
Abstract
Purpose
To compare the bioavailability (BA) and pharmacokinetic (PK) properties and to demonstrate the bioequivalence (BE) between two active product ingredient (API) sources of eslicarbazepine acetate (ESL) in healthy volunteers.
Design, subjects and methods
Forty healthy male and female subjects aged 18–40 years were randomized to treatment with 400 or 800 mg ESL marketed (MF) formulation [current active pharmaceutical ingredient (API) source] and 400 or 800 mg ESL to-be-marketed (TBM) formulation (new API source) under a gender-balanced, two-period, two-sequence crossover open-label study design. Subjects were assigned to receive either 400 or 800 mg ESL dose strengths, and each was randomly administered on two occasions—either a single oral tablet of MF or a single oral tablet of TBM—separated by a washout period of at least 7 days. Formulations were to be considered bioequivalent if, for both 400 or 800 mg ESL dosage strengths, the test (TBM)/reference (MF) geometric mean ratios (GMR) and 90 % confidence intervals (90 % CI) of the area under the plasma concentration-time curve (AUC) and peak plasma concentration (C max) were within the predetermined range of 80–125 %.
Results
Test/reference GMR (90 % CI) for the C max and AUC was respectively 100 % (94–109 %) and 96 % (94–98 %) following 400 mg ESL and 100 % (95–105 %) and 100 % (97–103 %) following 800 mg ESL.
Conclusion
Oral tablet formulations of either 400 or 800 mg ESL from the new API source were found to be bioequivalent to the corresponding marketed Zebinix® formulation according to the regulatory definition of bioequivalence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Eslicarbazepine acetate (ESL) is a once-daily anticonvulsant approved in 2009 by the European Medicines Agency (EMA) as adjunct therapy in adults with partial-onset seizures (POS), with or without secondary generalization. ESL is structurally distinct from carbamazepine (CBZ) and oxcarbazepine (OXC), although the three compounds are dibenz[b,f]azepine derivatives [1]. This molecular distinction results in differences in metabolism [2]. CBZ and ESL do not share any common metabolite and, contrarily to CBZ, ESL is not susceptible to metabolic auto-induction [3, 4].
Following oral administration, ESL undergoes extensive first pass hydrolysis to its major active metabolite eslicarbazepine [also known as (S)-licarbazepine] [5–9], which represents approximately 95 % of circulating active moieties and is believed to be responsible for its antiseizure effects [10–14], most likely through blockade of voltage-gated sodium channels and type T calcium channels [15, 16].
ESL is currently available in the form of tablets for oral administration. A new active pharmaceutical ingredient (API) source was brought on board, and since the tablets manufactured with it dissolve somewhat faster than those manufactured with the current API (data on file), the in vivo bioavailability (BA) of ESL and its metabolites was deemed uncertain by EMA.
The most important property of any non-intravenous dosage form (e.g., oral) is the ability to deliver the API to the bloodstream in an amount sufficient to cause the desired response. This property of a dosage form has historically been identified as bioavailability. BA captures two essential features, namely how fast the drug enters the systemic circulation (rate of absorption) and how much of the nominal strength enters the body (extent of absorption) [17]. Moreover, in the management of epilepsy that requires treatment for years, the BA of the anticonvulsant drug should not fluctuate. It may lead to intoxication or seizures may relapse [18].
The aim of this study was the assessment of the BA and pharmacokinetic (PK) properties of the ESL formulation with the new API source (Test) and to determine its bioequivalence (BE) to the current and marketed ESL formulation, Zebinix® (reference).
2 Methods
2.1 Study Design
This study (trial registration EudraCT No. 2010-022478-15) was a two-center (Biotrial SA, Rennes and Paris, France) phase 1 study to demonstrate the BE between two API sources of ESL at two dose strengths (400 and 800 mg) in 40 (20 per dosage strength) healthy male and female subjects under an open-label, randomized, gender-balanced, two-period, two-sequence, crossover study design.
The study design consisted of two treatment periods separated by a washout period of at least 7 days between doses. In one of the two treatment periods, subjects received either a single oral dose of 400 or 800 mg ESL of the marketed (MF) formulation—current API source (Zebinix®). In the other treatment period, a single oral dose of 400 or 800 mg ESL of the to-be-marketed (TBM) formulation—new API source—was administered. ESL tablets from both formulations were manufactured by BIAL-Portela & Co., SA, S. Mamede do Coronado, Portugal.
Subjects were required to attend the research facilities for a follow-up visit 7–14 days after clinical discharge (72 h post-dose) of the last treatment period or early discontinuation. Subjects were admitted to the research facilities for both treatment periods on the day before (Day−1) the dosing day (Day 1) and resided in the research facilities until at least the 24 h post-dose (Day 2) procedures. The Day 2 (36 h post-dose) to Day 4 (72 h post-dose) assessments were performed in an ambulatory way. Plasma levels of parent drug (ESL) are usually undetectable. In the present study an achiral method was used, thus not allowing to distinguish between eslicarbazepine and its minor metabolite, (R)-licarbazepine; in such cases, the mixture is reported as BIA 2-005 [19, 20].
ESL was administered as a single dose under a two-period, two-sequence crossover design because single-dose PK studies to demonstrate BE are generally more sensitive in assessing release of the drug substance from the drug product into the systemic circulation. Due to the fact that two formulations are to be compared a non-replicate crossover, a two-period and two-sequence design was chosen. The ESL dosage regimen was chosen from the Zebinix® dose strengths already marketed (400 and 800 mg).
The within-subject coefficient of variation of AUC0–∞ and C max observed in previous studies with ESL was <15 %. It was estimated for each dosage strength group that with 16 subjects an overall power above 0.8 is attained in an equivalence range of 80 to 125 % with a α value of 0.05 [21, 22]. Twenty subjects allowed for eventual dropouts and balancing for gender (i.e., 16 subjects completing each group).
The studies were conducted according to the Helsinki Declaration, ICH Good Clinical Practice recommendations and applicable local regulations. The studies were approved by an Independent Ethics Committee (CPP—Comité de Protection des Personnes, Ouest VI, Brest, France) and the French Medicines Agency (AFSSAPS). Written informed consent was obtained for each study participant.
2.2 Population
Potential male and female subjects were screened for eligibility within 28 and 2 days of admission to the first treatment period. Screening consisted of discussion of informed consent, medical history, physical examination, vital signs, 12-lead ECG, clinical laboratory tests (hematology, plasma biochemistry, coagulation, urinalysis, viral serology, alcohol and drugs of abuse screen, and urine pregnancy test) and review of the selection criteria. Subjects were to be aged 18–55 years, within 18–25 kg/m2 of body mass index (BMI) and non-smokers or smokers of <10 cigarettes per day; women had to be pre-menopausal and use double barrier or intrauterine device pregnancy protection. No medication other than the study drugs or necessary for the treatment of adverse events (AEs) was allowed from the initial day of screening until final discharge.
On dosing days, subjects had an overnight fast for at least 10 h before dosing and remained fasted until 4 h post-dose. Water drinking was allowed as desired except for 1 h before and after dosing. Products were administered, in the morning with approximately 240 mL of water.
Subjects were requested to abstain from strenuous physical activity, consumption of grapefruit juice, alcohol and stimulating beverages containing xanthine derivatives for 48 h prior to dosing and during each treatment period. Subjects were also instructed to abstain from smoking for 2 h prior to until 24 h after drug administration at each treatment period.
2.3 Blood Sampling and Plasma Drug Assays
Plasma concentrations of ESL and BIA 2-005 were determined using a validated liquid chromatography coupled to tandem mass spectrometry (LC MS/MS) method in compliance with Good Laboratory Practices (GLP).
Blood samples (4 mL of venous blood) were drawn by direct venipuncture or via an intravenous catheter into heparin-lithium vacutainers before the ESL dose and then 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48 and 72 hours post-dose.
After collection, blood samples were immediately centrifuged at approximately 1,500g for 10 min at 4 °C. Prior to shipment to the laboratory for the analytical assays (Swiss Bioanalytics AG, Birsfelden, Switzerland), the resulting plasma was separated into aliquots of 0.75 mL and stored at −20 °C. The lowest level of quantification (LLOQ) was at 10 ng/mL [19, 20].
2.4 Pharmacokinetic Assessments and Statistical Analysis
Plasma levels of parent drug (ESL) are usually below the limit of quantification at almost all sampling times. Therefore, pharmacokinetic analysis was to be done for the main metabolite (BIA 2-005).
The following pharmacokinetic parameters for BIA 2-005 were derived from the individual plasma concentration-time profiles: maximum observed plasma concentration (C max); time of occurrence of C max (t max); area under the plasma concentration versus time curve (AUC) from time zero to the last sampling time at which concentrations were at or above the limit of quantification (AUC0–t ) and AUC from time zero to infinity (AUC0–∞), calculated by the linear trapezoidal rule; apparent terminal rate constant, calculated by log-linear regression of the terminal segment of the concentration versus time curve (λz); apparent terminal half-life (t½), calculated from ln 2/λz. Descriptive statistics and individual pharmacokinetic were determined.
For the evaluation of the formulation bioequivalence, the parameters AUC0–∞, AUC0–t and C max of BIA 2-005 were the primary variables. The test procedure was analogous to equivalence testing. For each ESL dosage strength, an analysis of variance (ANOVA) was performed using log-transformed data for C max, AUC0–t and AUC0–∞ of BIA 2-005 with sequence, period and treatment as fixed effects and subject within sequence as random effect. The 90 % confidence intervals (90 % CI) for the test/reference geometric mean ratio (GMR) of BIA 2-005 C max, AUC0–t and AUC0–∞ was calculated. In accordance with the guidelines for bioequivalence testing, bioequivalence was assumed when the ratio test/reference fell within the 90 % CI 80–125 reference range. The alpha error was set at 0.05 to define statistical significance. The pharmacokinetic parameters and analyses were calculated using WinNonlin Version 5.2 (Pharsight Corporation, Mountain View, CA, USA). The statistical package SAS version 9.2 (SAS Institute Inc, Cary, NC, USA) was used in some computations.
2.5 Safety Assessments
Safety and tolerability assessments included routine laboratory tests (blood chemistries, hematological profile, coagulation and urinalysis), physical examination, ECG and vital signs. Any undesirable sign, symptom or medical condition occurring after starting the study, whether reported spontaneously or when prompted, was recorded regardless of suspected relation to the study medications.
3 Results
3.1 Population
A total of 40 healthy subjects were randomized to the study, 20 (20) in each dosage strength (400 and 800 mg ESL).
The overall mean ± SD (range) demographic data were as follows: age = 35.7 ± 10.6 (range 20–54) years; height = 171 ± 9 (156–191) cm; BMI = 22.1 ± 1.9 (18.1–24.7) kg/m2. All subjects were exposed to ESL.
Twenty (20) subjects (11 males and 9 females) received a single oral tablet of 400 mg ESL from both MF and TBM formulations. Thus, all subjects completed both periods of the 400 mg dosage strength and were available for PK analysis.
Twenty (20) subjects (10 males and 10 females) received a single oral tablet of 800 mg ESL of the MF formulation but only 18 subjects received a single oral tablet of 800 mg ESL of the TBM formulation. Two (2) subjects discontinued the study before dosing on their second treatment period (ESL 800 mg TBM): one subject presented a positive result for opiates due to the intake of antitussive syrup, and the other withdrew the informed consent for personal reasons. Thus, 18 (18) subjects (10 males and 8 females) completed both periods of the 800-mg dosage strength and were available for PK analysis.
3.2 Pharmacokinetics
3.2.1 ESL
ESL (parent) plasma concentrations were systematically found to be below the limit of quantification; therefore, the concentration-time profiles of ESL could not be displayed nor the PK parameters calculated. Thus, PK analysis was done exclusively for the main metabolite (BIA 2-005).
3.2.2 BIA 2-005
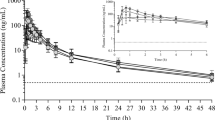
Mean plasma concentrations over time of BIA 2-005 following a single oral dose of ESL 400 mg MF and TBM formulations and ESL 800 mg MF and TBM formulations are presented in Fig. 1. Plasma drug concentration-time curves show that the mean concentrations of BIA 2-005 were similar for the two formulations (MF and TBM) over the entire sampling period and for both 400 and 800 mg dose strengths (Fig. 1).

Mean plasma concentration-time profiles of BIA 2-005 following a single oral dose of ESL 400 mg MF and TBM (n = 20) and ESL 800 mg MF and TBM (n = 20, n = 18 for ESL 800 mg TBM). a Linear scale; b semi-log scale
Following 400 mg ESL, the BIA 2-005 mean C max values of the test (ESL 400 mg TBM) and reference (ESL 400 mg MF) formulations were 6.4 and 6.3 µg/mL, respectively. The median T max values were 2.0 h for both. Results for the extent of absorption, as determined from mean AUC0–t and AUC0–∞ values, were 105.9 and 106.6 μg h/mL, respectively, after administration of the Test formulation and 110.3 and 111.1 μg h/mL, respectively, after administration of the reference formulation (Table 1).
Following 800 mg ESL, the BIA 2-005 mean C max values of the test (ESL 800 mg TBM) and reference (ESL 800 mg MF) formulations were 12.81 and 12.95 µg/mL, respectively. The mean t max values were 1.8 and 2.0 h, respectively. Results for the extent of absorption, as determined from mean AUC0–t and AUC0–∞ values, were 272.7 and 277.1 μg h/mL, respectively, after administration of the Test formulation and 273.4 and 277.3 μg h/mL, respectively, after administration of the reference formulation (Table 1).
The bioequivalence was evaluated by using the geometric means of C max, AUC0–t and AUC0–∞ values for BIA 2-005. The ratio (test/reference) of each parameter ranged from 96 to 101 % for both dose strengths (Table 2). Following 400 mg ESL, the 90 % confidence intervals for the ratios of C max, AUC0–t and AUC0–∞ were 94–109, 94–98 and 94–98 %, respectively, meeting the predetermined criteria for bioequivalence. Following 800 mg ESL, the 90 % confidence intervals for the ratios of C max, AUC0–t and AUC0–∞ were 95–105, 95–103 and 95–103 %, respectively, also meeting the predetermined criteria for bioequivalence (Table 2).
3.3 Tolerability
A total of 40 healthy subjects were randomized to the study with all subjects exposed to ESL. Twenty (20) subjects (11 males and 9 females) received a single oral tablet of 400 mg ESL from both MF and TBM formulations; 20 subjects (10 males and 10 females) received a single oral tablet of 800 mg ESL of the MF formulation, but only 18 subjects received a single oral tablet of 800 mg ESL of the TBM formulation. Two (2) subjects discontinued the study before dosing on their second treatment period (ESL 800 mg TBM): one subject presented a positive result for opiates due to the intake of antitussive syrup, and the other withdrew the informed consent for personal reasons.
Overall, 13 treatment-emergent AEs (TEAEs) were reported by 7 (17.5 %) subjects (2 of them presenting TEAEs in both treatment periods). No TEAEs were reported in the ESL 400 mg MF treatment period, two TEAEs were reported by one subject (5.0 %) in the ESL 400 mg TBM, five TEAEs by four subjects (20.0 %) in the ESL 800 mg MF and six TEAEs by four (22.2 %) subjects in the ESL 800 mg TBM (Table 3). The majority of AEs were mild in intensity and considered possibly related to treatment.
There was no serious AE (SAE) and no important medical event. No AE required the withdrawal of a subject, and all subjects with TEAEs had recovered at the end of the study. No clinically relevant difference was observed in the nature, the intensity of TEAEs or their relationship with ESL between both formulations.
4 Discussion
The present study demonstrated that pharmacokinetics properties of the new ESL formulation under evaluation were similar to the marketed ESL formulation.
When two distinct formulations of the same drug, which obeys a linear pharmacokinetics, are alike in the rate and extent to which its active product ingredient is absorbed and becomes equally available at the site of action, they are considered bioequivalent and thus assumed to be therapeutically equivalent since this is a function of its pharmacokinetic-pharmacodynamic relationship [18, 23–25]. Moreover, to demonstrate bioequivalence, it is generally accepted that the 90 % confidence interval for the ratio of means of logarithmically transformed AUC and C max should lie within the range of 80–125 %, with no differences in T max evaluated by a non-parametric test on the untransformed values [26, 27].
ESL presents a pharmacokinetic profile that can be considered linear [19, 28], and our study data revealed that the both formulations of ESL presented similar pharmacokinetic characteristics. The study results show that both ESL formulations are bioequivalent for the rate and extent of absorption. The 90 % confidence intervals were completely contained within the predefined bioequivalence criteria of 80–125 % for C max and AUC.
In general, ESL formulations were well tolerated at both doses (400 and 800 mg) and formulations (MF and TBM) tested, and the observed adverse events were typical of previous studies of ESL conducted in healthy subjects.
5 Conclusion
Oral tablet formulations of either 400 or 800 mg ESL from the new API source was found to be bioequivalent to the corresponding marketed Zebinix® formulation according to the regulatory definition of bioequivalence.
References
Benes J, Parada A, Figueiredo AA, Alves PC, Freitas AP, Learmonth DA, et al. Anticonvulsant and sodium channel-blocking properties of novel 10,11-dihydro-5H-dibenz[b,f]azepine-5-carboxamide derivatives. J Med Chem. 1999;42(14):2582–7.
Hainzl D, Parada A, Soares-da-Silva P. Metabolism of two new antiepileptic drugs and their principal metabolites S(+)- and R(−)-10,11-dihydro-10-hydroxy carbamazepine. Epilepsy Res. 2001;44(2–3):197–206.
Almeida L, Bialer M, Soares-da-Silva P. Eslicarbazepine acetate. In: Shorvon S, Perucca E, Engel J, editors. The treatment of epilepsy. 3rd ed. Oxford: Blackwell Publishing; 2009. p. 485–98.
Bialer M, Soares-da-Silva P. Pharmacokinetics and drug interactions of eslicarbazepine acetate. Epilepsia. 2012;53(6):935–46.
Falcao A, Maia J, Almeida L, Mazur D, Gellert M, Soares-da-Silva P. Effect of gender on the pharmacokinetics of eslicarbazepine acetate (BIA 2–093), a new voltage-gated sodium channel blocker. Biopharm Drug Dispos. 2007;28(5):249–56.
Almeida L, Potgieter JH, Maia J, Potgieter MA, Mota F, Soares-da-Silva P. Pharmacokinetics of eslicarbazepine acetate in patients with moderate hepatic impairment. Eur J Clin Pharmacol. 2008;64(3):267–73.
Almeida L, Minciu I, Nunes T, Butoianu N, Falcao A, Magureanu SA, et al. Pharmacokinetics, efficacy, and tolerability of eslicarbazepine acetate in children and adolescents with epilepsy. J Clin Pharmacol. 2008;48(8):966–77.
Maia J, Almeida L, Falcão A, Soares E, Mota F, Potgieter JH, et al. Effect of renal impairment on the pharmacokinetics of eslicarbazepine acetate. Int J Clin Pharmacol Ther. 2008;46(3):119–30.
Perucca E, Elger C, Halasz P, Falcao A, Almeida L, Soares-da-Silva P. Pharmacokinetics of eslicarbazepine acetate at steady-state in adults with partial-onset seizures. Epilepsy Res. 2011;96(1–2):132–9.
Pires N, Palma N, Loureiro AI, Bonifacio MJ, Wright LC, Soares-da-Silva P. Effects of eslicarbazepine acetate, eslicarbazepine, carbamazepine and oxcarbazepine in the maximal electroconvulsive shock test in the mice. Epilepsia. 2011;52(Suppl. 6):118.
Torrao L, Machado R, Pires N, Palma N, Bonifacio MJ, Wright LC, et al. Effects of eslicarbazepine acetate, eslicarbazepine, carbamazepine and oxcarbazepine in the 6-HZ psychomotor seizure model in the mice. Epilepsia. 2011;52(Suppl. 6):118–9.
Pekcec A, Potschka H, Soares-da-Silva P. Effects of eslicarbazepine acetate and its metabolites in the corneal kindling model of epilepsy. Epilepsia. 2011;52(Suppl. 6):257.
Soerensen J, Pekcec A, Potschka H, Soares-da-Silva P. The effects of eslicarbazepine acetate in the amygdala kindling model of temporal lobe epilepsy. Epilepsia. 2011;52(Suppl. 6):257.
Sierra-Paredes G, Sierra-Marcuno G, Loureiro AI, Wright LC, Soares-da-Silva P. Effects of eslicarbazepine acetate on acute and chronic latrunculin A-induced seizures and extracellular amino acid levels in the mouse hippocampus. Epilepsia. 2011;52(Suppl. 6):119.
Hebeisen S, Brady K, Konrad D, Soares-da-Silva P. Inhibitory effects of eslicarbazepine acetate and its metabolites against neuronal voltage-gated sodium channels. Epilepsia. 2011;52(Suppl. 6):257–8.
Brady K, Hebeisen S, Konrad D, Soares-da-Silva P. The effects of eslicarbazepine, R-licarbazepine, oxcarbazepine and carbamazepine on ion transmission Cav3.2 channels. Epilepsia. 2011;52(Suppl. 6):260.
Thiessen J. Bioavailability and bioequivalence. In: du Souich P, Orme M, Erill S, editors. The IUPHAR compendium of basic principles for pharmacological research in humans. IUPHAR; 2004. p. 55–66.
Shep D, Nimkar A, Shah R, Jaiswal V. Comparative bioavailability study with two sodium valproate tablet formulations in healthy subjects. Int J Pharma Sci Drug Res. 2011;3(2):101–3.
Almeida L, Soares-da-Silva P. Safety, tolerability, and pharmacokinetic profile of BIA 2-093, a novel putative antiepileptic, in a rising multiple-dose study in young healthy humans. J Clin Pharmacol. 2004;44(8):906–18.
Fontes-Ribeiro C, Macedo T, Nunes T, Neta C, Vasconcelos T, Cerdeira R, et al. Dosage form proportionality and food effect of the final tablet formulation of eslicarbazepine acetate: randomized, open-label, crossover, single-centre study in healthy volunteers. Drugs R D. 2008;9(6):447–54.
Chow SC, Wang H. On sample size calculation in bioequivalence trials. J Pharmacokinet Pharmacodyn. 2001;28(2):155–69.
Steinijans VW, Sauter R, Hauschke D, Diletti E, Schall R, Luus HG, et al. Reference tables for the intrasubject coefficient of variation in bioequivalence studies. Int J Clin Pharmacol Therapeut. 1995;33(8):427–30.
Hassan Y, Alfadly S, Azmin M, Peh K, Tan T, Noorizan A, et al. Bioequivalence evaluation of two different formulations of ciprofloxacin tablets in healthy volunteers. Singap Med J. 2007;48:819–23.
Midha K, McKay G. Bioequivalence; its history, practice, and future. AAPS J. 2009;11(4):664–70.
Rani S, Pargal A. Bioequivalence: an overview of statistical concepts. Indian J Pharmacol. 2004;36(4):209–16.
EMEA/CPMP. Note for guidance on the investigation of bioavailability and bioequivalence. CPMP/EWP/1401/98, European Agency for the Evaluation of Medicinal Products, Committee for Proprietary Medicinal Products (CPMP), 2001 [online]. Available from URL:http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003011.pdf. Accessed 11 Apr 2011.
FDA/CDER. Guidance for industry (draft). Food-effect bioavailability and fed bioequivalence studies. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), 2002 [online]. Available from URL:http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126833.pdf. Accessed 11 Apr 2011.
Almeida L, Falcao A, Maia J, Mazur D, Gellert M, Soares-da-Silva P. Single-dose and steady-state pharmacokinetics of eslicarbazepine acetate (BIA 2-093) in healthy elderly and young subjects. J Clin Pharmacol. 2005;45(9):1062–6.
Acknowledgments
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. This study was sponsored by BIAL-Portela & Co., SA.
Disclosure
This study was sponsored by BIAL-Portela & Co., SA. All authors were involved in the design or conduct of the study, the collection, management or analysis of the data, and the preparation or review of the manuscript. Dr. Falcão received consultancy honoraria from BIAL-Portela & Co., SA. Drs. Lima, Sousa, Nunes and Soares-da-Silva are or were employees of BIAL at the time of the study.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Falcão, A., Lima, R., Sousa, R. et al. Bioequivalence of Eslicarbazepine Acetate from Two Different Sources of its Active Product Ingredient in Healthy Subjects. Drugs R D 13, 137–143 (2013). https://doi.org/10.1007/s40268-013-0016-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-013-0016-6