
Abstract
Non-small-cell lung cancer (NSCLC) accounts for about 85% of all lung cancer cases and is the leading cause of cancer-related deaths. Most NSCLC patients are diagnosed with advanced disease and require systemic treatment. Despite emerging advances in chemotherapy and immunotherapy, the prognosis of stage IV patients remains poor. However, the discovery of oncogenic driver mutations including mutations in the epidermal growth factor receptor (EGFR), the anaplastic lymphoma kinase (ALK) and others, characterize a subset of patients with the opportunity of targeted therapies. Fusions between the ALK and echinoderm microtubule-associated protein-like 4 (EML4) are present in ∼ 3–5% of patients with NSCLC. Several first-, second-, and third-generation ALK tyrosine kinase inhibitors (TKIs) have been developed in the last decade and have tremendously changed treatment options and outcomes of ALK-positive NSCLC patients. With increasing treatment options, treatment sequence decisions have become more and more complex. ALK-mutations, fusion variants, or activation of by-pass pathways result in treatment resistance during the course of treatment in nearly all patients. Mutation-guided treatment sequencing can lead to better outcomes, and re-biopsy or liquid-biopsy should be performed whenever possible in case of disease progression in ALK-rearranged patients. In the future, combinational treatment of ALK TKIs with other pathway-inhibitors might further improve patients’ treatment options and outcomes. Here, we review the data for currently available ALK TKIs, discuss approaches of treatment sequencing, and give an outlook on emerging developments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
About 3–5% of all lung cancers are driven by a distinct gene-rearrangement involving the anaplastic lymphoma kinase (ALK). |
ALK tyrosine kinase inhibitors can be used to effectively treat these patients. |
Distinct resistance mechanisms lead to treatment failure of these drugs. |
Adapting the treatment sequence might improve patients’ outcome. |
1 Introduction
In 1994, Morris and colleagues identified a novel fusion partner in the t(2;5) chromosomal translocation in anaplastic large cell lymphoma (ALCL), and consequently named it anaplastic lymphoma kinase (ALK) [1]. Three years later the molecular structure of ALK was described as a full-length tyrosine kinase with an extracellular binding domain, a transmembrane domain, and an intracellular domain with high similarity to the insulin receptor [2, 3]. Mainly expressed in neuronal tissue, ALK activates several downstream pathways as PI3K-AKT, CRKL-C3G, MEKK2/3-MEK5-ERK5, JAK-STAT, and MAPK [4].
The oncogenic potential of ALK results mainly from ALK-fusion proteins. However, point mutations and overexpression of ALK also have an oncogenic role in several cancer types [5]. While more than 30 fusion partners of ALK have been reported in different cancer types, NPM (nucleophosmin-anaplastic lymphoma kinase)-ALK in ALCL and EML4 (echinoderm microtubule-associated protein-like 4)-ALK in non-small-cell lung cancer (NSCLC) are the most extensively studied ones [5].
Soda and colleagues first identified the EML4-ALK fusion protein as an oncogenic driver in NSCLC [6]. About 3–5% of non-squamous NSCLCs harbour an ALK translocation, enriched in younger, female, and light- or never-smoker patients [7].
There are different ways to detect ALK aberrations. Immunohistochemistry (IHC) for ALK expression is a quick and easy examination method. Positive results in ALK IHC are highly predictive for the existence of ALK translocations, so IHC can serve as a potent screening tool. Fluorescence in situ hybridization (FISH) or next-generation sequencing (NGS) with RNA-sequencing technologies are used to confirm the presence of an ALK translocation [8].
Since the development of effective treatment options, all international guidelines recommend testing for ALK translocations in all non-squamous NSCLC [9, 10].
2 Current Anaplastic Lymphoma Kinase (ALK) Tyrosine Kinase Inhibitors
Several ALK tyrosine-kinase inhibitors (TKIs) are already approved and others are in clinical development. Apart from ALK, varying targets are inhibited by these drugs.
2.1 Crizotinib
Crizotinib is a multikinase inhibitor initially developed to target c-MET [11]. Besides its activity against c-MET, additional inhibition of ALK and ROS-1 was found [11,12,13]. As ALK has been recognized as an important target in NSCLC since 2007, the clinical investigation of crizotinib focused initially on ALK-positive NSCLC patients. The first study evaluating a relevant number of ALK-positive NSCLC patients was the phase I/II PROFILE1001 study. Eighty-two patients were enrolled into an expanded cohort and were treated with 250 mg crizotinib twice a day. Most patients were heavily pre-treated. The overall response rate (ORR) was 57%, with 46 patients achieving a partial response (PR) and one patient a complete response (CR), while 27 patients had stable disease. Progression-free survival (PFS) after 6 months was 72% (95% confidence interval (CI), 61–83) [14]. Additional enrolment of 143 patients into the PROFILE1001 study led to an update of the initial data. Eighty-seven (61%) patients had an objective response. Median PFS was 9.7 months, and estimated overall survival (OS) was 88% at 6 months and 75% at 12 months [15].
For further evaluation of the activity of crizotinib in ALK-positive NSCLC, 901 patients were enrolled into the phase II PROFILE1005 study. Only 255 patients were evaluable for response and showed an ORR of 53% and a median PFS of 8.5 months, confirming the results of PROFILE1001 [16]. Due to the results of PROFILE1001 and PROFILE1005, the US Food and Drug Administration (FDA) granted accelerated approval for crizotinib in 2011.
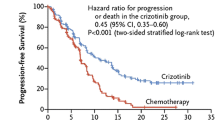
The results of these studies led to two phase III trials evaluating crizotinib in pre-treated (PROFILE1007) and previously untreated (PROFILE1014) patients with advanced ALK positive NSCLC. PROFILE1007 was an open-label, phase III study that compared crizotinib with chemotherapy (docetaxel or pemetrexed) in 347 patients with ALK-positive NSCLC who progressed after one line of platinum-based chemotherapy [17]. The ORR and the median PFS were significantly prolonged in the crizotinib group (65% vs. 20% and 7.7 vs. 3.0 months). Median OS showed no significant difference between the crizotinib and the chemotherapy group, probably due to the high rate of subsequent crizotinib treatment in the chemotherapy cohort [17].
PROFILE1014 was an open-label, phase III study comparing crizotinib versus chemotherapy (cisplatin/carboplatin and pemetrexed) in patients with advanced ALK-positive NSCLC without any previous systemic therapy [18]. Median PFS was significantly longer with crizotinib (10.9 months) than with chemotherapy (7.0 months). Also, ORR was improved in the crizotinib group compared to the chemotherapy group (74% vs. 45%; p < 0.001). As in PROFILE1007, there was no difference in OS, attributed to a high rate of crossover.
Crizotinib is generally well tolerated. Visual disorders, diarrhoea, oedema, vomiting, constipation, elevated liver transaminases, upper respiratory tract infection, dysgeusia and dizziness are frequent, but well manageable. The rate of de novo interstitial lung disease is rare (1.2%), but associated with high mortality rates (50%) [19].
These data underline the potent activity of crizotinib in ALK-positive NSCLC patients, and crizotinib was established as a first-line TKI treatment for these patients. Real-world data have been published in recent years, confirming the response rates and survival data from the pivotal studies [20, 21]. However, due to the poor intracranial activity and shorter PFS, crizotinib is now widely displaced in first-line by next-generation TKIs, and especially in patients with CNS involvement front-line use of alectinib, brigatinib or ceritinib is recommended [9].
2.2 Ceritinib
Ceritinib is a second-generation ALK-selective TKI [22]. Ceritinib was demonstrated to cross the blood-brain-barrier, closing an important gap after approval compared to crizotinib [22]. Results from the ASCEND study program provided the data on which basis ceritinib was approved in 2014 by the FDA and in 2015 by the European Medicines Agency (EMA).
The ASCEND-1 study enrolled 255 ALK-positive patients after at least one line of chemotherapy with or without previous TKI treatment. ORR was 72% in patients without prior TKI exposure and 56% in patients pre-treated with an ALK inhibitor. The median PFS was 18.4 months in ALK inhibitor-naive patients and 6.9 months in ALK inhibitor pre-treated patients [23, 24].
The single-arm, open-label, multicentre, phase II study ASCEND-2 was conducted to extend the results of ASCEND-1 in crizotinib pre-treated patients who had received at least one line of platinum-based chemotherapy. Many patients were heavily pre-treated (56% of patients had at least three lines of treatment). Results were consistent with the findings from ASCEND-1 with an ORR of 38.6%. The median PFS was 5.7 months, and the median OS was 14.9 months [25].
Another open-label, single-arm, phase II trial (ASCEND-3) was conducted in patients who had previously received up to three lines of chemotherapy but were TKI naive. The ORR was 67.7%, the median PFS was 16.6 months, and median OS was 51.3 months [26].
The ASCEND-4 trial was a phase III open-label study. Patients could be enrolled with ALK-rearranged NSCLC and no previous chemotherapy. Overall, 376 patients were randomized to receive either oral ceritinib 750 mg/day or platinum-based chemotherapy. Median PFS was significantly prolonged in patients receiving ceritinib compared to chemotherapy (16.6 months vs. 8.1 months). ORR was also significantly improved (72.5% vs 26.7%) [27].
The phase III ASCEND-5 study was designed to compare ceritinib treatment with chemotherapy in the third-line setting. 232 patients were randomized to receive either ceritinib or chemotherapy. Median PFS was significantly longer in the ceritinib group (5.4 months vs. 1.6 months), with an ORR of 39% versus 7% [28].
Despite good response rates and intracranial activity, ceritinib treatment is characterized by intense gastrointestinal (GI) side effects. Nausea, vomiting, diarrhoea and liver enzyme elevation are very common with the recommended dosage of 750 mg per day. Most patients need dose reduction and supportive treatment to tolerate ceritinib. Other investigations were able to show that a lower dose of ceritinib (450 mg) taken with a low-fat meal showed similar efficacy with less severe GI adverse events (AEs) versus the 750 mg dose taken without food [29]. According to these results, the lower dose with food is now approved.
There is an adjusted indirect comparison with external controls between crizotinib and ceritinib in ALK-TKI-naïve patients, which demonstrates a statistically significant and clinically relevant advantage of ceritinib regarding PFS (HR 0.52) and OS (HR 0.59) [30].
Ceritinib as a first type of second-generation ALK inhibitor provided efficacy for patients with brain metastases. It is approved for first-line treatment and after failure of treatment with crizotinib. However, after approval of other TKIs with potent intracranial activity, ceritinib was widely replaced in first- and second-line treatment, mainly due its unfavourable safety profile.
2.3 Alectinib
Alectinib is a second-generation ALK TKI that also has inhibitory activity against RET fusion proteins [31]. Alectinib demonstrated high efficacy with a favourable safety profile in several clinical trials.
Two single-arm, open-label phase II trials evaluated the efficacy and safety of alectinib in ALK-positive NSCLC patients who had progressed on crizotinib. Most patients in both studies had received chemotherapy before crizotinib (80% and 74%). In both studies, alectinib demonstrated meaningful clinical efficacy with an ORR of 51% and 52%, respectively. PFS was 8.9 months and 8.0 months, and OS was estimated with 26.0 months and 22.7 months, respectively [32, 33].
The phase III J-ALEX study enrolled only in Japan and showed the efficacy of alectinib as a first-line therapy for ALK-positive NSCLC. Median (PFS) in patients receiving alectinib was significantly higher compared to patients treated with crizotinib (not reached vs. 10.2 months) [34]. Nevertheless, alectinib dosage in this trial was 300 mg twice a day, which is not the recommended dose in the Caucasian population.
The efficacy of alectinib compared to that of crizotinib in previously untreated patients with advanced ALK-positive NSCLC was evaluated in the global randomized, open-label phase III ALEX trial. 303 patients were randomly assigned to either alectinib 600 mg twice daily or crizotinib 250 mg twice daily. The median PFS was not reached at the time of analysis. The 12-month PFS was 68.4% with alectinib versus 48.7% with crizotinib. ORR was 82.9% versus 75.5% [35]. An update was published by Camidge et al. in 2019 and by Mok et.al. in 2020, showing that the median PFS was estimated at 34.8 months versus 10.9 months. ORR remained unchanged. The 5-year OS rate was 62.5% with alectinib and 45.5% with crizotinib. Median OS data are still premature [36, 37].
The ALESIA trial was a randomized phase III study comparing alectinib versus crizotinib in untreated Asian patients (China, South Korea and Thailand). 187 ALK-positive patients were randomized (2:1) to receive either alectinib (600 mg twice daily) or crizotinib (250 mg twice daily). The independent review committee assessed the median PFS, which was significantly longer in the alectinib group compared to the crizotinib group (not reached vs. 10.7 months). ORR was 91% with alectinib, and 77% with crizotinib, with a longer duration of response for alectinib. Time to CNS progression and response to baseline brain metastases was significantly improved in the alectinib group. Furthermore, fewer patients had grade 3–5 adverse toxicities (29% vs. 48%) or serious adverse events (15% vs. 26%) [38].
The randomized, open-label, phase III ALUR study compared the efficacy of alectinib versus standard relapse chemotherapy in patients with stage IV ALK-positive NSCLC previously treated with two lines of systemic treatment: platinum-based doublet chemotherapy and crizotinib. 107 patients were randomized 2:1 to alectinib 600 mg twice daily or chemotherapy (pemetrexed 500 mg/m2 for 3 weeks or docetaxel 75 mg/m2 every 3 weeks). ORR was 37.5% versus 2.9% and median PFS was 9.6 months versus 1.4 months, demonstrating the clinical benefit of alectinib in later lines [39].
Most adverse events (AEs) with alectinib were of mild to moderate severity. AEs of grade ≥ 3 severity occur in < 4% of patients. The most common AEs of any grade were GI disorders (e.g. constipation 35%, nausea 19%, diarrhoea 16%, vomiting 11%); oedema (30%); increased levels of bilirubin (18%), aspartate aminotransferase (AST) (15%) and alanine aminotransferase (ALT) (14%); myalgia (28%), rash (18%), anaemia (17%) and increase in bodyweight (12%) [31]. The rate of alectinib-induced interstitial lung disease ranges between 1% and 8% [34, 35].
Alectinib was the first second-generation ALK TKI with a favourable safety profile and relevant intracranial efficacy to be approved for first-line treatment in ALK-positive patients. Thus, alectinib widely replaced ceritinib and also crizotinib in the first-line treatment. The first real-world data confirmed previous study results in second- as well as in first-line treatment [40, 41].
2.4 Brigatinib
Brigatinib is a multikinase inhibitor with activity against ALK, proto-oncogene tyrosine-protein kinase ROS-1 (ROS1), insulin-like growth factor-1 receptor (IGF-R)-1, and fms-like tyrosine kinase 3 (FLT-3), as well as epithelial growth factor receptor (EGFR) deletion and point mutations [42].
In a phase I/II study 137 patients were enrolled (79 with ALK-positive NSCLC of whom 71 had previously been treated with crizotinib). The phase I part of the study was designed to titrate the right dosage of brigatinib. The phase II part of the trial aimed at investigating the safety and efficacy of the drug. ORR was 63% with a median PFS of 13.2 months. Median PFS in crizotinib-naïve patients was 34.2 months [43].
The second-line ALTA trial was a randomized two-arm, open-label, multicentre clinical trial to investigate the efficacy and safety of brigatinib (Arm A: 90 mg/day; Arm B: 180 mg/day following a 7-day lead-in with 90 mg) in patients with crizotinib-refractory advanced ALK positive NSCLC. The 7-day lead-in reduced the pulmonary side effects markedly. 154 patients (69%) had brain metastases at baseline, and 164 patients (74%) had received prior chemotherapy. ORR at 8-month median follow-up was 45% in arm A and 54% in arm B. Median PFS was 9.2 months in arm A and 12.9 months in arm B. The 1-year OS probability was 71% in arm A and 80% in arms B, respectively [44]. These results were confirmed at the 2-year follow-up, including a high intracranial response rate [45]. As arm B was consistently more effective than arm A, 180 mg once daily with a 7-day lead-in of 90 mg was designated to be the recommended dose.
The open-label, phase III trial ALTA 1L randomly assigned 275 untreated ALK-positive NSCLC patients to receive either brigatinib or crizotinib. Twelve months’ PFS was significantly higher with brigatinib than with crizotinib (67% vs. 43%). The confirmed ORR was 71% with brigatinib and 60% with crizotinib [46]. Estimated median PFS for patients treated with brigatinib was 24 months (95% CI: 18.5, NE) compared with 11 months (95% CI 9.2, 12.9) for those treated with crizotinib (HR 0.49; 95% CI 0.35, 0.68; p < 0.0001). Confirmed ORR was 74% (95% CI 66, 81) and 62% (95% CI 53, 70), respectively [47].
Based on these data brigatinib was approved in 2017 by the FDA as second-line treatment after crizotinib failure, followed by approval for untreated ALK-positive NSCLC patients in 2019 [48]. Approvals were also granted by the EMA.
Brigatinib showed an acceptable safety profile with a risk of early pulmonary events in about 3% of patients receiving brigatinib as first-line therapy, according to results of the phase III ALTA-1L trial [46]. The toxicity can be reduced by a lead-in phase with 90 mg daily. Additionally, myositis may occur.
In summary, brigatinib showed good efficacy, including intracranial activity, and a favourable safety profile in first and later lines. Pulmonary toxicity is rare but potentially a severe event that can occur early after treatment initiation. The first real-world data have been published in the last year, confirming the promising results from the pivotal studies [49,50,51].
2.5 Lorlatinib
Lorlatinib is a third-generation small-molecule TKI with selective activity against ALK and ROS1 [52]. In Europe and the USA, lorlatinib is approved for ALK-positive NSCLC after failure of either alectinib or ceritinib, or crizotinib and another ALK-TKI.
In an international multicentre, open-label, single-arm, first-in-man phase I dose-escalation study, 41 patients with ALK-positive NSCLC were treated with lorlatinib. Most patients had received at least two TKIs and suffered from brain metastases. The ORR was 46%, and in patients who had received two or more TKIs the ORR was 42%. Median PFS in all ALK patients was 9.6 months [53].
Based on the promising phase I data, a phase II study was designed to include ALK-positive NSCLC patients in six different expansion cohorts depending on their previous treatments. All together 276 patients were enrolled: 30 of them were ALK positive and treatment naive (Exp. 1); 59 were ALK positive and received previous crizotinib without (n = 27; Exp. 2) or with (n = 32; Exp. 3A) previous chemotherapy; 28 were ALK positive and received one previous non-crizotinib ALK TKI, with or without chemotherapy (Exp. 3B); 112 were ALK positive with two (n = 66; Exp. 4) or three (n = 46; Exp. 5) previous ALK TKIs with or without chemotherapy [54]. Patients without prior therapy showed an ORR of 90%, and pre-treated patients (pooled Exps. 2–5) had an ORR of 47%, demonstrating the potent role of lorlatinib in later treatment lines.
The first results of the CROWN study have been presented at the European Society for Medical Oncology (ESMO) 2020 congress. Lorlatinib was compared to crizotinib in the first-line setting. Lorlatinib demonstrated a significantly longer PFS (HR 0.28) than crizotinib, along with intracranial efficacy and no new safety signals [55].
The most common AEs with lorlatinib treatment were hypercholesterolemia (82.4%), hypertriglyceridemia (60.7%), oedema (51.2%), peripheral neuropathy (43.7%), bodyweight gain (20.7%), cognitive effects (23.1%), mood effects (21%), fatigue (23.1%), diarrhoea (17.6%), and arthralgia (19.7%); rare but potentially fatal side effects were pneumonitis (3.4%) and respiratory failure (1.4%) [54, 56].
Lorlatinib has the potential to overcome TKI resistance in many, in part heavily pre-treated, patients. Furthermore, lorlatinib shows potent activity in patients with brain metastases. Thus, lorlatinib may be established as a good option in patients pre-treated with one or two ALK TKIs. Currently, the first real-world data confirm the meaningful results from the pivotal studies [57, 58].
Results of the discussed studies are summarized in Table 1.
3 New Options and Developments on the Horizon
The approval of potent ALK TKIs has tremendously changed the treatment options and outcomes of ALK-positive NSCLC patients. Nevertheless, mechanisms of resistance and the optimal sequencing of treatment needs further investigation. This includes development of further next-generation TKIs with potentially better efficacy and intracranial activity, as well as improved safety profiles. Two new substances are currently on the way to complement the approved ALK TKIs.
Ensartinib (X-393) is a novel next-generation ALK inhibitor demonstrating increased preclinical activity compared to crizotinib, alectinib and ceritinib against mutations leading to resistance to crizotinib [59]. The eXalt2 study is a phase II trial investigating the activity of 225 mg ensartinib daily in several cohorts of ALK-positive NSCLC patients (ALK-TKI naïve, relapsed after crizotinib or second-generation TKI, patients with stable untreated brain metastases) [59]. The ORR in this trial was 58%. In patients who were TKI naïve the ORR was 87% with a PFS of 23.8 months (95% CI 6.2–40.5). Recently, a Chinese phase II study aimed at investigating the efficacy and safety of ensartinib in 160 crizotinib-resistant patients [60]. The ORR was 52% (95% CI 43–60), median PFS was 9.6 months (95% CI 7.4–11.6). Furthermore, an objective intracranial response was noted in 70% (95% CI 53–83) of patients [60]. The first results of the randomised phase IIII eXalt3 trial in ALK-inhibitor-naïve patients were recently presented. A superior efficacy versus crizotinib was demonstrated in the brain. The OS was not different [61].
Repotrectinib is a rationally designed TKI developed to inhibit ALK, ROS-1 and TRKA-C, with activity especially in point-mutated tyrosine kinase fusion products [62]. Repotrectinib is smaller in size and more rigid in structure, which is supposed to be favourable in penetrating the blood-brain barrier and binding the mutated receptor kinases [62]. Repotrectinib demonstrated promising activity in ALK, ROS-1 and NTRK rearranged preclinical models [62], leading to an ongoing first-in-human dose-escalation phase I/II trial (NCT03093116) in patients with advanced ALK-, ROS1- or NTRK1–3-rearranged cancers.
Potentially, these and other ALK inhibitors will complement and probably change the treatment landscape of ALK-positive NSCLC patients. Nevertheless, much further effort is needed to find the best drug, treatment sequence, and biomarkers to provide an effective and tailored treatment for each individual ALK-positive patient.
4 Intracranial Activity of ALK-TKIs
Brain metastases are known to be a common complication in patients with ALK-translocated NSCLC. About 30% of patients harbour brain metastases and 15% even show more than four cerebral metastases at initial diagnosis [63]. Crizotinib was the first approved TKI in the first-line setting. Nevertheless, many patients treated with crizotinib progress over time with development of brain metastases because of poor intracranial efficacy of crizotinib [64]. The reduced intracranial control by crizotinib results from P-glycoprotein-mediated efflux across the blood-brain barrier [65]. Next-generation ALK inhibitors were designed to pass the blood-brain barrier; however, the development or progression of brain metastases pose significant problems in ALK-positive lung cancer patients.
Ceritinib was the first approved second-generation TKI demonstrating potent intracranial activity in patients with brain metastases previously treated with crizotinib. The intracranial response rate in patients from the ASCEND-2 trial was 45% (95% CI 23.1–68.5) [25].
The intracranial activity of brigatinib was evaluated in an exploratory study including all patients from the phase I/II ALTA trials [66]. Most patients with ALK-positive NSCLC having brain metastases at initial diagnosis were treated with crizotinib. Confirmed intracranial ORR was 53%. Intracranial response was similar in patients without prior radiation or progression after radiotherapy. Among patients with any baseline brain metastases, median intracranial PFS was 14.6 months (95% CI 12.7–36.8) [66].
Gadgeel and colleagues evaluated the cumulative incidence rates (CIRs) of central nervous system (CNS) progression in patients from the phase II alectinib trials. They found that in patients with baseline brain metastases, CIRs of CNS and non-CNS progression at 24 months were 43.9% and 31.0%, respectively. In patients without baseline CNS metastases, the CIR of CNS progression was 8.0% at 24 months. These patients progressed at a higher rate in organs other than the brain (50.9%) [67].
The good intracranial activity of alectinib compared to crizotinib was also shown in an explorative analysis of the J-ALEX study [68]. The hazard ratio for time to CNS progression in patients with and without baseline CNS metastases was 0.51 (95% CI 0.16–1.64; p = 0.2502) and 0.19 (95% CI 0.07–0.53; p = 0.0004), respectively. The CIRs of CNS progression and non-CNS progression were lower in the alectinib group compared to the crizotinib group. The 12-month CIRs of CNS progression were 16.8% and 5.9% with crizotinib and alectinib, respectively. The 1-year CIRs of non-CNS progression were 38.4% and 17.5% [68].
The third-generation ALK TKI lorlatinib was specifically designed to penetrate the blood-brain barrier and was proven to result in relevant drug concentrations in the cerebrospinal fluid [53]. Bauer et al. reported the results regarding the intracranial activity of lorlatinib from the ongoing phase II trial discussed above [54, 69]. 198 patients were evaluable, 59 only received previous crizotinib treatment and 139 received at least one second-generation TKI. The CIR of non-CNS-progression was consistently higher compared to the CIR of CNS-progression, indicating a potent and durable intracranial activity of lorlatinib in heavily pre-treated patients [69].
A current investigation presented at the ASCO 2020 assessed 22 patients with ALK-positive lung cancer with brain-only progression who received lorlatinib. The authors found an intracranial ORR of 59%. All patients had at least one line of ALK TKI and most patients (77%) had two or three prior TKIs [70].
5 Mechanisms of Treatment Resistance
Since the approval of crizotinib and subsequent generations of ALK TKIs, the treatment and the prognosis of ALK-positive NSCLC patients has changed tremendously. However, under TKI treatment all patients will inevitably progress sooner or later. Several mechanisms leading to resistance have been investigated over the past years.
Shortly after the approval of crizotinib, it has been shown that many patients developed somatic mutations in the tyrosine kinase domain of ALK leading to crizotinib resistance [71, 72]. Gainor and colleagues demonstrated that the rate of patients with resistance mutations is higher in patients treated with second-generation ALK inhibitors (ceritinib 54%, alectinib 53%, brigatinib 71%) compared to crizotinib (20%) [73]. Furthermore, each TKI induces a distinct pattern of ALK-resistance mutations, with predominant appearance of G1202R after second-generation TKIs [73]. Previous investigations additionally presented in vitro IC50 values for all available ALK TKIs regarding the different mutations, the findings illustrating that lorlatinib has the broadest activity, including against the G1202R mutation [73]. The in vitro data were confirmed in a cohort of 198 patients showing the efficacy of lorlatinib in patients with and without ALK resistance mutation after failure of one or more lines of TKI treatment [74]. In a recent study, Horn and colleagues presented comprehensive in vitro results of the activity of several ALK TKIs on different ALK mutations. These results are in line with the previous studies showing activity of lorlatinib and brigatinib against most resistance mutations. The study also demonstrated that IC50 values are higher in EML4-ALK fusion variant 4. Additionally, the authors could show that circulating tumour DNA (ctDNA) and detection of ALK mutations by liquid biopsy might serve as a longitudinal follow-up strategy [75].
Beside ALK mutations, ALK amplification was also found to be a rare event leading to crizotinib resistance [72].
EML4 is the most common fusion partner of ALK in NSCLC. However, more than 15 EML4-ALK fusion variants have been identified [76]. The most common are variant 1 (v1 exon 13 of EML4 fused to exon 20 of ALK [E13;A20]) and v3a/b (exon 6a/b of EML4 fused to exon 20 of ALK [E6a/b;A20]) [76]. Yoshida and colleagues could show that the median PFS was significantly longer in patients with variant 1 than in those with non-variant 1 when treated with crizotinib (11.0 months [95% CI 6.5–43.0] vs. 4.2 months [95% CI 1.6–10.2] [77].
A recent study evaluated the impact of ALK variants on the development of resistance mutations in 129 patients with ALK-positive NSCLC. Most patients had v1 (43%) or v3 (40%). ALK resistance mutations were identified in ten patients with v1 (30%) compared with 25 with v3 (57%). The G1202R mutation was detected in 0% of patients with v1 versus 32% of 44 patients with v3 [76]. Another study with 110 Chinese ALK-positive patients confirmed that patients harbouring EML4-ALK rearrangement variant 3 or 5 have significantly reduced PFS when treated with crizotinib [78]. The same study revealed that patients with fusion partners other than EML4 had the same benefit from crizotinib treatment [78].
Beside ALK mutations and fusion variants, co-occurring somatic mutations, fusions or amplifications may provide bypass mechanisms and thus result in resistance to ALK-TKIs.
Concomitant TP53 mutations characterize patients with poor prognosis [79, 80]. Co-occurring TP53 mutations in ALK-positive NSCLC patients have been found in 20–23.8% of all cases [81, 82]. Patients with concomitant TP53 mutation at baseline or acquisition of TP53 mutation during the course of treatment showed reduced PFS and OS compared to wild-type patients [79, 82]. The disadvantage regarding survival in TP53 co-mutated patients was shown to be independent of the treatment modality [81]. Patients with EML4-ALK fusion variant 3 and TP53 mutations therefore characterize a subgroup with very poor prognosis. In this group the risk of death was increased with a hazard ratio of 9.1 [82].
Furthermore, MET alterations are known to be common resistance mechanisms in EGFR-mutant NSCLC and have been shown to be overcome with concomitant MET-blockade [83,84,85]. However, MET alterations in ALK-positive NSCLC are currently not extensively studied but represent an attractive target due to several available MET TKIs [86]. A recent study could show that MET alterations are common events (15%) in patients relapsing after next-generation ALK TKIs [87]. MET amplifications were revealed in 13% of patients relapsing under next-generation TKIs. In some patients, tissue from earlier biopsies was available. None of them showed MET amplification, indicating that it developed during TKI treatment [87]. MET alterations were nearly exclusive with ALK-resistance mutations, indicating that MET serves as an independent resistance mechanism [87]. Patients treated with crizotinib were significantly less affected by MET-amplifications. In two patients of this study the ST7-MET-fusion was detected. In ALK TKI-resistant ST7-MET rearranged cell-lines concomitant treatment with MET TKIs resulted in potently suppressed cell proliferation [87]. Another study found a prevalence of MET-amplification in ALK positive NSCLC patients of 10%. When patients with MET-amplification were treated with crizotinib they showed a superior PFS compared to patients without amplification [88]. If ALK-positive patients with concomitant MET alteration might benefit from combined treatment of an ALK and MET TKI or if crizotinib with its additional activity against MET is beneficial in these patients needs to be investigated in further studies.
A few studies indicated that activation of the EGFR pathway may serve as a by-pass mechanism leading to resistance to first-, second- and third-generation ALK TKIs [72, 89, 90]. Furthermore, activation of the RAS-MAPK pathway as well as the IGF pathway demonstrated the ability to overcome ALK TKI effectiveness [91,92,93]. These results suggest that combinational treatment with other TKIs might be a possible strategy to prevent or overcome ALK-TKI resistance.
At present, treatment guided by specific resistance mutations, ALK fusion variants or activated by-pass pathways should be evaluated in specialized centres with lots of experience in treating ALK-positive NSCLC patients. However, increasing knowledge in this field will translate into to daily practice in the next years.
6 Thoughts on ALK TKI Treatment Sequence
Of course, it would be desirable to have sequencing trials with different drugs at the beginning to test the different impact factors that allow the best sequence for the individual tumour. At the moment the advantages of alectinib and brigatinib regarding survival and intracranial activity in patients with baseline brain metastases, as well as the protection of patients without brain metastases, make these drugs preferable in first-line treatment—especially in patients with already existing brain metastases.
Disease progression should be followed by re-biopsy whenever possible to detect resistance mutations or bypass mechanisms. As demonstrated by Horn et al., liquid biopsies can also serve as effective and less invasive methods to obtain new tumour DNA [75]. If a distinct mutation is found, the next TKI could be chosen according to the activity profile against the mutation. For most of the available TKIs, there are at least comprehensive in vitro data of activity patterns against distinct resistance mutations [73, 75]. Using resistance mutation-guided treatment could also lead to re-sensitization to TKIs of an earlier generation [94]. Nevertheless, it was shown that lorlatinib had stronger efficacy in patients with ALK mutations compared to patients without mutations after failure of at least one second-generation ALK TKI. This indicates that lorlatinib is a good choice in patients with ALK-resistance mutations [74].
Based on current data, no treatment recommendations of different fusion variants can be made. However, the knowledge that patients with variant 3 or 5 have poorer prognosis could lead to closer surveillance for early detection of treatment failure. Perhaps for variant 1 without brain metastases and without p53 mutation crizotinib could be used.
If an alternative bypass pathway is detected, targeted combination therapy, preferably in clinical trials, could be considered. With concurrent MET alterations, crizotinib could be an alternative option in patients without brain metastases to cover both pathways [87]. If additional activation of the EGFR pathway is detected during disease progression, concomitant treatment with an EGFR-TKI could considered.
In the case of oligo-progression, local treatment and TKI therapy beyond progression should be considered and interdisciplinary treatment discussed.
Otherwise, chemotherapy, especially pemetrexed-based regimens or chemo-immunotherapy, is known to be highly effective in patients with ALK mutations. There are only few data on checkpoint inhibitor monotherapy in ALK-rearranged patients, indicating a reduced efficacy. The addition of angiogenesis inhibitors as under investigation in EGFR-mutated tumours may be an option and has to be trialled further. This is also the case for the combination of ALK inhibition with chemotherapy in first-line therapy (see Fig. 1).

Shows a possible algorithm to structure a treatment sequence in ALK-positive NSCLC patients
7 Conclusions
The treatment of ALK-rearranged NSCLC with ALK TKIs has tremendously changed the outcome and quality of life of these patients. However, all patients will inevitably progress after a distinct time. To expand PFS and hopefully OS it is crucial to think about evidence-based treatment sequencing. Every ALK TKI has its own advantages and disadvantages. Regular re-biopsies during disease progression are important to guide treatment sequencing. At present, we are just starting to understand the relevance of ALK mutations, variants, co-mutations or bypass pathways for therapy sequencing. Therefore, more clinical and translational research is needed to expand our knowledge and thus the survival of patients with ALK-rearranged NSCLC.
Code availability
Not applicable.
Change history
26 May 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40265-021-01536-8
References
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–4.
Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14(4):439–49.
Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene. 1997;14(18):2175–88.
Hallberg B, Palmer RH. The role of the ALK receptor in cancer biology. Ann Oncol. 2016;27(Suppl 3):iii4–15.
Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13(10):685–700.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6.
Chia PL, Mitchell P, Dobrovic A, John T. Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin Epidemiol. 2014;6:423–32.
Kerr KM, Lopez-Rios F. Precision medicine in NSCLC and pathology: how does ALK fit in the pathway? Ann Oncol. 2016;27(Suppl 3):iii16–24.
Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv192–237.
Ettinger DS, Wood DE, Aggarwal C, Aisner DL, Akerley W, Bauman JR, et al. NCCN guidelines insights: non-small cell lung cancer, version 1.2020. J Natl Compr Canc Netw. 2019;17(12):1464–72.
Sahu A, Prabhash K, Noronha V, Joshi A, Desai S. Crizotinib: a comprehensive review. South Asian J Cancer. 2013;2(2):91–7.
Nwizu T, Kanteti R, Kawada I, Rolle C, Vokes EE, Salgia R. Crizotinib (PF02341066) as a ALK /MET inhibitor- special emphasis as a therapeutic drug against lung cancer. Drugs Future. 2011;36(2):91–9.
Yasuda H, de Figueiredo-Pontes LL, Kobayashi S, Costa DB. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J Thorac Oncol. 2012;7(7):1086–90.
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703.
Camidge DR, Bang Y-J, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–9.
Kim D-W, Ahn M-J, Shi Y, Pas TMD, Yang P-C, Riely GJ, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2012;30(15_suppl):7533.
Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–94.
Solomon BJ, Mok T, Kim D-W, Wu Y-L, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–77.
Yoneda KY, Scranton JR, Cadogan MA, Tassell V, Nadanaciva S, Wilner KD, et al. Interstitial lung disease associated with crizotinib in patients with advanced non-small cell lung cancer: independent review of four PROFILE trials. Clin Lung Cancer. 2017;18(5):472–9.
Liu C, Yu H, Long Q, Chen H, Li Y, Zhao W, et al. Real world experience of crizotinib in 104 patients with ALK rearrangement non-small-cell lung cancer in a Single Chinese Cancer Center. Front Oncol. 2019;9:1116.
Davis KL, Lenz C, Houghton K, Kaye JA. Clinical outcomes of crizotinib in real-world practice settings for patients with advanced ALK-positive non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2017;98(1):238–9.
Blakely CM, Riess JW. Interpretation of ceritinib clinical trial results and future combination therapy strategies for ALK-rearranged NSCLC. Expert Rev Anticancer Ther. 2019;19(12):1061–75.
Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–97.
Kim D-W, Mehra R, Tan DSW, Felip E, Chow LQM, Camidge DR, et al. Activity and safety of ceritinib in patients with ALK -rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016;17(4):452–63.
Crino L, Ahn MJ, De Marinis F, Groen HJ, Wakelee H, Hida T, et al. Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2. J Clin Oncol. 2016;34(24):2866–73.
Nishio M, Felip E, Orlov S, Park K, Yu CJ, Tsai CM, et al. Final overall survival and other efficacy and safety results from ASCEND-3: phase II study of ceritinib in ALKi-naive patients with ALK-rearranged NSCLC. J Thorac Oncol. 2020;15(4):609–17.
Soria J-C, Tan DSW, Chiari R, Wu Y-L, Paz-Ares L, Wolf J, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK -rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. The Lancet. 2017;389(10072):917–29.
Shaw AT, Kim TM, Crinò L, Gridelli C, Kiura K, Liu G, et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017;18(7):874–86.
Cho BC, Obermannova R, Bearz A, McKeage M, Kim DW, Batra U, et al. Efficacy and safety of ceritinib (450 mg/d or 600 mg/d) with food versus 750-mg/d fasted in patients with ALK receptor tyrosine kinase (ALK)-Positive NSCLC: primary efficacy results from the ASCEND-8 Study. J Thorac Oncol. 2019;14(7):1255–65.
Tan DS, Araujo A, Zhang J, Signorovitch J, Zhou ZY, Cai X, et al. Comparative efficacy of ceritinib and crizotinib as initial ALK-targeted therapies in previously treated advanced NSCLC: An adjusted comparison with external controls. J Thorac Oncol. 2016;11(9):1550–7.
Paik J, Dhillon S. Alectinib: a review in advanced, ALK-Positive NSCLC. Drugs. 2018;78(12):1247–57.
Barlesi F, Dingemans AMC, Yang JCH, Ou S-H, Ahn JS, Petris L, et al. Updated efficacy and safety from the global phase II NP28673 study of alectinib in patients (pts) with previously treated ALK+ non-small-cell lung cancer (NSCLC). Jpn Soc. Med Oncol. 2016;27:vi437.
Camidge D, Gadgeel S, Ou S-H, Gandhi L, Riely G, Cetnar J, et al. MA07.02 updated efficacy and safety data from the phase 2 NP28761 study of alectinib in ALK-positive non-small-cell lung cancer. J Thorac Oncol. 2017;12:S378.
Hida T, Nokihara H, Kondo M, Kim YH, Azuma K, Seto T, et al. Alectinib versus crizotinib in patients with ALK -positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. The Lancet. 2017;390(10089):29–39.
Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim D-W, et al. Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N Engl J Med. 2017;377(9):829–38.
Camidge DR, Dziadziuszko R, Peters S, Mok T, Noe J, Nowicka M, et al. Updated efficacy and safety data and impact of the EML4-ALK fusion variant on the efficacy of alectinib in untreated ALK-positive advanced non-small cell lung cancer in the global phase III ALEX study. J Thorac Oncol. 2019;14(7):1233–43.
Mok T, Camidge DR, Gadgeel SM, Rosell R, Dziadziuszko R, Kim DW, et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann Oncol. 2020;31(8):1056–64.
Zhou C, Kim S-W, Reungwetwattana T, Zhou J, Zhang Y, He J, et al. Alectinib versus crizotinib in untreated Asian patients with anaplastic lymphoma kinase-positive non-small-cell lung cancer (ALESIA): a randomised phase 3 study. Lancet Respir Med. 2019;7(5):437–46.
Novello S, Mazieres J, Oh IJ, de Castro J, Migliorino MR, Helland A, et al. Alectinib versus chemotherapy in crizotinib-pretreated anaplastic lymphoma kinase (ALK)-positive non-small-cell lung cancer: results from the phase III ALUR study. Ann Oncol. 2018;29(6):1409–16.
DiBonaventura MD, Wong W, Shah-Manek B, Schulz M. Real-world usage and clinical outcomes of alectinib among post-crizotinib progression anaplastic lymphoma kinase positive non-small-cell lung cancer patients in the USA. Onco Targets Ther. 2018;11:75–82.
Masuda N, Ohe Y, Gemma A, Kusumoto M, Yamada I, Ishii T, et al. Safety and effectiveness of alectinib in a real-world surveillance study in patients with ALK-positive non-small-cell lung cancer in Japan. Cancer Sci. 2019;110(4):1401–7.
Spencer SA, Riley AC, Matthew A, Di Pasqua AJ. Brigatinib: novel ALK inhibitor for non-small-cell lung cancer. Ann Pharmacother. 2019;53(6):621–6.
Gettinger SN, Bazhenova LA, Langer CJ, Salgia R, Gold KA, Rosell R, et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(12):1683–96.
Kim DW, Tiseo M, Ahn MJ, Reckamp KL, Hansen KH, Kim SW, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490–8.
Huber RM, Hansen KH, Paz-Ares Rodríguez L, West HL, Reckamp KL, Leighl NB, et al. Brigatinib in crizotinib-refractory ALK+ NSCLC: 2-year follow-up on systemic and intracranial outcomes in the phase 2 ALTA trial. J Thorac Oncol. 2020;15(3):404–15.
Camidge DR, Kim HR, Ahn MJ, Yang JC, Han JY, Lee JS, et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N Engl J Med. 2018;379(21):2027–39.
Camidge DR, Kim HR, Ahn MJ, Yang JCH, Han JY, Hochmair MJ, et al. Brigatinib versus crizotinib in advanced ALK inhibitor-naive ALK-positive non-small cell lung cancer: second interim analysis of the phase iII ALTA-1L trial. J Clin Oncol. 2020;38(31):3592–360.
Umbela S, Ghacha S, Matuknauth R, Gause S, Joshee S, Deshmukh RR. Brigatinib: New-generation ALK inhibitor for nonsmall cell lung cancer. Curr Probl Cancer. 2019;43(6):100477.
Descourt R, Perol M, Rousseau-Bussac G, Planchard D, Mennecier B, Wislez M, et al. Brigatinib in patients with ALK-positive advanced non-small-cell lung cancer pretreated with sequential ALK inhibitors: a multicentric real-world study (BRIGALK study). Lung Cancer. 2019;136:109–14.
Lin HM, Pan X, Hou P, Allen S, Baumann P, Hochmair MJ. Real-world treatment duration in ALK-positive non-small-cell lung cancer patients receiving brigatinib through the early access program. Future oncology. 2020;16(15):1031–41.
Hochmair M, Weinlinger C, Schwab S, Naber J, Setinek U, Krenbek D, et al. Treatment of ALK-rearranged non-small-cell lung cancer with brigatinib as second or later lines: real-world observations from a single institution. Anticancer Drugs. 2019;30(7):e0787.
Syed YY. Lorlatinib: first global approval. Drugs. 2019;79(1):93–8.
Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18(12):1590–9.
Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. 2018;19(12):1654–67.
Solomon B, Bauer TM, De Marinis F, Felip E, Goto Y, Liu G, et al. LBA2 Lorlatinib vs crizotinib in the first-line treatment of patients (pts) with advanced ALK-positive non-small cell lung cancer (NSCLC): Results of the phase III CROWN study. Ann Oncol. 2020;31:S1180–1.
Bauer TM, Felip E, Solomon BJ, Thurm H, Peltz G, Chioda MD, et al. Clinical management of adverse events associated with lorlatinib. Oncologist. 2019;24(8):1103–10.
Zhu VW, Lin YT, Kim DW, Loong HH, Nagasaka M, To H, et al. An international real-world analysis of the efficacy and safety of lorlatinib through early or expanded access programs in patients with tyrosine kinase inhibitor-refractory ALK-positive or ROS1-positive NSCLC. J Thorac Oncol. 2020;15(9):1484–96.
Peled N, Gillis R, Kilickap S, Froesch P, Orlov S, Filippova E, et al. GLASS: global Lorlatinib for ALK(+) and ROS1(+) retrospective Study: real world data of 123 NSCLC patients. Lung Cancer. 2020;148:48–54.
Singhi EK, Horn L. Background and rationale of the eXalt3 trial investigating X-396 in the treatment of ALK+ non-small-cell lung cancer. Future oncology. 2018;14(18):1781–7.
Yang Y, Zhou J, Zhou J, Feng J, Zhuang W, Chen J, et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib-resistant, ALK-positive non-small-cell lung cancer: a multicentre, phase 2 trial. Lancet Respir Med. 2020;8(1):45–53.
Horn L. Phase 3 Randomized Study of Ensartinib vs Crizotinib in Anaplastic Lymphoma Kinase (ALK)–Positive NSCLC Patients: eXalt3. In: 2020;Presidenital Symposium WCLC 2020, Abtract 2.
Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/Alk inhibitor that potently inhibits ROS1/TRK/ALK solvent- front mutations. Cancer Discov. 2018;8(10):1227–36.
Johung KL, Yeh N, Desai NB, Williams TM, Lautenschlaeger T, Arvold ND, et al. Extended survival and prognostic factors for patients with ALK-rearranged non-small-cell lung cancer and brain metastasis. J Clin Oncol. 2016;34(2):123–9.
Costa DB, Shaw AT, Ou SH, Solomon BJ, Riely GJ, Ahn MJ, et al. Clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J Clin Oncol. 2015;33(17):1881–8.
Costa DB, Kobayashi S, Pandya SS, Yeo WL, Shen Z, Tan W, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29(15):e443–5.
Camidge DR, Kim DW, Tiseo M, Langer CJ, Ahn MJ, Shaw AT, et al. Exploratory analysis of brigatinib activity in patients with anaplastic lymphoma kinase-positive non-small-cell lung cancer and brain metastases in two clinical trials. J Clin Oncol. 2018;36(26):2693–701.
Gadgeel S, Shaw AT, Barlesi F, Crino L, Yang JC, Dingemans AC, et al. Cumulative incidence rates for CNS and non-CNS progression in two phase II studies of alectinib in ALK-positive NSCLC. Br J Cancer. 2018;118(1):38–42.
Nishio M, Nakagawa K, Mitsudomi T, Yamamoto N, Tanaka T, Kuriki H, et al. Analysis of central nervous system efficacy in the J-ALEX study of alectinib versus crizotinib in ALK-positive non-small-cell lung cancer. Lung Cancer. 2018;121:37–40.
Bauer TM, Shaw AT, Johnson ML, Navarro A, Gainor JF, Thurm H, et al. Brain penetration of lorlatinib: cumulative incidences of CNS and non-CNS progression with lorlatinib in patients with previously treated ALK-positive non-small-cell lung cancer. Target Oncol. 2020;15(1):55–65.
Dagogo-Jack I, Oxnard GR, Fink J, Diubaldi G, Helms C, Gainor JF, et al. A phase II study of lorlatinib in patients (pts) with ALK-positive (ALK+) lung cancer with brain-only progression. J Clin Oncol. 2020;38(15_Suppl):9595.
Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–82.
Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4(120):120ra17.
Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–33.
Shaw AT, Solomon BJ, Besse B, Bauer TM, Lin CC, Soo RA, et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non-small-cell lung cancer. J Clin Oncol. 2019;37(16):1370–9.
Horn L, Whisenant JG, Wakelee H, Reckamp KL, Qiao H, Leal TA, et al. Monitoring therapeutic response and resistance: analysis of circulating tumor DNA in patients with ALK+ lung cancer. J Thorac Oncol. 2019;14(11):1901–11.
Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo-Jack I, et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J Clin Oncol. 2018;36(12):1199–206.
Yoshida T, Oya Y, Tanaka K, Shimizu J, Horio Y, Kuroda H, et al. Differential crizotinib response duration among ALK fusion variants in ALK-positive non-small-cell lung cancer. J Clin Oncol. 2016;34(28):3383–9.
Su Y, Long X, Song Y, Chen P, Li S, Yang H, et al. Distribution of ALK fusion variants and correlation with clinical outcomes in chinese patients with non-small cell lung cancer treated with crizotinib. Target Oncol. 2019;14(2):159–68.
Christopoulos P, Dietz S, Kirchner M, Volckmar AL, Endris V, Neumann O, et al. Detection of TP53 mutations in tissue or liquid rebiopsies at progression identifies ALK+ lung cancer patients with poor survival. Cancers. 2019;11(1).
Aisner DL, Sholl LM, Berry LD, Rossi MR, Chen H, Fujimoto J, et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations—the Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res. 2018;24(5):1038–47.
Kron A, Alidousty C, Scheffler M, Merkelbach-Bruse S, Seidel D, Riedel R, et al. Impact of TP53 mutation status on systemic treatment outcome in ALK-rearranged non-small-cell lung cancer. Ann Oncol. 2018;29(10):2068–75.
Christopoulos P, Kirchner M, Bozorgmehr F, Endris V, Elsayed M, Budczies J, et al. Identification of a highly lethal V3(+) TP53(+) subset in ALK(+) lung adenocarcinoma. Int J Cancer. 2019;144(1):190–9.
Bean J, Brennan C, Shih J-Y, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104(52):20932–7.
Li YQ, Song SS, Jiang SH, Zhang XY. Combination therapy of erlotinib/crizotinib in a lung adenocarcinoma patient with primary EGFR mutation plus secondary MET amplification and a novel acquired crizotinib-resistant mutation MET G1108C. Ann Oncol. 2017;28(10):2622–4.
Kauffmann-Guerrero D, Kahnert K, Kumbrink J, Syunyaeva Z, Tufman A, Huber RM. Successful treatment of a patient with NSCLC harboring an EGFR mutation and a concomitant Met Exon 14 skipping mutation combining afatinib and crizotinib. Clinical Lung Cancer. 2019;20(1):59–62.
Salgia R, Sattler M, Scheele J, Stroh C, Felip E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat Rev. 2020;87:102022.
Dagogo-Jack I, Yoda S, Lennerz JK, Langenbucher A, Lin JJ, Rooney MM, et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin Cancer Res. 2020;26(11):2535–45.
Chen RL, Zhao J, Zhang XC, Lou NN, Chen HJ, Yang X, et al. Crizotinib in advanced non-small-cell lung cancer with concomitant ALK rearrangement and c-Met overexpression. BMC Cancer. 2018;18(1):1171.
Tani T, Yasuda H, Hamamoto J, Kuroda A, Arai D, Ishioka K, et al. Activation of EGFR bypass signaling by TGFalpha overexpression induces acquired resistance to Alectinib in ALK-translocated lung cancer cells. Mol Cancer Ther. 2016;15(1):162–71.
Redaelli S, Ceccon M, Zappa M, Sharma GG, Mastini C, Mauri M, et al. Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018;78(24):6866–80.
Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med. 2015;21(9):1038–47.
Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20(9):1027–34.
Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat Med. 2018;24(4):512–7.
Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54–61.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this article.
Conflicts of interest
Diego Kauffmann-Guerrero has received honoraria from Pfizer, Roche, Takeda and AstraZeneca, Boehringer-Ingelheim, BMS. Kathrin Kahnert declares that she has no conflicts of interest that might be relevant to the contents of this article. Rudolf M. Huber has received honoraria from AbbVie, AstraZeneca, Bayer, BMS, Boehringer-Ingelheim, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, Tesaro
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Additional information
The original article has been updated: Due to retrospective open choice order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kauffmann-Guerrero, D., Kahnert, K. & Huber, R.M. Treatment Sequencing for Anaplastic Lymphoma Kinase-Rearranged Non-Small-Cell Lung Cancer. Drugs 81, 87–100 (2021). https://doi.org/10.1007/s40265-020-01445-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01445-2