
Abstract
Systemic autoimmune disorders are complex heterogeneous chronic diseases involving many different immune cells. A significant proportion of patients respond poorly to therapy. In addition, the high burden of adverse effects caused by “classical” anti-rheumatic or immune modulatory drugs provides a need to develop more specific therapies that are better tolerated. Bruton’s tyrosine kinase (BTK) is a crucial signaling protein that directly links B-cell receptor (BCR) signals to B-cell activation, proliferation, and survival. BTK is not only expressed in B cells but also in myeloid cells, and is involved in many different signaling pathways that drive autoimmunity. This makes BTK an interesting therapeutic target in the treatment of autoimmune diseases. The past decade has seen the emergence of first-line BTK small-molecule inhibitors with great efficacy in the treatment of B-cell malignancies, but with unfavorable safety profiles for use in autoimmunity due to off-target effects. The development of second-generation BTK inhibitors with superior BTK specificity has facilitated the investigation of their efficacy in clinical trials with autoimmune patients. In this review, we discuss the role of BTK in key signaling pathways involved in autoimmunity and provide an overview of the different inhibitors that are currently being investigated in clinical trials of systemic autoimmune diseases, including rheumatoid arthritis and systemic lupus erythematosus, as well as available results from completed trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Bruton’s tyrosine kinase (BTK) is a crucial signaling protein that links signals from the B-cell antigen receptor to the activation, proliferation, and survival of B cells. |
Together with the emergence of very specific small-molecule BTK inhibitors, this makes BTK an interesting therapeutic target in the treatment of autoimmune diseases. |
Here, we discuss the role of BTK in autoimmunity and the current status of BTK inhibition in clinical trials of systemic autoimmune diseases. |
1 Introduction
Autoimmune disorders are driven by a loss of self-tolerance, often involving aberrant selection and activation of autoreactive B cells and subsequent auto-antibody production [1]. Each B cell harbors a unique antigen receptor, the B-cell receptor (BCR), that can recognize a specific antigen, thereby activating the B cell. Bruton’s tyrosine kinase (BTK) is a crucial signaling protein that directly links BCR signals to B-cell proliferation and survival [2] (Fig. 1). The gene encoding BTK was discovered as the gene affected in X-linked agammaglobulinemia (XLA) patients [3, 4]. Because of the lack of functional BTK protein, mature B cells and antibodies are almost completely absent in these patients. The importance of BCR signaling in autoimmunity is underlined by studies in animal models showing that single-gene defects and changes in expression of BCR signaling molecules or inhibitory co-receptors can lead to autoimmunity [5, 6]. Furthermore, in human autoimmune diseases, genetic susceptibility loci include genes encoding BCR signaling molecules, for example LYN, BLK, BANK1, PTPN22, and PXK [7,8,9,10,11,12]. The efficacy of B-cell-depleting therapy in autoimmune patients has prompted the search for more specific B-cell-targeting therapies, leading to the development of small-molecule inhibitors of BCR signaling molecules including BTK.

Role of BTK in B-cell signaling. Overview of BCR signaling and other important signaling modules for B cells. Upon BCR engagement, LYN will activate and phosphorylate Ig-α and Ig-β, subsequently activating SYK. Together with CD19-mediated activation of PI3K, this leads to the activation of SLP65, BTK, and PLCγ2. This in turn activates downstream signaling pathways crucial for proliferation and survival, including engagement of ERK, NF-κB, and downstream mediators of AKT like S6, and anti-apoptotic proteins like BCL-2. Signaling downstream of TLRs and BAFFR also involves BTK phosphorylation, leading to activation of these same proliferation and survival factors. Other receptor signaling pathways like chemokine receptor signaling also contribute to migration, proliferation, and survival of B cells. BTK Bruton’s tyrosine kinase, BCR B-cell receptor, Ig immunoglobulin, PTPN22 protein tyrosine phosphatase non-receptor type 22, SYK spleen tyrosine kinase, PI3K phosphoinositide 3-kinase, SLP65 Src homology 2 domain-containing leukocyte adaptor protein of 65 kDa, CIN85 Cbl-interacting protein of 85 kDa, PLCγ2 phospholipase Cγ2, DAG diacylglycerol, IP3 inositol triphosphate, PKC-β protein kinase C β, TRAF3 TNF receptor-associated factor 3, NIK NF-κB-inducing kinase, IKKα inhibitor of NF-κB kinase, MyD88 myeloid differentiation factor 88, MAL MyD88 adaptor-like, IRAK2 interleukin-1 receptor-associated kinase 2, ERK extracellular signal-related kinase, NFAT nuclear factor of activated T cells, BCAP B-cell adaptor for PI3K, PTEN phosphatase and tensin homolog, SHIP1 SH-2 containing inositol 5' polyphosphatase 1, BCL-2 B-cell lymphoma-2
In addition to B cells, BTK is expressed by most hematopoietic cells, including the monocyte/macrophage/dendritic cell lineage, granulocytes, mast cells, platelets, osteoclasts, and erythroid cells [3, 4, 13,14,15,16,17,18,19,20]. Although rarely BTK expression has been reported in mouse T and natural killer (NK) cells [21, 22], it is generally accepted that these cells do not express BTK protein (search BTK at http://rstats.immgen.org/Skyline/skyline.html) [2,3,4, 20]. BTK also plays a role in many other key immune signaling pathways, including B-cell-activating factor receptor (BAFFR), Toll-like receptor (TLR), chemokine receptor, and Fc receptor (FcR) signaling (Fig. 1) [20].
The first-in-kind inhibitor of BTK kinase activity to reach the clinic was ibrutinib (Imbruvica, PCI-32765), which obtained US Food and Drug Administration (FDA) approval in 2013 for mantle cell lymphoma (MCL) patients, and subsequently for chronic lymphocytic leukemia (CLL) patients upon successful phase II trials [23, 24]. The efficacy of ibrutinib in CLL may not only be attributed to inhibition of BTK, affecting survival and homing of CLL cells, as changes in the tumor microenvironment through off-target effects, such as reducing T-cell exhaustion and promoting an anti-tumor Th1 phenotype, might contribute to its efficacy [25]. Off-target kinase inhibition may not be beneficial in autoimmunity and may be associated with significant side effects, making ibrutinib less suitable for treatment of chronic autoimmune diseases. These findings prompted the development of more specific BTK inhibitors with a favorable safety profile for the treatment of autoimmune diseases.
The first BTK inhibitor reported for the treatment of a chronic inflammatory disorder was evobrutinib (M2951), in a clinical trial with active relapsing-remitting multiple sclerosis (MS) patients [26]. At a dose of 75 mg once daily, this BTK inhibitor significantly reduced the number of enhancing lesions from 12 through 24 weeks of treatment compared to placebo. Efficacy was maintained for 2 years with a reduction in annual relapse rate and increased time until first relapse [27].
A large number of experimental models support the efficacy of BTK inhibition in MS. In experimental autoimmune encephalomyelitis (EAE) mouse models of MS, evobrutinib prophylactically reduced disease severity [28], and evobrutinib treatment in mice with established symptoms resulted in reduced leptomeningeal inflammation [29]. Hereby, the numbers of meningeal B cells were reduced, but myeloid cell infiltrates appeared to persist. BTK is expressed in microglia and astrocytes, and treatment with evobrutinib resulted in a significant reduction in astrocytosis in the cortex surrounding the areas of meningeal inflammation [29]. Likewise, ibrutinib treatment decreased the proliferative response of microglia cells upon injection of anti-myelin oligodendrocyte glycoprotein monoclonal antibodies in mice [30]. BTK inhibition resulted in improved remyelination in vitro in demyelinated cerebellar slices, as well as in vivo in a transgenic Xenopus laevis model [31]. Furthermore, evobrutinib was shown to inhibit activation of B cells, reducing pro-inflammatory cytokine RNA expression, and limiting expression of proteins involved in antigen presentation [28]. However, T cells were also indirectly affected, and evobrutinib reduced disease severity in a B-cell independent MS mouse model [32]. In addition, GM-CSF-induced M1 macrophage differentiation and cytokine expression were reduced in vitro [33]. Taken together, these findings suggest that the therapeutic efficacy of BTK inhibition in MS patients may partly be due a substantial impact on the myeloid compartment, and thus beyond effects on B-cell activity.
The positive results of BTK inhibition obtained in MS patients, together with the promising efficacy in pre-clinical animal models of rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) [34, 35], make BTK inhibition an interesting therapeutic option in systemic autoimmune disorders. This notion is further supported by various human in vitro studies. In this review, we discuss the role of BTK in key signaling pathways involved in autoimmunity, as well as the various inhibitors that are currently being investigated in clinical trials in patients with systemic autoimmune disease.
2 Bruton’s Tyrosine Kinase (BTK) in Signaling Pathways
2.1 BTK in B-Cell Receptor Signaling
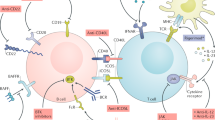
In the context of autoimmune disease, B cells can have various pathogenic characteristics (Fig. 2). Autoreactive B cells can initiate and/or enhance the autoimmune response by presenting auto-antigens on major histocompatibility complex II (MHC-II) molecules, and by providing activating signals through co-stimulatory factors, such as CD80 and CD86, to autoreactive T cells. In turn, B cells can differentiate, in a T-dependent or -independent manner, into plasma cells that produce autoreactive antibodies. B cells can be the source of large quantities of cytokines, including the pro-inflammatory cytokine IL-6. These support an inflammatory and autoimmune environment by activating surrounding cells and stimulating proliferation and survival. Finally, B cells are involved in stimulating the development and maintenance of tertiary lymphoid structures by producing soluble factors, such as lymphotoxin α1β2 [36, 37]. These structures may contain germinal centers and are often found in the affected organs of autoimmune disease patients.

The four main pathogenic roles of B cells in the context of systemic autoimmune disease. (I) Initiation or enhancement of autoimmune responses by presenting auto-antigens to T cells and concomitantly providing co-stimulatory signals. (II) B cells can differentiate into autoantibody-producing plasma cells. (III) B cells can produce pro-inflammatory cytokines. (IV) B cells are involved in stimulating the development and maintenance of these tertiary lymphoid structures
B cells are generated in the bone marrow, where gene rearrangement of immunoglobulin V, D, and J elements in developing B cells leads to the expression of a unique BCR. Only those B cells expressing a functional BCR survive and subsequently undergo negative selection as they are checked for autoreactivity. This selection mainly takes place during development in the bone marrow and during the transitional stage in the circulation and is dependent on BCR signaling strength [38]. The BCR converts extracellular activation by cognate antigen into intracellular signals, mostly dominated by phosphorylation of protein kinases (Fig. 1). Following antigen binding to the BCR, the recruitment of the Src-family tyrosine protein kinase Lck/yes novel tyrosine kinase (LYN) and phosphorylation of the IgM-associated heterodimers Igα and Igβ are early events in this cascade, leading to the phosphorylation of spleen tyrosine kinase (SYK) [39]. BTK is then recruited to the cell membrane by phosphoinositide 3-kinase (PI3K) through the formation of phosphatidylinositol-3,4,5-triphosphate (PIP3) [40]. This recruitment enables SYK to phosphorylate BTK at tyrosine Y551 in its kinase domain [41,42,43,44,45]. SYK concomitantly phosphorylates the B-cell linker (BLNK) protein, also known as SLP65, which serves as a scaffolding protein by interacting with various signaling molecules including BTK and PLCγ2 [46]. Upon BCR engagement, multiple SLP65 molecules are recruited to CIN85 trimers, resulting in the assembly of large CIN85-SLP65 macromolecular signaling complexes [47]. As a result, BTK is fully activated and autophosphorylation at Y223 follows, although this event does not appear to be crucial for BTK function in mouse B cells [48]. Active BTK in turn phosphorylates phospholipase Cγ2 (PLCγ2) [49], leading to cytoplasmic calcium influx and the activation of several downstream signaling pathways and transcription factors, important for survival, proliferation, and differentiation, including nuclear factor (NF)-κB [50, 51].
The rapid and stronger response of IgG-expressing memory B cells is thought to be, at least partially, dependent on the presence of the immunoglobulin tail tyrosine (ITT) motif in the cytoplasmic tail of membrane IgG [52]. The ITT motif can be phosphorylated by SYK, resulting in the recruitment of BTK via the adaptor protein Grb2. The dual activation pathway of BTK is thought to result in an amplification of IgG-induced calcium mobilization in memory B cells.
2.2 BTK and B-Cell-Activating Factor Receptor (BAFFR) Signaling
Survival and homeostatic proliferation of mature B cells are dependent on tonic BCR signaling and B-cell activating factor (BAFF), which is a cytokine that belongs to the tumor necrosis factor (TNF) ligand family [53, 54]. BAFF can bind three receptors: B-cell maturation antigen (BCMA), transmembrane activator and CAML interactor (TACI), and BAFF receptor (BAFFR). In addition to activating the non-canonical NFκB pathway, the latter transduces survival signals via the canonical NF-κB pathway by co-opting the BCR signaling pathway through SYK and BTK (Fig. 1) [55,56,57]. Activation of these downstream signaling pathways fuels B-cell survival in a direct and indirect fashion: directly by enhancing the expression of pro-survival factors and dampening the expression of pro-apoptotic factors [58, 59], and indirectly by driving the expression of the receptors for BAFF and their downstream signaling substrates [60,61,62]. In addition, during B-cell activation BAFF supports class switching and differentiation into plasma cells [63].
Murine studies have shown that a disbalance in either of these signals can cause an altered B-cell selection and can thereby result in an autoimmune phenotype. For example, overexpression of BAFF in mice leads to the development of a Sjögren/SLE-like phenotype [64]. Similarly, B-cell-specific overexpression of BTK leads to a systemic autoimmune phenotype in mice [6]. In human systemic autoimmune disease, increased BAFF levels in serum and enhanced BCR signaling in circulating B cells are also observed [63, 65], possibly allowing the escape of autoreactive B cells from negative selection. Increased levels of BAFF may rescue autoreactive B cells from anergy and promote the formation of a mature follicular dendritic cell network, allowing subsequent affinity maturation and differentiation in germinal centers (GC) [66, 67].
2.3 BTK in Other Signaling Pathways
Next to its well described role in BCR signaling, BTK is also involved in signaling downstream of TLRs (Fig. 1), specifically TLR3, 4, 7, 8, and 9 both in B cells and in other hematopoietic cell types [68]. Although evidence exists for the involvement of BTK kinase activity in downstream TLR signaling [69], BTK is mainly thought to function as a linker protein for the recruitment of myeloid differentiation primary response 88 (MyD88) and MyD88 adaptor-like (MAL) to the receptor [70]. TLR stimulation leads to the activation of interferon-regulatory factors and/or the NF-κB pathway, resulting in expression of co-stimulatory receptors and adhesion molecules, the production of pro-inflammatory cytokines and antigen presentation. In B cells, TLR activation leads to B-cell survival by similar mechanisms as previously described for BCR and BAFF signaling [71]. Therefore, TLR signaling can lead to synergistic effects when B cells are simultaneously stimulated by the BCR [72].
BTK is also involved in FcR signaling [73, 74]. The activation of the FcεR promotes histamine release in mast cells and FcγR activation leads to pro-inflammatory cytokine production and enhances antigen presentation by myeloid cells [75]. BTK links RANK (receptor activator of NF-κΒ) activation together with co-stimulation, for example by FcRs, to drive osteoclast differentiation [76]. This can play a pathogenic role in systemic autoimmune diseases such as RA [76, 77]. BTK is involved in downstream signaling of several chemokine receptors, including CXCR4 [78], and in the intracellular signal transduction required for integrin activation and upregulation [79,80,81]. Therefore, in leukocytes it is important for the attraction towards and infiltration into the site of inflammation. BTK may also play an antibody-independent role in autoimmunity through its interaction with transcription factor Bright/ARID3a, which is implicated in Ig heavy chain transcription [82]. Mice overexpressing Arid3a display autoimmunity, and SLE patients show increased ARID3a expression in B cells, although this did not correlate with autoantibody production [83,84,85]. In this context, it is of note that both ARID3a and interferon (IFN)α were induced in B cells upon TLR9 stimulation [86].
Taken together, the wide expression and functional profile of BTK implies that modulating BTK activity could be of benefit by restraining these inflammatory and autoimmune processes by targeting multiple pathways in multiple cell types simultaneously.
2.4 BTK Structure and Inhibitor Target Sites
BTK is a member of the highly conserved family of cytoplasmic TEC kinases, which also includes tyrosine kinase expressed in hepatocellular carcinoma (TEC), interleukin-2 inducible T-cell kinase (ITK), bone marrow-expressed kinase (BMX), and resting lymphocyte kinase (RLK) [19].
BTK consists of 659 amino acids and contains five domains with two phosphorylation sites. It is transiently recruited to the cell membrane upon BCR activation through the activity of PI3K, which results in the generation of PIP3, which interacts with the N-terminal pleckstrin homology (PH) domain of BTK. In addition, it contains a Tec homology (TH), a SRC homology 3 (SH3), and an SH2 domain, as well as a C-terminal kinase domain. The proline-rich TH domain is important for optimal activity and stability of the protein, but also has the capacity to bind other kinases including LYN. The SH3 domain contains the Y223 autophosphorylation site and interacts with various signaling molecules via binding to proline-rich regions. The SH2 domain has the capacity to bind to phosphorylated tyrosine residues of signaling molecules, in particular SLP65.
Because many human diseases, particularly malignancies, involve dysregulation of one of the 518 identified protein kinases [87], these enzymes have become important drug targets. Nearly all currently FDA-approved small-molecule kinase inhibitors have a competitive mechanism of action. Such reversible inhibitors are classified as type I-V, based on their interaction with the ATP-binding pocket of an active or inactive kinase conformation or binding to a site distinct from the active site [88]. In contrast, many BTK inhibitors, including the prototypic irreversible inhibitor ibrutinib [34, 35], are classified as type VI, and form a covalent bond with Cys-481 within the ATP-binding site of the kinase domain. They generally have a high specificity, because the Cys-481 amino acid residue is poorly conserved among kinases (Table 1). More recently developed inhibitors, such as acalabrutinib, poseltinib, spebrutinib, and tirabrutinib, also interact irreversibly with Cys-481 [89] (Table 1). Although rilzabrutinib (PRN-1008) forms a covalent bond with Cys-481, inhibition is reversible [90].
Other reversible BTK inhibitors, including RN486, CGI1746, and fenebrutinib [75, 91, 92], are classified as type II inhibitors, because they stabilize an inactive non-phosphorylated form of BTK [93]. These inhibitors target the H3 pocket (a network of three hydrogen bonds involving the residues K430, G414, and T474), which is immediately adjacent to the region occupied by ATP. Thereby, these inhibitors sequester the Y551 residue and prevent its phosphorylation [94]. Interestingly, the ability to sequester Y551 was shown to determine the potency for inhibition of FcγR and FcεR signaling, but not of BCR signaling, perhaps providing an added benefit in autoimmune and allergic indications, respectively [94]. Very recently, another BTK inhibitor was developed, remibrutinib (LOU064), which also stabilizes an inactive BTK conformation by selectively targeting the H3 pocket by forming a covalent bond [95]. An obvious important benefit of covalent inhibitors is their ability to reach higher and more sustained BTK occupancy, compared with reversible inhibitors that need to be present in the circulation at high levels to allow for effective inhibition.
It has been well documented that many CLL patients treated with ibrutinib develop drug resistance due to a C481S mutation that disrupts the binding of this BTK inhibitor to the cysteine residue [96]. Several BTK inhibitors can effectively counteract ibrutinib-resistant C481S-BTK. These include the type II inhibitors CGI-1746 and fenebrutinib, as well as XMU‐MP‐3, which has a unique binding mode and interacts with the M477, E445, and S538 residues of BTK. Likewise, the irreversible inhibitor ZYBT1 inhibits both wild-type and C481S-BTK but binds to the same site as ibrutinib [75, 92, 97, 98]. Altogether, the current number of BTK inhibitors and the obtained knowledge on their biological activity is overwhelming, given that already by the end of February 2017 almost 1,500 small-molecule inhibitors had been developed, involving over 300 BTK-inhibitor-related patents [89].
3 Evidence For a Critical Role of BTK in Autoimmunity
3.1 Animal Models
Many animal studies have indicated a critical role of BTK in autoimmunity, in both B cells and myeloid cells. Btk-deficient Xid mice are protected against the development of (full blown) autoimmunity in several disease models, including collagen-induced arthritis (CIA) for RA and the NZBxNZW model for SLE [20, 99, 100]. Vice versa, overexpression of BTK specifically in murine B cells is sufficient to induce a spontaneous autoimmune phenotype resembling human SLE and Sjögren’s syndrome (SjS), featuring spontaneous GC and plasma cell formation, autoantibody production and immune infiltration in lungs, kidneys, and salivary glands [6]. This phenotype depends on B–T cell interaction and TLR signaling, and can be ameliorated by in vivo Btk inhibition [6, 101, 102].
In murine CIA, Btk inhibition in vivo reduces disease severity in a dose-dependent fashion, even after onset of arthritis, comparable to standard-of-care therapeutics such as dexamethasone (Table 2) [35, 92, 103, 104]. Furthermore, Btk inhibition enhances the effects of standard-of-care therapeutics when administered together [105]. Inhibitors may target multiple cell types involved in the pathogenesis of disease, including B cells, myeloid cells, and osteoclasts, thereby reducing autoantibody formation, pro-inflammatory cytokine production and bone destruction. Similar to CIA, Btk inhibition also protects against severe progressive disease in a number of murine lupus models, including mice with a mutation in Fas (MRL/lpr) [2]. The efficacy depends on inhibition of both B cells and myeloid cells. Interestingly, lupus nephritis or kidney damage is often reduced, despite persistence of autoantibodies in the serum, particularly anti-RNA antibodies [106].
Other non-autoimmune disease models provide evidence of a role for BTK in cell types and pathogenic mechanisms that may be relevant in autoimmune diseases. For example, Btk is essential in the crosstalk between TLR4 and FcγRIIa signaling leading to activation of alveolar neutrophils in an LPS/immune complex-mediated model of acute respiratory distress syndrome (ARDS) [107]. Furthermore, Btk inhibition inhibited neutrophil extracellular trap release into the lungs upon influenza infection in mice [108]. BTK may also be protective in some diseases. In bone marrow-derived macrophages in mice (and monocytes in humans), Btk deficiency or Btk inhibition leads to increased NLR family pyrin domain-containing 3 (NLRP3) activation, increasing susceptibility for inflammatory bowel disease (IBD) [109]. Together these studies emphasize the therapeutic potential of BTK inhibition in autoimmunity and may explain side effects observed in patients treated with BTK inhibitors.
3.2 Human Studies
BTK protein and phosphorylated BTK levels are increased in circulating B cells of RA, SjS, and granulomatosis with polyangiitis (GPA) patients [65, 110]. This increase was already evident in new emerging B cells, i.e., transitional and naïve B cells, and was associated with active disease. BTK protein and phosphorylation levels correlated with autoantibodies and with numbers of pathogenic T cells in the circulation, as well as with immune infiltrates in the salivary glands of SjS patients [110, 111].
In vitro treatment of macrophages in synovial explants of RA and psoriatic arthritis patients with RN486, a highly selective, reversible inhibitor of BTK, showed that BTK is required for cytokine production in macrophages upon FcR or CD40 stimulation [112]. Enhanced BTK expression, through BTK-encoding adenovirus transduction of macrophages, increased IL-6 production upon CD40 or FcγR stimulation [112]. In a co-culture system of activated platelets with primary human fibroblast-like synoviocytes (FLS), cytokine production was inhibited by RN486 in a dose-dependent manner [113].
Similarly, pro-inflammatory cytokine production by effector B cells of systemic sclerosis (SSc) patients is inhibited by ibrutinib treatment in vitro, as is anti-topoisomerase autoantibody production [114]. In B cells from GPA patients, in vitro BTK inhibition with BMS-986142 also reduced BCR-mediated cytokine production, but appeared to be less effective in B cells from patients with active GPA, compared to B cells from patients in remission and healthy controls [65]. Furthermore, in vitro memory B cell and plasma cell formation and antibody production were inhibited in patients in remission, but not in patients with active GPA. These findings indicate that although BTK inhibition can inhibit effector functions of B cells in autoimmune patients, B cells from patients with active disease may be less susceptible to BTK inhibition.
Based on the observation that BTK inhibition protects against fatal lung injury in acute respiratory distress syndrome mouse models [107, 108], as well as the rather mild course of Coronavirus disease 2019 (COVID-19) in XLA patients and BTK inhibitor-treated leukemia patients [115,116,117,118,119], acalabrutinib was administered in a prospective study in COVID-19 patients. Clinical recovery of COVID-19 was seen and inflammatory markers normalized, indicating that BTK inhibition has the capacity to correct the dysregulated and damaging inflammatory response by macrophages [120]. Although a randomized phase II clinical trial of acalabrutinib in hospitalized patients (NCT04346199, available at clinicaltrials.gov) failed to meet the primary endpoint according to an Astra Zeneca press release on 12 November 2020, other BTK inhibitors, including abivertinib (NCT04528667; NCT04440007) and ibrutinib (NCT04439006), are currently under investigation in clinical trials. Interestingly, very recently, evidence was provided that during severe COVID-19, the SYK inhibitor fostamatinib can counteract the critical pro-inflammatory role of aberrantly glycosylated anti-spike IgG that activates macrophages via FcγRs [121]. Blocking signaling of the FcγRIIa-SYK axis in platelets using fostamatinib may additionally prevent platelet aggregation and vascular complications in COVID-19 patients [122]. These findings may have implications for the treatment of autoimmune disorders, since changes in antibody glycosylation are also observed during active autoimmune disease [123] and BTK inhibition abolishes FcγRIII-induced TNFα, IL-1β, and IL-6 production in macrophages in the context of myeloid cell-mediated RA [75].
4 BTK Inhibitors in Clinical Trials of Systemic Autoimmune Diseases
Although several trials with BTK inhibitors in autoimmune diseases have been completed, few have been published in research articles. When no publications were available, the data in this review were largely obtained from clinicaltrials.gov (search for stated NTC number) or from published abstracts of scientific meetings.
4.1 Acalabrutinib
Acalabrutinib, an irreversible, covalent inhibitor of BTK, was the second BTK inhibitor to reach the clinic for B-cell malignancies [124]. This inhibitor has a much higher specificity for BTK than the first-in-kind inhibitor ibrutinib, making it more suitable for autoimmune patients. A clinical trial was conducted with RA patients with stable methotrexate (MTX) treatment, but to date results have not been published (NCT02387762).
4.2 BMS-986142
This reversible, covalent BTK inhibitor reduced FcR-mediated cytokine production by healthy human peripheral blood mononuclear cells (PBMCs) and BCR-induced cytokine production by healthy human B cells in vitro (Table 2) [105]. As described above, in GPA, BMS-986142 inhibited plasma cell differentiation and antibody production in vitro, although B cells from patients with active disease appeared less susceptible to treatment than B cells from remission patients or healthy individuals. In animal models of RA, this inhibitor has shown promising efficacy by dose dependently reducing joint inflammation and destruction, synergistically with standard-of-care treatment [105]. However, in a clinical trial with active RA patients, no significant improvement of symptoms was observed after 12 weeks of combined treatment with BMS-986142 and MTX compared to placebo and MTX (NCT02638948) (Table 3). A monotherapy study with primary SjS patients was terminated due to inability to reach protocol objectives (NCT02843659).
4.3 Elsubrutinib
In animal models of autoimmunity, this irreversible, non-covalent BTK inhibitor showed dose-dependent improvement in disease severity, organ damage, and mortality [125]. Importantly, antibody formation following model antigen immunization or vaccination was not hampered in mice, suggesting that normal antibody responses can be mounted upon infection [125]. In vitro, elsubrutinib inhibited activation and pro-inflammatory cytokine production by B cells and PBMCs. However, in a clinical trial with RA patients, elsubrutinib did not enhance the efficacy of the JAK-1 inhibitor upadacitinib, and appeared to have limited efficacy as monotherapy (NCT03682705). A phase II clinical trial with SLE patients is currently recruiting patients (NCT03978520; long-term extension study NCT04451772).
4.4 Evobrutinib
This irreversible covalent BTK inhibitor showed promising results in animal models for RA, SLE, and MS [28, 126]. In addition, it showed clinical efficacy in MS patients, as described above [26]. Nevertheless, evobrutinib did not appear to improve symptoms in RA patients on stable MTX or with inadequate response to MTX and SLE patients (NCT02784106, NCT03233230, and NCT02975336).
4.5 Fenebrutinib
Fenebrutinib is a highly specific, reversible, non-covalent inhibitor of BTK. It was effective in CIA in mice and inhibits BCR and CD40L-mediated signaling and activation of B cells [92]. In a cohort of RA patients with an inadequate response to MTX, fenebrutinib increased the proportion of patients with an American College of Rheumatology (ACR) 50 response (50% improvement of disease symptoms defined by the ACR [127]) after 12 weeks of treatment with a dose of 150 mg daily compared to placebo, and reached an effectiveness comparable to adalimumab treatment with a high dose of 200 mg twice daily [128]. In addition, at this high dose, it increased the ACR50 response rate in another patient cohort, characterized by an inadequate response to TNFα inhibition, although not significantly. However, ACR20 response and changes in disease activity score 28-C-reactive protein (DAS28-CRP; a disease activity score including 28 swollen joint counts and CRP levels [129]) at 12 weeks compared to baseline were significant in these patients [128]. All doses were well tolerated, with (serious) adverse events occurring in fenebrutinib-treated patients at similar rates as in adalimumab-treated patients. Fenebrutinib moderately reduced total IgM, IgG, and rheumatoid factor-IgM in the serum of patients in a dose-dependent fashion, and in the second cohort it also reduced serum anti-citrullinated protein antibody (ACPA)-IgG. Despite the high efficacy in RA patients, a similar study in moderate to severe SLE patients did not improve symptoms (NCT02908100; long-term extension study NCT03407482).
4.6 Poseltinib
Poseltinib, an irreversible, non-covalent BTK inhibitor, reduced disease severity in several animal models of autoimmunity after onset of disease [103, 130]. Safety assessment in RA patients with mildly active disease showed favorable results [131]. However, the second part of this trial, with moderately to severely active RA patients, was discontinued when interim analysis showed no significant improvement of symptoms after 12 weeks of treatment with any of the three doses tested [131].
4.7 Rilzabrutinib
Rilzabrutinib is a reversible, covalent BTK inhibitor that potently inhibits B-cell activation and proliferation upon BCR stimulation, and antibody production upon CD40/IL-21R or TLR9 stimulation in vitro [132]. In addition, it showed efficacy in a wide range of (auto)immune animal models [132]. Of note, in contrast to first-line BTK inhibitors, rilzabrutinib does not interfere with normal platelet aggregation. An open-label study with pemphigus vulgaris patients yielded promising results, with 52% and 70% of patients showing control of disease activity at 4 weeks and 8 weeks, respectively, which further increased and was maintained after treatment was stopped [133]. A phase III, placebo-controlled trial is currently ongoing (NCT03762265).
4.8 Spebrutinib
Spebrutinib can effectively inhibit the activation of several immune cells through various receptors and signaling pathways in vitro and reduces disease severity in CIA in mice [104]. In a clinical trial with female RA patients on stable MTX, naïve B cells increased in the circulation of treated patients, and transitional B cells decreased. In addition, levels of CTX-1, a collagen-degradation product, were decreased, suggesting reduced osteoclastogenesis, as were CXCL13 levels [134]. However, the ACR20 response rate was not significantly increased in treated patients versus placebo-treated controls at 4 weeks.
4.9 Tirabrutinib
This irreversible, covalent inhibitor of BTK showed significant effectiveness in the inhibition of human osteoclasts in vitro and in CIA in vivo [135,136,137,138]. Nevertheless, in a clinical trial with SjS patients, no clinical improvement was observed (NCT03100942).
4.10 BTK Inhibitors in Clinical Trials of Autoimmune Diseases with Results Pending
Branebrutinib is an irreversible, covalent inhibitor of BTK that has shown efficacy in RA and SLE mouse models, reducing clinical signs of disease dose dependently [139]. This inhibitor was well tolerated in a phase I study with healthy individuals [140], and these promising results have led to a clinical trial with active SLE, primary SjS, and RA patients, which is currently recruiting patients (NCT04186871).
Orelabrutinib is an irreversible, covalent inhibitor of BTK that was reported to be effective in animal models of RA and SLE, and is currently in a clinical trial with SLE patients (NCT04305197).
Remibrutinib is a highly specific, irreversible, non-covalent BTK inhibitor that reduced antigen-specific IgM formation in rats upon sheep red blood-cell immunization and reduced arthritis development and joint destruction in CIA in rats [95]. A clinical trial with primary SjS patients is currently recruiting (NCT04035668).
TAS5315 is a covalent inhibitor of BTK that showed high selectivity and promising efficacy in animal models of RA and SLE [141,142,143,144]. A clinical trial with RA patients with inadequate response to MTX has been completed, but no data have been reported yet (NCT03605251).
5 Concluding Remarks and Future Perspectives
The last decade has seen the emergence of a large number of small-molecule inhibitors targeting BTK, many of which demonstrated promising efficacy in preclinical studies with autoimmune animal models. However, in contrast to the convincing clinical efficacy of BTK inhibitors in patients with B-cell malignancies, many clinical trials conducted in autoimmune patients to date have reported negative results, indicating that BTK inhibition as monotherapy may not be sufficient in (many of) these patients. However, the observed reduction of enhancing lesions in evobrutinib-treated patients with multiple sclerosis is an important and very promising exception. Differences in the pathogenic mechanisms of disease and cell types involved may underlie the differences in efficacy. For example, it is of note that BTK is expressed in astrocytes and microglia cells and that animal models for MS have demonstrated that BTK inhibition affects the numbers of astrocytes and microglia cells in the brain and supports remyelination [30, 31].
The reasons for a limited efficacy of BTK inhibition in systemic autoimmunity remain largely unknown, but several mechanisms may contribute to this phenomenon. First, BTK inhibitor treatment in humans may be associated with low BTK occupancy. Effects of BTK inhibition in affected organs in humans are mostly studied in vitro, and BTK occupancy is often not measured in vivo, even in preclinical models. For example, in vitro experiments have shown an effect of BTK inhibition on synovial macrophages and the platelet–fibroblast-like synoviocyte interaction in RA [112, 113], but whether these effects also occur in vivo remains unclear. For MS, in vivo data in humans are limited, but in EAE mice high BTK occupancy following evobrutinib treatment was observed in the brain [145]. Limited BTK occupancy might also be supported by the observation that there is no profound loss of circulating B cells even after prolonged inhibitor treatment, in contrast to the severe B-cell deficiency found in patients with XLA [146].
Secondly, it is conceivable that BTK inhibition affects critical immunosuppressive capacities of B cells. For example, BTK inhibition in vitro inhibited the differentiation of B cells with a regulatory function and their production of IL-10 and IL-35, which are essential mediators of immunosuppressive B cells [147]. However, small doses of ibrutinib were shown to preserve IL-10 production, while reducing the profibrotic cytokines TNFα and IL-6 in B cells of patients with SSc [114].
Thirdly, BTK inhibitor efficacy may be related to differential involvement of BTK in other signaling pathways in various immune cells. As mentioned above, BTK inhibition can be particularly effective in specific cells types, for example strongly limiting the activity of astrocytes and microglia cells. BTK is required for TLR-induced IL-10 production in macrophage and DC populations [148, 149]. In this context, it was shown that BTK-deficient DCs or inhibitor-treated DCs gain a more mature phenotype with a stronger T-cell-stimulatory ability than wild-type cells [149, 150]. Furthermore, the absence of BTK in XLA patients is associated with impaired TLR9-induced production of IL-6, IL-12, TNF-α, and IL-10, whereby it is of note that TLR9 is protective in autoimmune diseases such as SLE [151, 152].
Finally, in particular signaling pathways BTK functions as a linker molecule, as was shown for interaction with Myd88 and MAL [70], which would render kinase inhibition ineffective. Efficacy may also be hampered by the induction of rewiring of signaling pathways by BTK inhibition in B cells. In this context, we recently showed that upon BTK inhibition in mice, phosphorylation of several proximal BCR signaling molecules, such as CD79a, Syk, and PI3K, as well as the key Btk-effector PLCγ2 and the more downstream kinase extracellular signal-regulated kinase (Erk) were significantly increased [153].
Taken together, these findings suggest that the suppressive activity of BTK inhibition in autoreactive B cells might partly be counteracted by inhibitor-induced activation of autoreactive T cells or proinflammatory myeloid cells. On the other hand, it is possible that the high specificity of second-generation BTK inhibitors, while greatly improving their safety profile, is accompanied by a loss of beneficial off-target effects that may well be essential in complex autoimmune diseases. To improve the effectiveness of BTK inhibitors, therapeutic strategies should be explored that combine BTK inhibition with other highly specific inhibitors, thereby retaining the favorable safety profile.
Selection or outgrowth of resistant clones is a major problem in clinical management of tumors. As most small-molecule inhibitors have fewer side effects than chemotherapy, treatment with multiple small-molecule inhibitors combined with B-cell-depleting agents is gaining broad interest [154]. Combinations explored in this field may also be of use in autoimmunity. Some strategies aim to target several signaling proteins in order to breach the microenvironmental niche involved in CLL disease. Treatment with the dual SYK/JAK inhibitor cerdulatinib showed induction of apoptosis in CLL cells and reduced capacity to produce CCL3 and CCL4 [155, 156], two chemokines that are induced upon interaction with nurse-like cells, which are supporting tumor-associated macrophages in CLL. It is also attractive to explore combination therapies of BTK inhibitors with histone deacetylase inhibitors (HDACis), as they have demonstrated potential in autoimmune animal models, and use of isoform selective HDACis may overcome adverse effects [157]. Combination therapy with ibrutinib and HDACis affected the survival of CLL cells in the EμTCL-1 CLL mouse model and induced synergistic cytotoxicity in primary cells from CLL patients [158]. Treatment with CUDC-907, which is a dual inhibitor of both PI3K and HDAC activity, resulted in increased apoptosis of MCL cells [159].
BTK inhibition may also enhance the effect of standard-of-care anti-rheumatic drugs, as shown with BMS-986142 in CIA [105]. However, this synergistic effect was not observed in a trial with combined treatment with the BTK inhibitor elsubrutinib and JAK-1 inhibitor upadacitinib in RA patients (NCT03682705). A similar study in SLE patients is currently ongoing (NCT03978520). Future studies investigating combination strategies with different BTK inhibitors and other specific therapeutics may lead to combination therapies that induce long-term remission of disease and facilitate personalized medicine.
References
Hofmann K, Clauder AK, Manz RA. Targeting B cells and plasma cells in autoimmune diseases. Front Immunol. 2018;9:835.
Corneth OBJ, Klein Wolterink RGJ, Hendriks RW. BTK Signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. 2016;393:67–105.
Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361(6409):226–33.
Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72(2):279–90.
Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol. 2010;6(6):348–57.
Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. 2012;119(16):3744–56.
Lu R, Vidal GS, Kelly JA, Delgado-Vega AM, Howard XK, Macwana SR, et al. Genetic associations of LYN with systemic lupus erythematosus. Genes Immun. 2009;10(5):397–403.
Zheng J, Ibrahim S, Petersen F, Yu X. Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes Immun. 2012;13(8):641–52.
Martin JE, Assassi S, Diaz-Gallo LM, Broen JC, Simeon CP, Castellvi I, et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum Mol Genet. 2013;22(19):4021–9.
Burbelo PD, Ambatipudi K, Alevizos I. Genome-wide association studies in Sjogren’s syndrome: What do the genes tell us about disease pathogenesis? Autoimmun Rev. 2014;13(7):756–61.
Jiang SH, Athanasopoulos V, Ellyard JI, Chuah A, Cappello J, Cook A, et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat Commun. 2019;10(1):2201.
Bolin K, Imgenberg-Kreuz J, Leonard D, Sandling JK, Alexsson A, Pucholt P, et al. Variants in BANK1 are associated with lupus nephritis of European ancestry. Genes Immun. 2021;22(3):194–202.
de Weers M, Verschuren MC, Kraakman ME, Mensink RG, Schuurman RK, van Dongen JJ, et al. The Bruton’s tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol. 1993;23(12):3109–14.
Smith CI, Baskin B, Humire-Greiff P, Zhou JN, Olsson PG, Maniar HS, et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol. 1994;152(2):557–65.
Genevier HC, Hinshelwood S, Gaspar HB, Rigley KP, Brown D, Saeland S, et al. Expression of Bruton’s tyrosine kinase protein within the B cell lineage. Eur J Immunol. 1994;24(12):3100–5.
Kawakami Y, Yao L, Miura T, Tsukada S, Witte ON, Kawakami T. Tyrosine phosphorylation and activation of Bruton tyrosine kinase upon Fc epsilon RI cross-linking. Mol Cell Biol. 1994;14(8):5108–13.
Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–40.
Schmidt U, van den Akker E, Parren-van Amelsvoort M, Litos G, de Bruijn M, Gutierrez L, et al. Btk is required for an efficient response to erythropoietin and for SCF-controlled protection against TRAIL in erythroid progenitors. J Exp Med. 2004;199(6):785–95.
Tomlinson MG, Kane LP, Su J, Kadlecek TA, Mollenauer MN, Weiss A. Expression and function of Tec, Itk, and Btk in lymphocytes: evidence for a unique role for Tec. Mol Cell Biol. 2004;24(6):2455–66.
Rip J, Van Der Ploeg EK, Hendriks RW, Corneth OBJ. The Role of Bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit Rev Immunol. 2018;38(1):17–62.
Xia S, Liu X, Cao X, Xu S. T-cell expression of Bruton’s tyrosine kinase promotes autoreactive T-cell activation and exacerbates aplastic anemia. Cell Mol Immunol. 2020;17(10):1042–52.
Bao Y, Zheng J, Han C, Jin J, Han H, Liu Y, et al. Tyrosine kinase Btk is required for NK cell activation. J Biol Chem. 2012;287(28):23769–78.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42.
Deeks ED. Ibrutinib: a review in chronic lymphocytic leukaemia. Drugs. 2017;77(2):225–36.
Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, et al. Placebo-controlled trial of an Oral BTK Inhibitor in Multiple Sclerosis. N Engl J Med. 2019;380(25):2406–17.
Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Martin EC, et al. Evobrutinib efficacy is maintained over two years in an open-label phase II study extension in patients with relapsing multiple sclerosis (4124). AAN Enterprises; 2021.
Torke S, Pretzsch R, Hausler D, Haselmayer P, Grenningloh R, Boschert U, et al. Inhibition of Bruton’s tyrosine kinase interferes with pathogenic B-cell development in inflammatory CNS demyelinating disease. Acta Neuropathol. 2020;140(4):535–48.
Bhargava P, Kim S, Reyes AA, Grenningloh R, Boschert U, Absinta M, et al. Imaging meningeal inflammation in CNS autoimmunity identifies a therapeutic role for BTK inhibition. Brain. 2021;144(5):1396–408.
Pellerin K, Rubino SJ, Burns JC, Smith BA, McCarl CA, Zhu J et al. MOG autoantibodies trigger a tightly-controlled FcR and BTK-driven microglia proliferative response. Brain. 2021;44(8):2361–74.
Martin E, Aigrot M-S, Grenningloh R, Stankoff B, Lubetzki C, Boschert U, et al. Bruton’s tyrosine kinase inhibition promotes myelin repair. Brain Plasticity. 2020;5(2):123–33.
Boschert U, Crandall T, Pereira A, Higginbotham G, Wu Y,Grenningloh R, Savinainen A, Bender A. Poster Session 1; P678. Multiple Scler J. 2017;23(3_suppl):85–426.
Alankus Y, Grenningloh R, Haselmayer P, Bender A, Bruttger J. BTK inhibition prevents inflammatory macrophage differentiation: a potential role in MS. Multiple Scler J. 2018 (Sage Publications Ltd 1 Olivers Yard, 55 City Road, London EC1Y 1SP, England; 2018. p. 264).
Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2(1):58–61.
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107(29):13075–80.
Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. 2001;167(8):4710–8.
Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med. 2011;208(1):125–34.
Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–7.
Kurosaki T, Takata M, Yamanashi Y, Inazu T, Taniguchi T, Yamamoto T, et al. Syk activation by the Src-family tyrosine kinase in the B cell receptor signaling. J Exp Med. 1994;179(5):1725–9.
Salim K, Bottomley MJ, Querfurth E, Zvelebil MJ, Gout I, Scaife R, et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J. 1996;15(22):6241–50.
Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science. 1996;271(5250):822–5.
de Weers M, Brouns GS, Hinshelwood S, Kinnon C, Schuurman RK, Hendriks RW, et al. B-cell antigen receptor stimulation activates the human Bruton’s tyrosine kinase, which is deficient in X-linked agammaglobulinemia. J Biol Chem. 1994;269(39):23857–60.
Cheng G, Ye ZS, Baltimore D. Binding of Bruton’s tyrosine kinase to Fyn, Lyn, or Hck through a SRC homology 3 domain-mediated interaction. Proc Natl Acad Sci USA. 1994;91(17):8152–5.
Aoki Y, Isselbacher KJ, Pillai S. Bruton tyrosine kinase is tyrosine phosphorylated and activated in pre-B lymphocytes and receptor-ligated B cells. Proc Natl Acad Sci USA. 1994;91(22):10606–9.
Saouaf SJ, Mahajan S, Rowley RB, Kut SA, Fargnoli J, Burkhardt AL, et al. Temporal differences in the activation of three classes of non-transmembrane protein tyrosine kinases following B-cell antigen receptor surface engagement. Proc Natl Acad Sci USA. 1994;91(20):9524–8.
Chiu CW, Dalton M, Ishiai M, Kurosaki T, Chan AC. BLNK: molecular scaffolding through ’cis’-mediated organization of signaling proteins. EMBO J. 2002;21(23):6461–72.
Kuhn J, Wong LE, Pirkuliyeva S, Schulz K, Schwiegk C, Funfgeld KG, et al. The adaptor protein CIN85 assembles intracellular signaling clusters for B cell activation. Sci Signal. 2016;9(434):ra66.
Middendorp S, Dingjan GM, Maas A, Dahlenborg K, Hendriks RW. Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J Immunol. 2003;171(11):5988–96.
Takata M, Kurosaki T. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med. 1996;184(1):31–40.
Bajpai UD, Zhang K, Teutsch M, Sen R, Wortis HH. Bruton’s tyrosine kinase links the B cell receptor to nuclear factor kappaB activation. J Exp Med. 2000;191(10):1735–44.
Petro JB, Rahman SM, Ballard DW, Khan WN. Bruton’s tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J Exp Med. 2000;191(10):1745–54.
Engels N, Konig LM, Schulze W, Radtke D, Vanshylla K, Lutz J, et al. The immunoglobulin tail tyrosine motif upgrades memory-type BCRs by incorporating a Grb2-Btk signalling module. Nat Commun. 2014;21(5):5456.
Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90(6):1073–83.
Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173(4):2245–52.
Shinners NP, Carlesso G, Castro I, Hoek KL, Corn RA, Woodland RT, et al. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol. 2007;179(6):3872–80.
Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, et al. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity. 2013;38(3):475–88.
Anderson JS, Teutsch M, Dong Z, Wortis HH. An essential role for Bruton’s [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc Natl Acad Sci USA. 1996;93(20):10966–71.
Castro I, Wright JA, Damdinsuren B, Hoek KL, Carlesso G, Shinners NP, et al. B cell receptor-mediated sustained c-Rel activation facilitates late transitional B cell survival through control of B cell activating factor receptor and NF-kappaB2. J Immunol. 2009;182(12):7729–37.
Craxton A, Draves KE, Gruppi A, Clark EA. BAFF regulates B cell survival by downregulating the BH3-only family member Bim via the ERK pathway. J Exp Med. 2005;202(10):1363–74.
Smith SH, Cancro MP. Cutting edge: B cell receptor signals regulate BLyS receptor levels in mature B cells and their immediate progenitors. J Immunol. 2003;170(12):5820–3.
Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ 3rd, et al. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat Immunol. 2008;9(12):1379–87.
Yu L, Mohamed AJ, Simonson OE, Vargas L, Blomberg KE, Bjorkstrand B, et al. Proteasome-dependent autoregulation of Bruton tyrosine kinase (Btk) promoter via NF-kappaB. Blood. 2008;111(9):4617–26.
Moisini I, Davidson A. BAFF: a local and systemic target in autoimmune diseases. Clin Exp Immunol. 2009;158(2):155–63.
Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20(6):785–98.
von Borstel A, Abdulahad WH, Sanders JS, Rip J, Neys SFH, Hendriks RW, et al. Evidence for enhanced Bruton’s tyrosine kinase activity in transitional and naive B cells of patients with granulomatosis with polyangiitis. Rheumatology (Oxford). 2019;58(12):2230–9.
Liu Z, Davidson A. BAFF and selection of autoreactive B cells. Trends Immunol. 2011;32(8):388–94.
Rahman ZSM, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling–deficient mice. J Exp Med. 2003;198(8):1157–69.
Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front Immunol. 2017;8:1454.
Doyle SL, Jefferies CA, Feighery C, O’Neill LA. Signaling by Toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. J Biol Chem. 2007;282(51):36953–60.
Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem. 2003;278(28):26258–64.
Abu-Rish EY, Amrani Y, Browning MJ. Toll-like receptor 9 activation induces expression of membrane-bound B-cell activating factor (BAFF) on human B cells and leads to increased proliferation in response to both soluble and membrane-bound BAFF. Rheumatology (Oxford). 2013;52(7):1190–201.
Kenny EF, Quinn SR, Doyle SL, Vink PM, van Eenennaam H, O’Neill LA. Bruton’s tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS ONE. 2013;8(8):e74103.
Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, et al. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther. 2011;13(4):R115.
Fiorcari S, Maffei R, Audrito V, Martinelli S, Ten Hacken E, Zucchini P, et al. Ibrutinib modifies the function of monocyte/macrophage population in chronic lymphocytic leukemia. Oncotarget. 2016;7(40):65968–81.
Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7(1):41–50.
Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132(5):794–806.
Ariza Y, Murata M, Ueda Y, Yoshizawa T. Bruton’s tyrosine kinase (Btk) inhibitor tirabrutinib suppresses osteoclastic bone resorption. Bone Rep. 2019;10:100201.
de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, et al. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26(1):93–104.
Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S, et al. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med. 2003;198(10):1539–50.
Volmering S, Block H, Boras M, Lowell CA, Zarbock A. The Neutrophil Btk signalosome regulates integrin activation during sterile inflammation. Immunity. 2016;44(1):73–87.
Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood. 2010;116(3):485–94.
Rajaiya J, Hatfield M, Nixon JC, Rawlings DJ, Webb CF. Bruton’s tyrosine kinase regulates immunoglobulin promoter activation in association with the transcription factor Bright. Mol Cell Biol. 2005;25(6):2073–84.
Shankar M, Nixon JC, Maier S, Workman J, Farris AD, Webb CF. Anti-nuclear antibody production and autoimmunity in transgenic mice that overexpress the transcription factor Bright. J Immunol. 2007;178(5):2996–3006.
Oldham AL, Miner CA, Wang HC, Webb CF. The transcription factor Bright plays a role in marginal zone B lymphocyte development and autoantibody production. Mol Immunol. 2011;49(1–2):367–79.
Ward JM, James JA, Zhao YD, Webb CF. Antibody reactivity of B cells in Lupus patients with increased disease activity and ARID3a expression. Antibodies (Basel). 2015;4:354–68.
Ward JM, Ratliff ML, Dozmorov MG, Wiley G, Guthridge JM, Gaffney PM, et al. Human effector B lymphocytes express ARID3a and secrete interferon alpha. J Autoimmun. 2016;75:130–40.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34.
Roskoski R Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res. 2016;103:26–48.
Liang C, Tian D, Ren X, Ding S, Jia M, Xin M, et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur J Med Chem. 2018;10(151):315–26.
Hill R, Smith P, Krishnarajah J, Bradshaw J, Masjedizadeh M, Bisconte A, et al. Discovery of PRN1008, a novel, reversible covalent BTK inhibitor in clinical development for rheumatoid arthritis: abstract number: 1671. Arthritis Rheumatol. 2015;67:2062–3.
Xu D, Kim Y, Postelnek J, Vu MD, Hu DQ, Liao C, et al. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J Pharmacol Exp Ther. 2012;341(1):90–103.
Crawford JJ, Johnson AR, Misner DL, Belmont LD, Castanedo G, Choy R, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton’s Tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61(6):2227–45.
Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2(7):358–64.
Bender AT, Gardberg A, Pereira A, Johnson T, Wu Y, Grenningloh R, et al. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-Cell receptor signaling. Mol Pharmacol. 2017;91(3):208–19.
Angst D, Gessier F, Janser P, Vulpetti A, Walchli R, Beerli C, et al. Discovery of LOU064 (Remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s Tyrosine Kinase. J Med Chem. 2020;63(10):5102–18.
Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–94.
Gui F, Jiang J, He Z, Li L, Li Y, Deng Z, et al. A non-covalent inhibitor XMU-MP-3 overrides ibrutinib-resistant Btk(C481S) mutation in B-cell malignancies. Br J Pharmacol. 2019;176(23):4491–509.
Ghoshdastidar K, Patel H, Bhayani H, Patel A, Thakkar K, Patel D, et al. ZYBT1, a potent, irreversible Bruton’s Tyrosine Kinase (BTK) inhibitor that inhibits the C481S BTK with profound efficacy against arthritis and cancer. Pharmacol Res Perspect. 2020;8(4):e00565.
Jansson L, Holmdahl R. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clin Exp Immunol. 1993;94(3):459–65.
Steinberg BJ, Smathers PA, Frederiksen K, Steinberg AD. Ability of the xid gene to prevent autoimmunity in (NZB X NZW)F1 mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. J Clin Invest. 1982;70(3):587–97.
Corneth OB, de Bruijn MJ, Rip J, Asmawidjaja PS, Kil LP, Hendriks RW. Enhanced expression of Bruton’s Tyrosine kinase in B cells drives systemic autoimmunity by disrupting T Cell homeostasis. J Immunol. 2016;197(1):58–67.
Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-like receptor signaling drives Btk-mediated autoimmune disease. Front Immunol. 2019;10:95.
Park JK, Byun JY, Park JA, Kim YY, Lee YJ, Oh JI, et al. HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: a potential drug for rheumatoid arthritis. Arthritis Res Ther. 2016;18(18):91.
Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–28.
Gillooly KM, Pulicicchio C, Pattoli MA, Cheng L, Skala S, Heimrich EM, et al. Bruton’s tyrosine kinase inhibitor BMS-986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard-of-care. PLoS ONE. 2017;12(7):e0181782.
Bender AT, Pereira A, Fu K, Samy E, Wu Y, Liu-Bujalski L, et al. Btk inhibition treats TLR7/IFN driven murine lupus. Clin Immunol. 2016;164:65–77.
Krupa A, Fol M, Rahman M, Stokes KY, Florence JM, Leskov IL, et al. Silencing Bruton’s tyrosine kinase in alveolar neutrophils protects mice from LPS/immune complex-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307(6):L435–48.
Florence JM, Krupa A, Booshehri LM, Davis SA, Matthay MA, Kurdowska AK. Inhibiting Bruton’s tyrosine kinase rescues mice from lethal influenza-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2018;315(1):L52–8.
Mao L, Kitani A, Hiejima E, Montgomery-Recht K, Zhou W, Fuss I, et al. Bruton tyrosine kinase deficiency augments NLRP3 inflammasome activation and causes IL-1beta-mediated colitis. J Clin Invest. 2020;130(4):1793–807.
Corneth OBJ, Verstappen GMP, Paulissen SMJ, de Bruijn MJW, Rip J, Lukkes M, et al. Enhanced Bruton’s tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol. 2017;69(6):1313–24.
Wang SP, Iwata S, Nakayamada S, Niiro H, Jabbarzadeh-Tabrizi S, Kondo M, et al. Amplification of IL-21 signalling pathway through Bruton’s tyrosine kinase in human B cell activation. Rheumatology (Oxford). 2015;54(8):1488–97.
Hartkamp LM, Fine JS, van Es IE, Tang MW, Smith M, Woods J, et al. Btk inhibition suppresses agonist-induced human macrophage activation and inflammatory gene expression in RA synovial tissue explants. Ann Rheum Dis. 2015;74(8):1603–11.
Hsu J, Gu Y, Tan SL, Narula S, DeMartino JA, Liao C. Bruton’s Tyrosine Kinase mediates platelet receptor-induced generation of microparticles: a potential mechanism for amplification of inflammatory responses in rheumatoid arthritis synovial joints. Immunol Lett. 2013;150(1–2):97–104.
Einhaus J, Pecher AC, Asteriti E, Schmid H, Secker KA, Duerr-Stoerzer S, et al. Inhibition of effector B cells by ibrutinib in systemic sclerosis. Arthritis Res Ther. 2020;22(1):66.
Soresina A, Moratto D, Chiarini M, Paolillo C, Baresi G, Foca E, et al. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr Allergy Immunol. 2020;31(5):565–9.
Quinti I, Lougaris V, Milito C, Cinetto F, Pecoraro A, Mezzaroma I, et al. A possible role for B cells in COVID-19? Lesson from patients with agammaglobulinemia. J Allergy Clin Immunol. 2020;146(1):211-3 e4.
Milosevic I, Jovanovic J, Stevanovic O. Atypical course of COVID-19 in patient with Bruton agammaglobulinemia. J Infect Dev Ctries. 2020;14(11):1248–51.
Treon SP, Castillo JJ, Skarbnik AP, Soumerai JD, Ghobrial IM, Guerrera ML, et al. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19-infected patients. Blood. 2020;135(21):1912–5.
Thibaud S, Tremblay D, Bhalla S, Zimmerman B, Sigel K, Gabrilove J. Protective role of Bruton tyrosine kinase inhibitors in patients with chronic lymphocytic leukaemia and COVID-19. Br J Haematol. 2020;190(2):e73–6.
Roschewski M, Lionakis MS, Sharman JP, Roswarski J, Goy A, Monticelli MA, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol. 2020;5(48).
Hoepel W, Chen H-J, Geyer CE, Allahverdiyeva S, Manz XD, de Taeye SW et al. High titers and low fucosylation of early human anti-SARS-CoV-2 IgG promote inflammation by alveolar macrophages. Sci Transl Med. 2021;13(596):eabf8654.
Apostolidis SA, Sarkar A, Giannini HM, Goel RR, Mathew D, Suzuki A, et al. Signaling through FcgammaRIIA and the C5a-C5aR pathway mediates platelet hyperactivation in COVID-19. bioRxiv. 2021.
Seeling M, Bruckner C, Nimmerjahn F. Differential antibody glycosylation in autoimmunity: sweet biomarker or modulator of disease activity? Nat Rev Rheumatol. 2017;13(10):621–30.
Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–32.
Goess C, Harris CM, Murdock S, McCarthy RW, Sampson E, Twomey R, et al. ABBV-105, a selective and irreversible inhibitor of Bruton’s tyrosine kinase, is efficacious in multiple preclinical models of inflammation. Mod Rheumatol. 2019;29(3):510–22.
Haselmayer P, Camps M, Liu-Bujalski L, Nguyen N, Morandi F, Head J, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202(10):2888–906.
Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727–35.
Cohen S, Tuckwell K, Katsumoto TR, Zhao R, Galanter J, Lee C et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: a randomized, double-blind, Phase II Trial (ANDES Study). Arthritis Rheumatol. 2020;72(9):1435–41.
Prevoo ML, van 't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38(1):44–8.
Kim YY, Park KT, Jang SY, Lee KH, Byun JY, Suh KH, et al. HM71224, a selective Bruton’s tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res Ther. 2017;19(1):211.
Genovese M, Spindler A, Sagawa A, Park W, Dudek A, Kivitz A et al. Safety and efficacy of Poseltinib, Bruton's Tyrosine kinase inhibitor, in patients with rheumatoid arthritis: a randomized, double-blind, placebo-controlled, 2-part Phase II Study. J Rheumatol. 2021;48(7):969–76.
Langrish CL, Bradshaw JM, Francesco MR, Owens TD, Xing Y, Shu J, et al. Preclinical Efficacy and anti-inflammatory mechanisms of action of the Bruton tyrosine kinase inhibitor Rilzabrutinib for immune-mediated disease. J Immunol. 2021;206(7):1454–68.
Murrell DF, Patsatsi A, Stavropoulos P, Baum S, Zeeli T, Kern JS, et al. Proof-of-concept for clinical effects of oral rilzabrutinib, the first Bruton tyrosine kinase inhibitor for pemphigus vulgaris: the phase 2 BELIEVE study. Br J Dermatol. 2021.
Schafer PH, Kivitz AJ, Ma J, Korish S, Sutherland D, Li L, et al. Spebrutinib (CC-292) affects markers of B cell activation, chemotaxis, and osteoclasts in patients with rheumatoid arthritis: results from a mechanistic study. Rheumatol Ther. 2020;7(1):101–19.
Ariza Y, Yoshizawa T, Ueda Y, Hotta S, Narita M, Kawabata K. ONO-4059-A Novel Small Molecule Bruton's Tyrosine Kinase (Btk) inhibitor, suppresses osteoclast differentiation and activation. Abstract nr. 1799. Arthritis Rheum. 2012;64.
Ariza Y, Yoshizawa T, Ueda Y, Hotta S, Yasuhiro T, Narita M, et al. ONO-4059-A Novel Small Molecule Dual Inhibitor Of Bruton's Tyrosine Kinase (Btk) and Tec kinase-suppresses osteoclastic bone resorption and inflammation: Abstract nr. 1824. Arthritis Rheum. 2013;65.
Ariza Y, Yoshizawa T, Ueda Y, Murata M, Kawabata K. ONO-4059-a Highly Potent and Dual Oral Inhibitor of Bruton's Tyrosine Kinase (Btk) and Tec Kinase: improves anti-nuclear antibodies-mediated SLE in Mice.: Abstract nr. 646. Arthritis Rheumatol. 2014;66.
Yoshizawa T, Ariza Y, Ueda Y, Hotta S, Narita M, Kawabata K. Development of a Bruton's Tyrosine Kinase (Btk) Inhibitor, ONO-4059: Efficacy in a collagen induced arthritis (CIA) model indicates potential treatment for rheumatoid arthritis (RA). Arthritis Rheum; 2012 (Wiley-BLACKWELL 111 RIVER ST, HOBOKEN 07030-5774, NJ USA; 2012. p. S709-S).
Watterson SH, Liu Q, Beaudoin Bertrand M, Batt DG, Li L, Pattoli MA, et al. Discovery of Branebrutinib (BMS-986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s Tyrosine Kinase (BTK). J Med Chem. 2019;62(7):3228–50.
Catlett IM, Nowak M, Kundu S, Zheng N, Liu A, He B, et al. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS-986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: randomised phase I, placebo-controlled trial in healthy participants. Br J Clin Pharmacol. 2020;86(9):1849–59.
Yoshiga Y, Hosoi F, Iguchi S, Kaneko R, Nakachi Y, Akasaka D, et al. TAS5315, a Novel Bruton's Tyrosine kinase inhibitor, demonstrates anti-inflammatory effect in autoimmune disease models. Arthritis Rheumatol. 2016 (Wiley 111 River St, Hoboken 07030-5774, NJ USA).
Kumagai Y, Tanaka Y, Murata M, Takeuchi T. A phase 1, single and multiple ascending dose study of TAS5315—a novel highly selective inhibitor of Bruton’s tyrosine kinase—in healthy male volunteers. Ann Rheum Dis. 78(Suppl 2):750.1–750. Poster FRI0156.
Hosoi F, Yoshiga Y, Iguchi S, Kaneko R, Nakachi Y, Akasaka D, et al. TAS5315, a Novel Bruton’s Tyrosine Kinase (BTK) inhibitor, demonstrates potent efficacy in an animal model of rheumatoid arthritis: abstract number: 2561. Arthritis Rheumatol. 2015;67:3095–6.
Yoshiga Y, Hosoi F, Iguchi S, Kaneko R, Nakachi Y, Akasaka D, et al. TAS5315, a Novel Bruton's tyrosine kinase inhibitor, ameliorates inflammation and bone erosion in murine model for rheumatoid arthritis. Arthritis Rheumatol. 2017 (Wiley 111 River St, Hoboken 07030-5774, NJ USA).
Boschert U, Crandall T, Pereira A, Higginbotham G, Wu Y, Grenningloh R, et al. T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by Evobrutinib (M2951) a novel Bruton’s tyrosine kinase inhibitor. Multiple Scler J. 2017;2017:327 (Sage Publications Ltd 1 Olivers Yard, 55 City Road, London EC1Y 1SP, England).
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–32.
Das S, Bar-Sagi D. BTK signaling drives CD1d(hi)CD5(+) regulatory B-cell differentiation to promote pancreatic carcinogenesis. Oncogene. 2019;38(17):3316–24.
Schmidt NW, Thieu VT, Mann BA, Ahyi AN, Kaplan MH. Bruton’s tyrosine kinase is required for TLR-induced IL-10 production. J Immunol. 2006;177(10):7203–10.
Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, et al. Regulation of dendritic cell maturation and function by Bruton’s tyrosine kinase via IL-10 and Stat3. Proc Natl Acad Sci USA. 2006;103(1):153–8.
Natarajan G, Oghumu S, Terrazas C, Varikuti S, Byrd JC, Satoskar AR. A Tec kinase BTK inhibitor ibrutinib promotes maturation and activation of dendritic cells. Oncoimmunology. 2016;5(6):1151592.
Lougaris V, Baronio M, Vitali M, Tampella G, Cattalini M, Tassone L, et al. Bruton tyrosine kinase mediates TLR9-dependent human dendritic cell activation. J Allergy Clin Immunol. 2014;133(6):1644-50 e4.
Fillatreau S, Manfroi B, Dorner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17(2):98–108.
Rip J, de Bruijn MJW, Neys SFH, Pal Singh S, Willar J, van Hulst JAC et al. Bruton’s tyrosine kinase inhibition induces rewiring of proximal and distal B-cell receptor signaling in mice. Eur J Immunol. 2021;51(9):2251–65.
Sharman JP, Egyed M, Jurczak W, Skarbnik A, Pagel JM, Flinn IW, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278–91.
Blunt MD, Koehrer S, Dobson RC, Larrayoz M, Wilmore S, Hayman A, et al. The Dual Syk/JAK inhibitor cerdulatinib antagonizes B-cell receptor and microenvironmental signaling in chronic lymphocytic leukemia. Clin Cancer Res. 2017;23(9):2313–24.
Guo A, Lu P, Coffey G, Conley P, Pandey A, Wang YL. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: Cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget. 2017;8(8):12953–67.
Hamminger P, Rica R, Ellmeier W. Histone deacetylases as targets in autoimmune and autoinflammatory diseases. Adv Immunol. 2020;147:1–59.
Bottoni A, Rizzotto L, Lai TH, Liu C, Smith LL, Mantel R, et al. Targeting BTK through microRNA in chronic lymphocytic leukemia. Blood. 2016;128(26):3101–12.
Guo H, Zeng D, Zhang H, Bell T, Yao J, Liu Y, et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene. 2019;38(11):1802–14.
Barf T, Covey T, Izumi R, van de Kar B, Gulrajani M, van Lith B, et al. Acalabrutinib (ACP-196): a covalent bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363(2):240–52.
Zhang B, Zhao R, Liang R, Gao Y, Liu R, Chen X, et al. Abstract CT132: Orelabrutinib, a potent and selective Bruton’s tyrosine kinase inhibitor with superior safety profile and excellent PK/PD properties. Can Res. 2020;80(16 Supplement):CT132-CT.
Liclican A, Serafini L, Xing W, Czerwieniec G, Steiner B, Wang T, et al. Biochemical characterization of tirabrutinib and other irreversible inhibitors of Bruton’s tyrosine kinase reveals differences in on - and off - target inhibition. Biochim Biophys Acta Gen Subj. 2020;1864(4):129531.
Caldwell RD, Qiu H, Askew BC, Bender AT, Brugger N, Camps M, et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 2019;62(17):7643–55.
Smith PF, Krishnarajah J, Nunn PA, Hill RJ, Karr D, Tam D, et al. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol. 2017;83(11):2367–76.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This project was supported by grants from the Target2B! consortium, The Dutch Arthritis Foundation (grant no. 19-1-201) and Erasmus MC MRace.
Conflict of Interest
The authors declare no conflicts of interest.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author Contributions
SFHN, JR, RWH, and OBJC wrote the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Neys, S.F.H., Rip, J., Hendriks, R.W. et al. Bruton’s Tyrosine Kinase Inhibition as an Emerging Therapy in Systemic Autoimmune Disease. Drugs 81, 1605–1626 (2021). https://doi.org/10.1007/s40265-021-01592-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01592-0