD patients has not been studied. It has been suggested in the literature that NAM derivatives may be uremic toxins, with increased oxidative stress and disturbance of the cellular repair processes caused by inhibition of PARP-1 activity [41]. These potential side effects need to be clinically evaluated.
1.6 Pharmaco-Economic Considerations
Cost-effectiveness is an important issue for all currently available phosphate binders, although the cost of daily treatment varies from one compound to another. For example, a cost-effectiveness analysis performed by the UK National Health Service in new dialysis patients found that the total 5-year discounted treatment cost was £24,216 in a sevelamer group and £17,985 in a calcium acetate group [57].
In France, the average daily dose (ADD) of NAM (1.5 g) is 16, 15, and 2 times less expensive than those of sevelamer hydrochloride (ADD 7.2 g), lanthanum carbonate (ADD 3 g), and calcium carbonate (ADD 4.62 g). Hence, if used instead of binders, NAM would be a cost-effective treatment for hyperphosphatemia in dialysis patients. There is also a need to evaluate the cost-effectiveness of NAM when it is used as an adjunct to phosphate binders.
2 Conclusion
Although hyperphosphatemia is not an approved indication for NAM, recent clinical studies have confirmed the drug’s effectiveness in reducing blood phosphate levels in dialysis patients. In fact, NAM may be an interesting alternative to phosphate binders for the treatment of hyperphosphatemia, given (1) the drug’s attractive mechanism of action (blockade or inhibition of the intestinal transport); (2) its potential cost-effectiveness; and (3) the limited number of tablets required to achieve good compliance.
In terms of adverse drug reactions, NAM-related gastrointestinal adverse events appear only at a daily dose of between 1 and 2 g and can often be resolved while therapy continues. Thrombocytopenia is a serious adverse event requiring treatment discontinuation and needs to be evaluated more precisely. The balance between NAM’s potential benefits and harmful effects must be assessed before widespread use of this drug in the management of hyperphosphatemia in dialysis patients can be considered. Previous studies have been limited by short follow-up periods and small sample sizes. Thus, long-term studies are needed to validate NAM’s tolerance, safety, and efficacy in dialysis patients.
References
Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol. 2005;16:520–8.
Six I, Maizel J, Barreto FC, Rangrez AY, Dupont S, Slama M, et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc Res. 2012;96:130–9.
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis. 1998;31:607–17.
Ganesh SK, Stack AG, Levin NW, Hulbert-Shearon T, Port FK. Association of elevated serum PO(4), Ca × PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol. 2001;12:2131–8.
Mathew S, Tustison KS, Sugatani T, Chaudhary LR, Rifas L, Hruska KA. The mechanism of phosphorus as a cardiovascular risk factor in CKD. J Am Soc Nephrol. 2008;19:1092–105.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl. 2009;(113):S1–130.
Lumlertgul D, Burke TJ, Gillum DM, Alfrey AC, Harris DC, Hammond WS, et al. Phosphate depletion arrests progression of chronic renal failure independent of protein intake. Kidney Int. 1986;29:658–66.
Haut LL, Alfrey AC, Guggenheim S, Buddington B, Schrier N. Renal toxicity of phosphate in rats. Kidney Int. 1980;17:722–31.
Hsu CH. Are we mismanaging calcium and phosphate metabolism in renal failure? Am J Kidney Dis. 1997;29:641–9.
Ramirez JA, Emmett M, White MG, Fathi N, Santa Ana CA, Morawski SG, et al. The absorption of dietary phosphorus and calcium in hemodialysis patients. Kidney Int. 1986;30:753–9.
Tonelli M, Pannu N, Manns B. Oral phosphate binders in patients with kidney failure. N Engl J Med. 2010;362:1312–24.
Coladonato JA. Control of hyperphosphatemia among patients with ESRD. J Am Soc Nephrol. 2005;16(Suppl 2):S107–14.
Hutchison AJ, Smith CP, Brenchley PEC. Pharmacology, efficacy and safety of oral phosphate binders. Nat Rev Nephrol. 2011;7:578–89.
Bellasi A, Kooienga L, Block GA. Phosphate binders: new products and challenges. Hemodial Int. 2006;10:225–34.
Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012;23:1407–15.
Di Iorio B, Bellasi A, Russo D, INDEPENDENT Study Investigators. Mortality in kidney disease patients treated with phosphate binders: a randomized study. Clin J Am Soc Nephrol. 2012;7:487–93.
Chase P, Dupre J, Mahon J, Ehrlich R, Gale E, Kolb H, et al. Nicotinamide and prevention of diabetes. Lancet. 1992;339:1051–2.
Gale EAM, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363:925–31.
Denekamp J, Fowler JF. ARCON—current status: summary of a workshop on preclinical and clinical studies. Acta Oncol. 1997;36:517–25.
Smythies JR. Letter: nicotinamide treatment of schizophrenia. Lancet. 1973;2:1450–1.
Pozzilli P, Browne PD, Kolb H. Meta-analysis of nicotinamide treatment in patients with recent-onset IDDM. The Nicotinamide Trialists. Diabetes Care. 1996;19:1357–63.
Karpe F, Frayn KN. The nicotinic acid receptor—a new mechanism for an old drug. Lancet. 2004;363:1892–4.
Guyton JR. Niacin in cardiovascular prevention: mechanisms, efficacy, and safety. Curr Opin Lipidol. 2007;18:415–20.
Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am J Cardiol. 2008;101:S20–6.
Sampathkumar K. Niacin and analogs for phosphate control in dialysis–perspective from a developing country. Int Urol Nephrol. 2009;41:913–8.
Kirkland JB. Niacin status impacts chromatin structure. J Nutr. 2009;139:2397–401.
Bodor ET, Offermanns S. Nicotinic acid: an old drug with a promising future. Br J Pharmacol. 2008;153(Suppl 1):S68–75.
Berndt TJ, Pfeifer JD, Knox FG, Kempson SA, Dousa TP. Nicotinamide restores phosphaturic effect of PTH and calcitonin in phosphate deprivation. Am J Physiol. 1982;242:F447–52.
Kempson SA, Colon-Otero G, Ou SY, Turner ST, Dousa TP. Possible role of nicotinamide adenine dinucleotide as an intracellular regulator of renal transport of phosphate in the rat. J Clin Invest. 1981;67:1347–60.
Eto N, Miyata Y, Ohno H, Yamashita T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol Dial Transplant. 2005;20:1378–84.
Katai K, Tanaka H, Tatsumi S, Fukunaga Y, Genjida K, Morita K, et al. Nicotinamide inhibits sodium-dependent phosphate cotransport activity in rat small intestine. Nephrol Dial Transplant. 1999;14:1195–201.
Sabbagh Y, O’Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009;20:2348–58.
Schiavi SC, Tang W, Bracken C, O’Brien SP, Song W, Boulanger J, et al. Npt2b deletion attenuates hyperphosphatemia associated with CKD. J Am Soc Nephrol. 2012;23:1691–700.
Petley A, Macklin B, Renwick AG, Wilkin TJ. The pharmacokinetics of nicotinamide in humans and rodents. Diabetes. 1995;44:152–5.
Stratford MR, Dennis MF, Hoskin P, Phillips H, Hodgkiss RJ, Rojas A. Nicotinamide pharmacokinetics in humans: effect of gastric acid inhibition, comparison of rectal vs oral administration and the use of saliva for drug monitoring. Br J Cancer. 1996;74:16–21.
Dragovic J, Kim SH, Brown SL, Kim JH. Nicotinamide pharmacokinetics in patients. Radiother Oncol. 1995;36:225–8.
Rutkowski P, Slominska EM, Wołyniec W, Smoleński RT, Szolkiewicz M, Swierczyński J, et al. Nicotinamide metabolites accumulate in the tissues of uremic rats. J Ren Nutr. 2008;18:56–9.
Gillmor HA, Bolton CH, Hopton M, Moore WP, Perrett D, Bingley PJ, et al. Measurement of nicotinamide and N-methyl-2-pyridone-5-carboxamide in plasma by high performance liquid chromatography. Biomed Chromatogr. 1999;13:360–2.
Rutkowski B, Slominska E, Szolkiewicz M, Smolenski RT, Striley C, Rutkowski P, et al. N-methyl-2-pyridone-5-carboxamide: a novel uremic toxin? Kidney Int Suppl. 2003;(84):S19–21.
Slominska EM, Kowalik K, Smolenski RT, Szolkiewicz M, Rutkowski P, Rutkowski B, et al. Accumulation of poly(ADP-ribose) polymerase inhibitors in children with chronic renal failure. Pediatr Nephrol. 2006;21:800–6.
Rutkowski B, Rutkowski P, Słomińska E, Smolenski RT, Swierczyński J. Cellular toxicity of nicotinamide metabolites. J Ren Nutr. 2012;22:95–7.
Knip M, Douek IF, Moore WPT, Gillmor HA, McLean AEM, Bingley PJ, et al. Safety of high-dose nicotinamide: a review. Diabetologia. 2000;43:1337–45.
Beyer KH, Russo HF. Renal tubular elimination of N1-methylnicotinamide. Am J Physiol. 1950;160:311–20.
Sampathkumar K, Selvam M, Sooraj YS, Gowthaman S, Ajeshkumar RNP. Extended release nicotinic acid—a novel oral agent for phosphate control. Int Urol Nephrol. 2006;38:171–4.
Müller D, Mehling H, Otto B, Bergmann-Lips R, Luft F, Jordan J, et al. Niacin lowers serum phosphate and increases HDL cholesterol in dialysis patients. Clin J Am Soc Nephrol. 2007;2:1249–54.
Restrepo Valencia CA, Cruz J. Safety and effectiveness of nicotinic acid in the management of patients with chronic renal disease and hyperlipidemia associated to hyperphosphatemia. Nefrologia. 2008;28:61–6.
Maccubbin D, Tipping D, Kuznetsova O, Hanlon WA, Bostom AG. Hypophosphatemic effect of niacin in patients without renal failure: a randomized trial. Clin J Am Soc Nephrol. 2010;5:582–9.
Takahashi Y, Tanaka A, Nakamura T, Fukuwatari T, Shibata K, Shimada N, et al. Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int. 2004;65:1099–104.
Cheng SC, Young DO, Huang Y, Delmez JA, Coyne DW. A randomized, double-blind, placebo-controlled trial of niacinamide for reduction of phosphorus in hemodialysis patients. Clin J Am Soc Nephrol. 2008;3:1131–8.
Young DO, Cheng SC, Delmez JA, Coyne DW. The effect of oral niacinamide on plasma phosphorus levels in peritoneal dialysis patients. Perit Dial Int. 2009;29:562–7.
Shahbazian H, Zafar Mohtashami A, Ghorbani A, Abbaspour MR, Belladi Musavi SS, Hayati F, et al. Oral nicotinamide reduces serum phosphorus, increases HDL, and induces thrombocytopenia in hemodialysis patients: a double-blind randomized clinical trial. Nefrologia. 2011;31:58–65.
Vasantha J, Soundararajan P, Vanitharani N, Kannan G, Thennarasu P, Neenu G, et al. Safety and efficacy of nicotinamide in the management of hyperphosphatemia in patients on hemodialysis. Indian J Nephrol. 2011;21:245–9.
Young EW, Albert JM, Satayathum S, Goodkin DA, Pisoni RL, Akiba T, et al. Predictors and consequences of altered mineral metabolism: the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2005;67:1179–87.
Delanaye P, Weekers L, Krzesinski JM. Diarrhea induced by high doses of nicotinamide in dialysis patients. Kidney Int. 2006;69:1914.
Winter SL, Boyer JL. Hepatic toxicity from large doses of vitamin B3 (nicotinamide). N Engl J Med. 1973;289:1180–2.
Rottembourg JB, Launay-Vacher V, Massard J. Thrombocytopenia induced by nicotinamide in hemodialysis patients. Kidney Int. 2005;68:2911–2.
Taylor MJ, Elgazzar HA, Chaplin S, Goldsmith D, Molony DA. An economic evaluation of sevelamer in patients new to dialysis. Curr Med Res Opin. 2008;24:601–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Lenglet, A., Liabeuf, S., Guffroy, P. et al. Use of Nicotinamide to Treat Hyperphosphatemia in Dialysis Patients. Drugs R D 13, 165–173 (2013). https://doi.org/10.1007/s40268-013-0024-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-013-0024-6