
Abstract
Risankizumab (Skyrizi®; risankizumab-rzaa) is a humanized immunoglobulin (Ig) G1 monoclonal antibody that specifically targets the p19 subunit of interleukin (IL)-23, thereby inhibiting IL-23-dependent cell signaling. Subcutaneous risankizumab is approved for the treatment of adults with moderate to severe plaque psoriasis who are candidates for systemic therapy (in the EU), those who are candidates for systemic therapy or phototherapy (in the USA) and those who have an inadequate response to conventional therapies (in Japan). In pivotal phase III trials (UltIMMa-1, UltIMMa-2, IMMvent and IMMhance), risankizumab was more effective than placebo, ustekinumab and adalimumab with regard to the proportion of patients achieving ≥ 90% improvement from baseline in Psoriasis Area and Severity Index score (PASI 90) and a static Physician’s Global Assessment score of 0 or 1 at week 16, with these benefits maintained over the longer term. In supportive head-to-head trials, risankizumab was also superior to secukinumab and fumaric acid esters in terms of PASI 90 response rate. In an ongoing open-label extension study (LIMMitless), risankizumab was associated with durable and improved efficacy after switching from ustekinumab or adalimumab, as well as durable maintenance of efficacy through > 2.5 years of continuous exposure. Treatment with risankizumab improved health-related quality of life and was generally well tolerated, both in the short- and longer-term. In conclusion, risankizumab represents a useful new treatment option for patients with moderate to severe plaque psoriasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
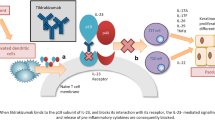
Humanized IgG1 monoclonal antibody that binds to and blocks the proinflammatory effects of IL-23 |
More effective than placebo, ustekinumab, adalimumab, secukinumab and fumaric acid esters in reducing the severity and extent of plaque psoriasis |
Improves health-related quality of life |
Generally well tolerated |
1 Introduction
Psoriasis is a chronic immune-mediated inflammatory skin condition that affects 2–3% of the population [1]. Plaque psoriasis is characterized by well-delineated, red, scaly plaques, with disease severity defined partially by the total body surface area (BSA) involved [2]. However, the disease may be severe regardless of BSA involvement, and can have a significant negative impact on patients’ quality of life (QOL) [2]. Conventional systemic treatments for plaque psoriasis such as phototherapy, methotrexate, ciclosporin, acitretin and apremilast [3, 4] are often inadequate for patients with moderate to severe disease [2]. An improved understanding of the pathophysiology of psoriasis over the last 20 years has led to the development of biological agents targeting specific molecules such as tumour necrosis factor (TNF)-α, interleukin (IL)-12/23, IL-17 and IL-23 [1]. Such agents have demonstrated sustained efficacy and a favourable safety profile [5], and are now a mainstay in the treatment of moderate to severe plaque psoriasis.
Risankizumab (Skyrizi®; risankizumab-rzaa) is a humanized immunoglobulin (Ig) G1 monoclonal antibody that selectively targets and binds to IL-23, thereby inhibiting the proinflammatory effects of IL-23 [5]. It is approved for the treatment of moderate to severe plaque psoriasis in multiple countries, including the USA [6], Japan [7] and those of the EU [8]. This article provides an overview of the pharmacological properties of risankizumab and reviews the clinical data relevant to its use in moderate to severe plaque psoriasis. Discussion of the use of risankizumab in other approved indications (i.e. psoriatic arthritis, erythrodermic psoriasis and generalized pustular psoriasis in Japan [7]) is beyond the scope of this article.
2 Pharmacodynamic Properties of Risankizumab
Risankizumab specifically binds with high affinity (dissociation constant < 10 pmol/L) to the p19 subunit of IL-23 [5], thereby preventing its interaction with the IL-23 receptor complex and subsequently inhibiting IL-23-dependent cell signaling and the release of pro-inflammatory cytokines and chemokines [6, 8]. In vitro, risankizumab was 2- to 3-fold more potent than guselkumab and 4- to 10-fold more potent than ustekinumab and tildrakizumab in inhibiting IL-23 signaling [9].
In human B-lymphoblastoid cell lines derived from human diffuse large cell lymphoma, risankizumab significantly inhibited IL-23-dependent phosphorylation of STAT3 [7]. At doses of 1–25 mg/kg, risankizumab inhibited ear swelling in a murine model of human IL-23-mediated psoriasiform dermatitis [9]. In patients with moderate to severe plaque psoriasis, single doses of intravenous (0.1–5 mg/kg) or subcutaneous (0.25 or 1 mg/kg) risankizumab reduced hyperkeratosis with parakeratosis, epidermal acanthosis and generalized inflammation in the dermis and epidermis [10]. Risankizumab was also associated with reduced expression of markers associated with keratinocyte layer thickening and hyperproliferation; dermal infiltration by T cells, neutrophils and dendritic cells; and tissue inflammation. Treatment with risankizumab resulted in significant (p < 0.005 vs placebo) reductions in the expression of lesional skin genes associated with IL-23/IL-17 signaling pathways, as well as normalization of the gene expression profile of psoriatic lesions to a profile closely resembling that of non-lesional skin. Reduced expression of a set of 79 genes was correlated with Psoriasis Area Severity Index (PASI) scores at week 8 (r = 0.73; p < 0.00002) [10].
3 Pharmacokinetic Properties of Risankizumab
According to an integrated analysis of seven phase I–III trials, the pharmacokinetics of risankizumab in healthy subjects (n = 67) and patients with moderate to severe plaque psoriasis (n = 1844) can be characterized using a two-compartment model with first-order absorption and elimination [11]. The pharmacokinetics of risankizumab were typical of those of other IgG1 monoclonal antibodies, with no apparent target-mediated drug disposition [11]. In exposure-response analyses using data from five phase II and III trials in patients with moderate to severe chronic plaque psoriasis (n = 1732), the approved regimen of subcutaneous risankizumab 150 mg at weeks 0 and 4, then every 12 weeks was shown to maximize short-term (week 16) and long-term (week 52) efficacy [12]. There was no apparent relationship between risankizumab exposure and adverse events (AEs), including serious AEs and infections [12].
Subcutaneous risankizumab exhibited linear pharmacokinetics across dose ranges of 0.25–1 mg/kg and 18–300 mg in healthy subjects and patients with psoriasis, with dose-proportional increases in exposure [6, 8]. The maximum plasma concentration of risankizumab was reached after 3–14 days [6,7,8] and steady-state concentrations were reached by week 16 [6]. The absolute bioavailability of subcutaneous risankizumab was estimated to be 89% and the steady-state volume of distribution (Vd) was 11.2–11.4 L [6, 8]. Given that risankizumab is a humanized IgG1 monoclonal antibody, it is expected to be degraded into small peptides and amino acids in the same manner as endogenous IgG. The estimated systemic clearance of risankizumab was 0.3 L/day and the terminal elimination half-life was 28 days [6, 8].
Age, sex and race (including Japanese or Chinese ethnicity [13]) did not affect the pharmacokinetics of risankizumab [6, 8, 13]. The effects of renal or hepatic impairment on the pharmacokinetics of risankizumab have not been studied [6, 8]. However, as risankizumab is predominantly eliminated via intracellular catabolism and is not expected to undergo renal elimination or metabolism by hepatic enzymes, the effects of renal or hepatic impairment on the pharmacokinetics of risankizumab are not expected to be clinically relevant [8]. Therefore, dosage adjustments are not required in patients with renal or hepatic impairment. Serum creatinine levels, creatinine clearance and markers of hepatic function (i.e. alanine aminotransferase, aspartate aminotransferase, bilirubin) are not expected to have a clinically meaningful effect on risankizumab clearance in patients with psoriasis [8]. The Vd and clearance of risankizumab increased (and plasma concentrations decreased [6]) with increasing bodyweight; however, no dosage adjustments for bodyweight are recommended [6, 8].
Risankizumab has no potential to cause drug interactions via CYP enzymes [14]. In patients with moderate to severe plaque psoriasis (n = 21), administration of subcutaneous risankizumab 150 mg once every 4 weeks (i.e. more frequently than the approved regimen) did not affect the in vivo activity of CYP1A2, CYP2C9, CYP2C19, CYP2D6 or CYP3A enzymes [14]. In population pharmacokinetic analyses, coadministration of risankizumab with the CYP substrates acetylsalicylate, amlodipine, atorvastatin, ibuprofen, levothyroxine, lisinopril or metformin had no effect on risankizumab exposure in patients with plaque psoriasis [8].
4 Therapeutic Efficacy of Risankizumab
The efficacy of subcutaneous risankizumab for the treatment of moderate to severe plaque psoriasis was primarily evaluated in four pivotal, large (n > 450), randomized, double-blind, multinational, phase III trials: UltIMMa-1 and UltIMMa-2 [15], IMMvent [16] and IMMhance [17], which are the main focus of discussion in this section. These data are supported by a phase II dose-ranging study [18], which is not discussed further; as well as a phase II/III trial in Japanese patients (SustaIMM) [19] and two head-to-head phase III trials comparing risankizumab with secukinumab (IMMerge) [20] and fumaric acid esters [21], which are briefly discussed in Sect. 4.3.
The pivotal phase III trials enrolled patients aged ≥ 18 years with stable moderate to severe chronic plaque psoriasis (with or without psoriatic arthritis) who were candidates for systemic therapy or phototherapy [15,16,17]. All patients had BSA involvement of ≥ 10%, a PASI score of ≥ 12 and a static Physician’s Global Assessment (sPGA) score of ≥ 3. Key exclusion criteria included non-plaque forms of psoriasis (guttate, erythrodermic, pustular) and known chronic or relevant acute infections, including tuberculosis (TB) [15,16,17]. Across all trials, the mean age of patients was 46–49 years (median 51 years in IMMhance [17]) and most (68–71%) patients were male [15,16,17].
All four trials were conducted in two parts (A and B); design details and treatment regimens evaluated are summarized in Table 1. In part A of each trial, the co-primary endpoints were the proportions of patients achieving ≥ 90% improvement in the PASI score (PASI 90) and an sPGA score of 0 (clear) or 1 (almost clear) at week 16 [15,16,17]. Statistical comparisons for the co-primary and ranked secondary endpoints were assessed using a prespecified hierarchical testing procedure [15,16,17].
4.1 Short-Term Treatment
Subcutaneous risankizumab was efficacious in the treatment of moderate to severe plaque psoriasis in pivotal phase III trials [15,16,17]. At week 16, the proportion of patients achieving a PASI 90 response and an sPGA score of 0 or 1 was significantly higher with risankizumab than with placebo [15, 17], ustekinumab [15] and adalimumab [16] (Table 2). These findings were supported by sensitivity analyses based on the per-protocol populations [15,16,17]. In the UltIMMa and IMMhance trials, significant differences in PASI 90 and sPGA 0 or 1 response rates between risankizumab and placebo were seen as early as week 4 (p < 0.001) [15, 17]. Significant differences between risankizumab and ustekinumab [15] and between risankizumab and adalimumab [16] were seen from week 4 for sPGA 0 or 1 (p < 0.05) and from week 8 for PASI 90 (p < 0.01).
Secondary endpoints also favoured risankizumab over placebo, ustekinumab and adalimumab [15,16,17]. Risankizumab was significantly more effective than placebo [15, 17], ustekinumab [15] and adalimumab [16] with regard to the proportion of patients achieving complete skin clearance (i.e. PASI 100 and sPGA 0) and the proportion of patients with a PASI 75 response (Table 2).
Risankizumab improved health-related QOL (HR-QOL), as assessed by the Dermatology Life Quality Index (DLQI) [15,16,17], the Psoriasis Symptom Scale (PSS) [15] and the Work Limitations Questionnaire (WLQ) [16]. At week 16, significantly more risankizumab recipients had a DLQI score of 0 or 1 (i.e. no effect on the patient’s life) than did patients receiving placebo [15, 17], ustekinumab [15] or adalimumab [16] (Table 2). In the UltIMMa trials, significantly more risankizumab than placebo and ustekinumab recipients achieved a PSS score of 0 at week 16 (Table 2) [15]. The mean change from baseline in PSS total score (minimal clinically important difference 2.0) was − 5.6 with risankizumab versus 0.2 with placebo in UltIMMa-1, and − 6.4 with risankizumab versus 0.0 with placebo in UltIMMa-2 (both p < 0.0001) [15]. In IMMvent, the least squares mean change from baseline at week 16 in WLQ score was − 2.8 with risankizumab versus − 1.9 with adalimumab (p = 0.0123) [16].
4.2 Long-Term Treatment
Risankizumab was associated with a durable response, with efficacy maintained over the longer term [15,16,17].
Among patients initially randomized to risankizumab in the UltIMMa trials, PASI and sPGA response rates continued to increase after week 16 [15]. At week 52, PASI, sPGA, DLQI and PSS response rates were significantly higher in risankizumab recipients than in ustekinumab recipients (Table 3) [15]. Similar results were seen for PASI 90 (81 vs 47%) and PASI 100 (58 vs 26%) using integrated data from both UltIMMa trials (both p < 0.001) [22]. In another integrated analysis, PASI 90 response rates at week 52 were significantly (p < 0.01) higher with risankizumab than with ustekinumab regardless of baseline characteristics (i.e. age, gender, body mass index and bodyweight), patient characteristics (i.e. baseline PASI, baseline sPGA and psoriatic arthritis status) and prior therapy exposure (i.e. naïve to biologicals, any biological exposure and failed any biological) [23]. Similar results were seen for sPGA 0 or 1 and PASI 100 response rates [23]. Among patients initially randomized to placebo who switched to risankizumab at week 16, PASI, sPGA, DLQI and PSS response rates at week 52 were similar to those in patients initially randomized to risankizumab (Table 3) [15]. Among patients who achieved a PASI 90 response at week 16, 88% of risankizumab recipients maintained this response through week 52 compared with 73% of ustekinumab recipients (p = 0.0009) [15].
In IMMvent, PASI, sPGA and DLQI response rates at week 44 were significantly higher in adalimumab intermediate responders (≥ 50 to < 90% improvement in PASI score) who were re-randomized to risankizumab at week 16 than in those who were re-randomized to adalimumab (Table 3) [16]. At week 44, the improvement from baseline in mean PASI score was 93% in patients who were re-randomized to risankizumab versus 72% in those who were re-randomized to adalimumab (p < 0.0001) [16], with similar results seen in the subgroup of patients who achieved ≥ 75 to 90% improvement in the PASI score at week 16 (95 vs 71%) and in the subgroup of patients who achieved 50 to < 75% improvement in the PASI score at week 16 (87 vs 71%) [24]. The change from baseline in the WLQ score at week 44 was − 2.1 in patients re-randomized to risankizumab and − 1.3 in those re-randomized to adalimumab [16]. Clinical responses with risankizumab were also observed in the other treatment groups. Among adalimumab non-responders (< 50% improvement in PASI score) who switched to risankizumab at week 16, 61% achieved a PASI 90 response and 63% achieved an sPGA score of 0 or 1 at week 44. Among patients who received risankizumab continuously throughout the study, PASI 90 and sPGA 0 or 1 responses were generally maintained up to week 44 (data not shown) [16].
In IMMhance, significantly more risankizumab responders (i.e. those who achieved an sPGA score of 0 or 1) who were re-randomized to risankizumab at week 28 maintained PASI and sPGA response rates than those who were re-randomized to placebo (i.e. withdrawal; Table 3) [17]. Similar results were seen at week 104 for sPGA 0 or 1 response (81 vs 7%; p < 0.001) as well as PASI 90, PASI 100 and sPGA 0 responses (all nominal p < 0.001). Starting at week 40, more patients re-randomized to risankizumab achieved PASI 90 (92 vs 82%) and sPGA 0 or 1 (88 vs 75%) than those re-randomized to placebo (both p < 0.01). The median time to loss of PASI 90 response was significantly (p < 0.001) different between patients re-randomized to placebo (210 days) and those re-randomized to risankizumab [not determinable due to the low rate of loss (22%)]. Likewise, the median time to relapse (defined as an sPGA score of ≥ 3) was significantly (p < 0.001) different between patients re-randomized to placebo (295 days) and those re-randomized to risankizumab [not determinable due to the low relapse rate (5%)]. Of the 153 risankizumab responders who were re-randomized to placebo and experienced relapse after treatment withdrawal, 128 (84%) regained an sPGA 0 or 1 response after 16 weeks of re-treatment with risankizumab. Many of these patients also achieved PASI 100 (43%), PASI 90 (76%), PASI 75 (95%) and sPGA 0 (42%) responses after 16 weeks of re-treatment [17].
4.2.1 Open-Label Extension
In an ongoing open-label extension study (LIMMitless), risankizumab was associated with durable and improved efficacy after switching from ustekinumab [25] or adalimumab [26], as well as durable maintenance of efficacy through > 2.5 years of continuous exposure [27]. Patients who completed one of seven phase II/III trials (including UltIMMa-1, UltIMMa-2 and IMMvent) were eligible to enter LIMMitless, during which all patients received long-term treatment with risankizumab 150 mg every 12 weeks [25,26,27].
Among patients who were initially randomized to ustekinumab in UltIMMa-1 or UltIMMa-2 (n = 199), 184 completed their originator study, and 172 enrolled in LIMMitless and were switched to risankizumab [25]. The PASI 90 response rate (observed cases; no imputation of missing data) was 83% at week 84 compared with 47% at entry/week 0 (i.e. while on ustekinumab). Corresponding PASI 75 response rates were 98 versus 78% and PASI 100 response rates were 57 versus 27%. It should be noted that similar results were seen when PASI response rates were analysed by last observation carried forward (LOCF) and modified non-responder imputation. The proportion of patients achieving a DLQI score of 0 or 1 (LOCF) was 81% at week 72, compared with 50% at entry/week 0 [25].
Among patients who were initially randomized to adalimumab in IMMvent (n = 304), 276 completed the trial and 260 entered LIMMitless, including adalimumab non-responders (n = 33) or intermediate responders (n = 48) who switched to risankizumab at week 16 of IMMvent and adalimumab responders (n = 130) or intermediate responders (n = 49) who switched to risankizumab at week 44 (i.e. the start of LIMMitless) [26]. At all timepoints up to week 84, PASI 75, 90 and 100 response rates and the proportion of patients achieving a DLQI score of 0 or 1 were numerically higher after switching to risankizumab compared with adalimumab, regardless of prior response to adalimumab [26].
Among patients who were randomized to receive risankizumab in UltIMMa-1, UltIMMa-2, IMMvent, SustaIMM or the head-to-head comparison with fumaric acid esters (n = 1014), 955 completed their base study and 897 enrolled in LIMMitless [27]. At week 136 (observed cases), 87% of patients achieved a PASI 90 response, 61% achieved a PASI 100 response and 86% achieved an sPGA score of 0 or 1. The proportion of patients achieving a DLQI score of 0 or 1 at week 124 was 83% [27].
4.3 Supportive Trials
4.3.1 In Japanese Patients
Beneficial effects of risankizumab were also seen in Japanese patients, with results reflective of those seen in the pivotal phase III trials. The phase II/III SustaIMM trial enrolled Japanese patients aged ≥ 20 years with stable moderate to severe chronic plaque psoriasis (with or without psoriatic arthritis) who were candidates for systemic therapy or phototherapy [19]. All patients had a PASI score of ≥ 12, a sPGA score of ≥ 3 and BSA involvement of ≥ 10%. Patients were randomized to receive subcutaneous risankizumab 75 mg (n = 58), risankizumab 150 mg (n = 55) or placebo (n = 58) at weeks 0 and 4, then every 12 weeks. At week 16, patients initially randomized to placebo were switched to risankizumab 75 mg (n = 27) or 150 mg (n = 27) for a further 36 weeks, while patients initially randomized to risankizumab continued to receive their current treatment [19]. Discussion in this section focuses on the approved risankizumab dose of 150 mg.
The proportion of patients with a PASI 90 response at week 16 (primary endpoint) was 72% with risankizumab 150 mg versus 2% with placebo (p < 0.001) [19]. Similar results were seen for PASI 75 (95 vs 9%), PASI 100 (33 vs 0%), sPGA 0 or 1 (93 vs 10%) and DLQI 0 or 1 (58 vs 5%) response rates at week 16 (all p < 0.001). Through week 52, PASI 90, PASI 100 and DLQI 0 or 1 response rates continued to increase in risankizumab 150 mg recipients (to 93, 42 and 80%, respectively). PASI and DLQI response rates also increased from week 16 to 52 in patients who switched from placebo to risankizumab [19].
4.3.2 Comparison with Secukinumab
Risankizumab was superior to secukinumab in patients with moderate to severe plaque psoriasis participating in a multicentre, randomized, open-label, phase III trial (IMMerge) [20]. Eligible patients were adults with moderate to severe chronic plaque psoriasis (with or without psoriatic arthritis) and were candidates for systemic therapy. They had a PASI score of ≥ 12, a sPGA score of ≥ 3 and BSA involvement of ≥ 10%. Patients were randomized to receive subcutaneous risankizumab 150 mg at weeks 0 and 4, then every 12 weeks (n = 164) or subcutaneous secukinumab 300 mg at weeks 0, 1, 2, 3 and 4, then every 4 weeks (n = 163). The primary endpoints were the proportion of patients with a PASI 90 response at week 16 to assess non-inferiority of risankizumab versus secukinumab (non-inferiority margin of 12%) and the proportion of patients with a PASI 90 response at week 52 to assess superiority of risankizumab versus secukinumab [20].
At week 16, 74% of risankizumab recipients and 66% of secukinumab recipients achieved a PASI 90 response; the adjusted between-group difference was 8.2% (96.25% CI − 2.2, + 18.6), indicating that non-inferiority was met [20]. The PASI 90 response rate at week 52 was 87% with risankizumab and 57% with secukinumab (p < 0.001). Risankizumab was significantly (p < 0.001) more effective than secukinumab for all ranked secondary endpoints at week 52, including PASI 75, PASI 100 and sPGA 0 or 1 [20].
4.3.3 Comparison with Fumaric Acid Esters
Risankizumab was superior to fumaric acid esters in patients with moderate to severe plaque psoriasis who were naïve to systemic therapy [21]. This multicentre, randomized, open-label, phase III trial enrolled patients aged ≥ 18 years who had stable moderate to severe chronic plaque psoriasis, a PASI score of > 10, BSA involvement of > 10% and a DLQI score of > 10, and were candidates for systemic therapy. Patients were randomized to receive subcutaneous risankizumab 150 mg at weeks 0, 4 and 16 (n = 60) or oral fumaric acid esters at increasing doses from 30 mg/day at week 0 up to 720 mg/day at weeks 8–24 (n = 60) [21].
The proportion of patients achieving a PASI 90 response at week 24 (primary endpoint) was 81% with risankizumab versus 10% with fumaric acid esters (p < 0.001) [21]. Similar results were seen for PASI 50 (100 vs 53%), PASI 75 (98 vs 33%), PASI 100 (50 vs 5%), sPGA 0 or 1 (93 vs 38%), sPGA 0 (52 vs 5%) and DLQI 0 or 1 (67 vs 10%) response rates at week 24 (all p < 0.001) [21].
5 Tolerability of Risankizumab
Subcutaneous risankizumab was generally well tolerated in patients with moderate to severe plaque psoriasis. In a pooled analysis of 16-week data (n = 2232) from the phase II dose-ranging trial and the four pivotal phase III trials discussed in Sect. 4.1 (i.e. UltIMMa-1, UltIMMa-2, IMMvent and IMMhance), treatment-emergent AEs (TEAEs) were reported in 49, 52, 57 and 48% of patients receiving risankizumab, ustekinumab, adalimumab and placebo, respectively [28]. The most common (incidence ≥ 10%) adverse drug reaction with risankizumab was upper respiratory infection (URTI; 13%) [6, 8]. Serious TEAEs occurred in 2, 5, 3 and 4% of risankizumab, ustekinumab, adalimumab and placebo recipients, respectively [28]. The most common serious TEAEs with risankizumab were cellulitis (0.2%) and squamous cell carcinoma (0.2%). TEAEs leading to discontinuation occurred in 0.7, 1.3, 2.0 and 3.0% of patients receiving risankizumab, ustekinumab, adalimumab and placebo, respectively [28].
In the supportive head-to-head comparison trials discussed in Sect. 4.3.2 and Sect. 4.3.3, the safety profile of risankizumab was similar to that of secukinumab (with no new safety concerns identified) [20] and more favourable than that of fumaric acid esters [21].
The longer-term tolerability profile of risankizumab was generally consistent with that observed over the shorter term. In a pooled analysis of all risankizumab data (n = 2673) from 11 completed and ongoing phase I–III trials [including UltIMMa-1, UltIMMa-2, IMMvent, IMMhance, LIMMitless (Sect. 4.2.1), SustaIMM (Sect. 4.3.1) and the head-to-head comparison with fumaric acid esters (Sect. 4.3.3)], the exposure-adjusted incidence rate (EAIR) of TEAEs did not increase over time [29]. With long-term (up to 58.6 months) exposure [5582.8 patient-years (PY)], the EAIR of overall TEAEs with risankizumab was 184.3 per 100 PY. The corresponding EAIR of serious TEAEs was 8.6 per 100 PY [29]. In the open-label extension (LIMMitless), the rates of AEs over 84 weeks in patients who switched from ustekinumab [25] or adalimumab [26] to risankizumab were generally comparable to those previously reported. Risankizumab was well tolerated through > 2.5 years of continuous exposure, with no new safety signals [27].
5.1 Adverse Events of Special Interest
Treatment with risankizumab may increase the risk of infections [6,7,8]. In the short-term pooled analysis, serious infections were reported in 0.4, 1.7, 0.3 and 0.3% of risankizumab, ustekinumab, adalimumab and placebo recipients, respectively [28]. The most common infections with risankizumab were viral URTI (6%), URTI (4%) and sinusitis (1%), all of which were of mild to moderate severity [28]. The EAIR of infection did not increase with long-term risankizumab exposure (90.8 per 100 PY through week 16 and 61.8 per 100 PY up to 58.6 months) [29]. The EAIR of serious infections (most commonly sepsis, cellulitis and pneumonia) remained stable over time (1.7 per 100 PY through week 16 and 1.4 per 100 PY up to 58.6 months) [29]. Caution is advised when using risankizumab in patients with chronic infections, a history of recurrent infection or known risk factors for infection [6, 8]. Treatment with risankizumab should not be initiated in patients with any clinically important active infection until the infection resolves or is adequately treated. Patients who develop an infection while receiving risankizumab should be closely monitored [6, 8].
In the pooled analyses, there were no cases of TB (including reactivation of latent TB) [29]. Of the patients in IMMhance with latent TB who did not receive TB prophylaxis during the trial (n = 31), none developed active TB [8]. Nevertheless, patients should be evaluated for TB infection prior to initiating treatment with risankizumab, and should be monitored for symptoms of active TB during treatment [6, 8]. Risankizumab should not be given to patients with active TB [6].
In the pooled analyses, the EAIRs for a number of other AEs of special interest were low and stable over time, namely malignant tumours excluding non-melanoma skin cancer (0.7 per 100 PY through week 16 and 0.6 per 100 PY up to 58.6 months), adjudicated major adverse cardiac events (MACE; 0.2 per 100 PY through week 16 and 0.4 per 100 PY up to 58.6 months), depression (1.0 per 100 PY through week 16 and 0.8 per 100 PY up to 58.6 months) and suicidal ideation and behaviour (0.5 per 100 PY through week 16 and < 0.1 per 100 PY up to 58.6 months) [29].
There were no reports of serious hypersensitivity in the short-term pooled analysis [29]. With long-term exposure (up to 58.6 months), the EAIR of serious hypersensitivity was < 0.1 per 100 PY. All cases of hypersensitivity were considered to be unrelated to risankizumab and did not lead to discontinuation of treatment [29]. Serious hypersensitivity reactions to risankizumab should be treated with appropriate therapy, and the drug should be discontinued immediately [7, 8]. Through week 16, injection-site reactions (ISRs) were reported in 2, 4, 6 and 1% of risankizumab, ustekinumab, adalimumab and placebo recipients, respectively [28]. The incidence of ISRs with risankizumab remained low over the longer term (4%), with the most common (> 2 patients) being injection-site erythema, reaction, pain, pruritus, haematoma, swelling, haemorrhage, bruising and induration [29]. All ISRs were of mild to moderate severity, and none led to risankizumab discontinuation [29].
Like all therapeutic proteins, risankizumab has the potential for immunogenicity [6, 8]. Among patients treated with risankizumab 150 mg for up to 52 weeks in psoriasis clinical trials (n = 1079), treatment-emergent anti-drug antibodies (ADAs) and neutralizing antibodies were detected in 263 (24%) and 150 (14%) patients. In most cases, the emergence of ADAs was not associated with changes in clinical response or safety. High antibody titres in ≈ 1% of risankizumab-treated patients were associated with a reduced clinical response [6, 8]. The incidence of ISRs was 3% in ADA-positive patients versus 1% in ADA-negative patients through week 16, with corresponding rates of 5 versus 3% at > 52 weeks [8].
6 Dosage and Administration of Risankizumab
Risankizumab is approved in multiple countries, including the USA [6] and those of the EU [8], for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy (or phototherapy [6]). In Japan, risankizumab is approved for the treatment of psoriasis in adults who have an inadequate response to conventional therapies [7]. The recommended dosage of risankizumab is 150 mg (two 75 mg subcutaneous injections) administered at weeks 0 and 4 and every 12 weeks thereafter [6,7,8]. Discontinuation of risankizumab should be considered if no response is seen after 16 weeks of treatment; patients with an initial partial response may subsequently improve with continued treatment beyond 16 weeks [8].
The efficacy and tolerability of risankizumab in paediatric patients has not been established [6,7,8]. There are limited data regarding the use of risankizumab in pregnant women [6,7,8]; however, it is preferable to avoid its use during pregnancy and effective contraception should be used during and for ≥ 21 weeks after risankizumab treatment in women of childbearing potential [8]. It is unknown whether risankizumab is excreted in human milk [6,7,8]. The benefits of breastfeeding for the infant and the clinical benefits of risankizumab for the mother should be considered [6, 8]. Local prescribing information should be consulted for detailed information regarding drug interactions, warnings and precautions, and use in special patient populations.
7 Place of Risankizumab in the Management of Moderate to Severe Plaque Psoriasis
In the USA [2], the UK [30] and Japan [31], currently available biological agents for the treatment of plaque psoriasis include the TNF-α inhibitors infliximab, adalimumab, etanercept (US and UK only) and certolizumab pegol (US and UK only); the IL-12/23 inhibitor ustekinumab; the IL-17 inhibitors secukinumab, ixekizumab and brodalumab; and the IL-23 inhibitors guselkumab, tildrakizumab (US and UK only) and risankizumab [2, 31]. European S3 guidelines published prior to the EU approval of risankizumab strongly recommend adalimumab, etanercept, infliximab, ustekinumab and secukinumab for the second-line treatment of plaque psoriasis [3]. The UK National Institute for Health and Care Excellence (NICE) recommends risankizumab as an option for the treatment of adults with severe plaque psoriasis who have not responded to other systemic treatments or in whom these agents are contraindicated or not tolerated [32].
In clinical trials, subcutaneous risankizumab was more effective than placebo, ustekinumab, adalimumab, secukinumab and fumaric acid esters in reducing the severity and extent of moderate to severe plaque psoriasis (Sect. 4). In four pivotal phase III trials (UltIMMa-1, UltIMMa-2, IMMvent and IMMhance) in adults with moderate to severe plaque psoriasis, PASI 90 and sPGA 0 or 1 response rates at week 16 were significantly higher with risankizumab than with placebo, ustekinumab and adalimumab (Sect. 4.1), with these benefits maintained over the longer term (Sect. 4.2). In an ongoing open-label extension, risankizumab was associated with durable and improved efficacy after switching from ustekinumab or adalimumab, as well as durable maintenance of efficacy through ≈ 2.5 years of continuous exposure (Sect. 4.2.1). Results in Japanese patients were reflective of those seen in the pivotal phase III trials (Sect. 4.3.1).
PASI 75 has previously been the benchmark for success when treating patients with moderate to severe plaque psoriasis [16]. However, a better understanding of the pathogenesis of psoriasis and the advent of new, improved biological agents has led to the use of more rigorous treatment targets [15, 16, 33]. Complete or near complete skin clearance is achievable for many patients receiving newer biological agents, with PASI 90 or PASI 100 now proposed as the new standards for optimal treatment response [15, 16, 33]. Notably, PASI 90 was used as a primary endpoint in all risankizumab clinical trials, with many patients also achieving a PASI 100 response (Sect. 4). Even residual disease can have a negative impact on HR-QOL in patients with psoriasis [34]. Therefore, the ability of a psoriasis treatment to attain the more rigorous treatment target of complete or near complete skin clearance is expected to result in improved HR-QOL [15, 16]. Indeed, in the pivotal phase III trials of risankizumab, improvements in skin symptoms were accompanied by sustained improvements in HR-QOL, as assessed by DLQI, PSS and WLQ scores (Sect. 4).
In patients with psoriasis, missed doses are a common occurrence in clinical practice [17]. Therefore, the durability of response without treatment and the ability to regain a response after relapse are important treatment considerations [17]. In IMMhance, efficacy outcomes were more favourable in patients receiving continuous risankizumab than in those re-randomized to placebo (i.e. withdrawal) at week 28 (Sect. 4.2). However, in patients re-randomized to placebo, the median times to loss of PASI 90 response and relapse were 30 weeks and 42 weeks, respectively, demonstrating the durability of the risankizumab response following treatment withdrawal [17]. It should also be noted that the majority of patients who relapsed during treatment withdrawal regained a response following re-treatment with risankizumab (Sect. 4.2).
Risankizumab was generally well tolerated in clinical trials, with URTI being the most commonly reported adverse reaction (Sect. 5). No new safety signals were identified through > 2.5 years of continuous exposure, with the longer-term tolerability profile of risankizumab in the pivotal phase III trials during this period consistent with that observed during the shorter term (Sect. 5). The rates of AEs of special interest (severe infections, malignant tumours, MACE, depression, suicidal ideation and behaviour, serious hypersensitivity and ISRs) with risankizumab were generally low and stable over time (Sect. 5.1).
A number of systematic reviews and network meta-analyses have indirectly compared the efficacy and safety of biological agents in patients with plaque psoriasis [35,36,37,38,39,40,41,42]. Generally, IL-17 inhibitors (e.g. brodalumab, ixekizumab) and IL-23 inhibitors (e.g. risankizumab, guselkumab, tildrakizumab) were associated with greater clinical benefits than TNF-α inhibitors and the IL-12/23 inhibitor ustekinumab. Some analyses have demonstrated apparent differences in efficacy and/or safety between IL-17 and IL-23 inhibitors [35,36,37,38,39,40,41,42]. However, results of these indirect comparisons should be treated with caution. A randomized, multicentre, phase III trial is currently underway to compare the efficacy and safety of risankizumab with those of methotrexate (NCT03219437) in patients with moderate to severe plaque psoriasis [43]. Results from this trial, as well as final results from the LIMMitless open-label extension, are awaited with interest.
According to current guidelines for psoriasis, the choice of treatment is individualized based on treatment-related factors (i.e. efficacy and safety) and patient-related factors (e.g. disease severity, comorbidities, personal preferences), as well as drug-related factors such as the route and frequency of administration [2, 30, 31]. All of the newer biological agents approved for plaque psoriasis are administered via subcutaneous injection. Like ustekinumab and tildrakizumab, risankizumab has a relatively favourable dosing profile, with maintenance doses administered once every 12 weeks (Sect. 6) as compared with once every 8 weeks for guselkumab, once every 4 weeks for secukinumab and ixekizumab, and once every 2 weeks for brodalumab [42]. This less frequent dosing regimen may be preferred by some patients [2], potentially resulting in better treatment adherence. However, despite its convenience for the patient, the 12-week dosing interval may also be associated with fluctuations in efficacy between doses [15]. Of note, efficacy between doses in UltIMMa-1 and UltIMMa-2 was better maintained with risankizumab than with ustekinumab, providing support for the appropriateness of the risankizumab dosing schedule of once every 12 weeks [15].
Cost is another factor that can influence the choice of treatment [2]. In the NICE guidance, the total costs associated with risankizumab were reported to be similar to or lower than those associated with guselkumab [32]. A modelled pharmacoeconomic study from a Japanese health system perspective suggests that risankizumab is cost-effective compared with other biological agents (i.e. adalimumab, infliximab, ustekinumab, secukinumab, brodalumab, ixekizumab and guselkumab) in patients with moderate to severe plaque psoriasis, based on typical willingness-to-pay thresholds of ¥5–6.7 million per quality-adjusted life year [44]. Results of another Markov model from the perspective of a public payer in Poland suggest that risankizumab is more cost-effective than ustekinumab for the treatment of severe psoriasis over a 5-year period [45]. In the Brazilian private healthcare system, the cost per responder (based on PASI 75, 90 and 100) across three time horizons (12–16 weeks, 1 year and 2 years) was lowest for risankizumab compared with other biological therapies (i.e. adalimumab, etanercept, guselkumab, infliximab, ixekizumab, secukinumab and ustekinumab) [46]. Further robust pharmacoeconomic data would be beneficial.
In conclusion, risankizumab is an effective and generally well-tolerated treatment that represents a useful new addition to the options available for the management of moderate to severe plaque psoriasis.
Data Selection Risankizumab: 285 records identified
Duplicates removed | 148 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 82 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 9 |
Cited efficacy/tolerability articles | 18 |
Cited articles not efficacy/tolerability | 28 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were Skyrizi, risankizumab, plaque psoriasis. Records were limited to those in English language. Searches last updated 22 June 2020 | |
Change history
02 September 2020
The original article can be found online.
References
Gisondi P, Geat D, Pizzolato M, et al. State of the art and pharmacological pipeline of biologics for chronic plaque psoriasis. Curr Opin Pharmacol. 2019;46:90–9.
Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80(4):1029–72.
Nast A, Gisondi P, Ormerod AD, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris–update 2015–short version–EDF in cooperation with EADV and IPC. J Eur Acad Dermatol Venereol. 2015;29(12):2277–94.
Menter A, Gelfand JM, Connor C, et al. Joint AAD-NPF guidelines of care for the management of psoriasis with systemic non-biological therapies. J Am Acad Dermatol. 2020;82(6):1445–86.
Torres T. Selective interleukin-23 p19 inhibition: another game changer in psoriasis? Focus on risankizumab. Drugs. 2017;77(14):1493–503.
AbbVie. Skyrizi® (risankizumab-rzaa) injection, for subcutaneous use: US prescribing information. 2019. http://www.rxabbvie.com. Accessed 29 Jun 2020.
AbbVie. Risankizumab (Skyrizi): Japanese prescribing information. 2019. http://www.pmda.go.jp. Accessed 29 Jun 2020.
European Medicines Agency. Skyrizi (risankizumab): summary of product characteristics. 2019. http://www.ema.europa.eu/en/medicines/human/EPAR/skyrizi. Accessed 29 Jun 2020.
Zhou L, Wang Y, Wan Q, et al. IL-23 antibodies in psoriasis - a non-clinical perspective [abstract plus poster]. In: 49th Annual Meeting of the European Society for Dermatological Research. 2019.
Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136(1):116–24.
Suleiman AA, Minocha M, Khatri A, et al. Population pharmacokinetics of risankizumab in healthy volunteers and subjects with moderate to severe plaque psoriasis: integrated analyses of phase I-III clinical trials. Clin Pharmacokinet. 2019;58(10):1309–21.
Khatri A, Suleiman AA, Polepally AR, et al. Exposure-response relationships for efficacy and safety of risankizumab in phase II and III trials in psoriasis patients. Clin Pharmacol Ther. 2019;107(2):378–87.
Khatri A, Eckert D, Oberoi R, et al. Pharmacokinetics of risankizumab in Asian healthy subjects and patients with moderate to severe plaque psoriasis, generalized pustular psoriasis, and erythrodermic psoriasis. J Clin Pharmacol. 2019;59(12):1656–68.
Khatri A, Cheng L, Camez A, et al. Lack of effect of 12-week treatment with risankizumab on the pharmacokinetics of cytochrome P450 probe substrates in patients with moderate to severe chronic plaque psoriasis. Clin Pharmacokinet. 2019;58(6):805–14.
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–61.
Reich K, Gooderham M, Thaci D, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. 2019;394(10198):576–86.
Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156(6):649–58.
Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376(16):1551–60.
Ohtsuki M, Fujita H, Watanabe M, et al. Efficacy and safety of risankizumab in Japanese patients with moderate to severe plaque psoriasis: results from the SustaIMM phase 2/3 trial. J Dermatol. 2019;46(8):686–94.
Warren RB, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): results from a phase 3, randomised, open-label, efficacy assessor-blinded clinical trial. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19341.
Thaci D, Eyerich K, Pinter A, et al. Direct comparison of risankizumab and fumaric acid esters in patients with moderate-to-severe plaque psoriasis who were naive to systemic therapy [abstract no. 718 plus poster]. In: 28th European Academy of Dermatology and Venerology Congress. 2019.
Gooderham M, Lebwohl M, Gordon K, et al. High and durable clearance through 52 weeks of risankizumab treatment in patients with moderate-to-severe plaque psoriasis [abstract no. P030]. J Eur Acad Dermatol Venereol. 2019;33(Suppl 3):20.
Strober B, Menter A, Leonardi C, et al. Efficacy of risankizumab in patients with moderate-to-severe plaque psoriasis by baseline demographics, disease characteristics and prior biologic therapy: an integrated analysis of the phase III UltIMMa-1 and UltIMMa-2 studies. J Eur Acad Dermatol Venereol. 2020. https://doi.org/10.1111/jdv.16521.
Ryan C, Crowley J, Valdecantos W, et al. Efficacy of switching to risankizumab compared with continued adalimumab treatment in patients with moderate-to-severe plaque psoriasis [abstract no. 8110]. J Am Acad Dermatol. 2019;81(4 Suppl 1):AB55.
Strober B, Eyerich K, Hong HC-H, et al. Long-term efficacy and safety of switching from ustekinumab to risankizumab: results from the open-label extension LIMMitless [abstract no. P1714 plus poster]. In: 28th European Academy of Dermatology and Venereology. 2019.
Reich K, Ryan C, Crowley JJ, et al. Long-term efficacy and safety of switching from adalimumab to risankizumab: results from the open-label extension LIMMitless [abstract no. P1713 plus poster]. In: 28th European Academy of Dermatology and Venereology. 2019.
Papp K, Lebwohl M, Ohtshuki M, et al. Long-term efficacy and safety of continuous Q12W risankizumab: results from the open-label extension study LIMMitless [abstract no. 14027 plus poster]. In: 78th Annual Meeting of the American Academy of Dermatology. 2020.
Bachelez H, Gordon KB, Blauvelt A, et al. Safety of risankizumab in patients with moderate to severe psoriasis: analysis of pooled clinical trial data [abstract no. P1764 plus poster]. In: 28th European Academy of Dermatology and Venereology. 2019.
Gordon KB, Bachelez H, Blauvelt A, et al. Pooled long-term safety analysis of risankizumab in patients with moderate to severe psoriasis [abstract no. 16332 plus poster]. In: 78th Annual Meeting of the American Academy of Dermatology. 2020.
Smith CH, Yiu ZZ, Bale T, et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2020—a rapid update. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19039.
Saeki H, Terui T, Morita A, et al. Japanese guidance for use of biologics for psoriasis (the 2019 version). J Dermatol. 2020;47(3):201–22.
National Institute for Health and Care Excellence. Risankizumab for treating moderate to severe plaque psoriasis. 2019. http://www.nice.org.uk. Accessed 29 Jun 2020.
Hsu SH, Tsai TF. Evolution of the inclusion/exclusion criteria and primary endpoints in pivotal trials of biologics and small oral molecules for the treatment of psoriasis. Expert Rev Clin Pharmacol. 2020;13(3):211–32.
Takeshita J, Callis Duffin K, Shin DB, et al. Patient-reported outcomes for psoriasis patients with clear versus almost clear skin in the clinical setting. J Am Acad Dermatol. 2014;71(4):633–41.
Armstrong AW, Puig L, Joshi A, et al. Comparison of biologics and oral treatments for plaque psoriasis: a meta-analysis. JAMA Dermatol. 2020;156(3):258–69.
Sawyer LM, Malottki K, Sabry-Grant C, et al. Assessing the relative efficacy of interleukin-17 and interleukin-23 targeted treatments for moderate-to-severe plaque psoriasis: a systematic review and network meta-analysis of PASI response. PLoS One. 2019;14(8):e0220868.
Xu S, Zhang X, Pan M, et al. Treatment of plaque psoriasis with IL-23p19 blockers: a systematic review and meta-analysis. Int Immunopharmacol. 2019;75:105841.
Witjes H, Khatri A, Diderichsen PM, et al. Meta-analyses of clinical efficacy of risankizumab and adalimumab in chronic plaque psoriasis: supporting evidence of risankizumab superiority. Clin Pharmacol Ther. 2019;107(2):435–42.
Yasmeen N, Sawyer LM, Malottki K, et al. Targeted therapies for patients with moderate-to-severe psoriasis: a systematic review and network meta-analysis of PASI response at 1 year. J Dermatol Treat. 2020. https://doi.org/10.1080/09546634.2020.1743811.
Warren RB, See K, Burge R, et al. Rapid response of biologic treatments of moderate-to-severe plaque psoriasis: a comprehensive investigation using Bayesian and Frequentist network meta-analyses. Dermatol Ther. 2020;10(1):73–86.
Bai F, Li GG, Liu Q, et al. Short-term efficacy and safety of IL-17, IL-12/23, and IL-23 inhibitors brodalumab, secukinumab, ixekizumab, ustekinumab, guselkumab, tildrakizumab, and risankizumab for the treatment of moderate to severe plaque psoriasis: a systematic review and network meta-analysis of randomized controlled trials. J Immunol Res. 2019. https://doi.org/10.1155/2019/2546161.
Sbidian E, Chaimani A, Afach S, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev. 2020. https://doi.org/10.1002/14651858.CD011535.pub3.
US National Institutes of Health. 2020. http://www.clinicaltrials.gov. Accessed 29 Jun 2020.
Saeki H, Ishii K, Joshi A, et al. An economic evaluation of risankizumab versus other biologic treatments of moderate to severe plaque psoriasis in Japan. J Dermatol. 2020. https://doi.org/10.1080/09546634.2020.1744505.
Dzik M, Nadzieja-Koziol A, Owczarek W. Cost-effectiveness comparison of risankizumab and ustekinumab for the treatment of severe psoriasis [abstract no. PB131]. Value Health. 2019;22(Suppl 3):S423.
de Carvalho AVE, Vieira Duarte G, Ianhez A, et al. Biologic therapies for moderate-to-severe psoriasis: cost per responder analysis considering private healthcare system in Brazil [abstract no PBI18]. Value Health. 2020;23(Suppl 1):S17.
Acknowledgements
During the peer review process, the manufacturer of risankizumab was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Hannah Blair is a salaried employee of Adis International Ltd/Springer Nature, is responsible for the article content and declares no relevant conflicts of interest.
Additional information
The original version of this article was revised due to a retrospective Open Access request.
Enhanced material for this Adis Drug Evaluation can be found at https://doi.org/10.6084/m9.figshare.12580334.
The manuscript was reviewed by: S. R. Feldman, Department of Dermatology, Pathology & Public Health Sciences, Wake Forest University School of Medicine, Winston-Salem, NC, USA; L. Puig, Department of Dermatology, Hospital de la Santa Creu I Sant Pau, Barcelona, Spain; T. Torres, Department of Dermatology, Centro Hospitalar Universitario do Porto, Porto, Portugal.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Blair, H.A. Risankizumab: A Review in Moderate to Severe Plaque Psoriasis. Drugs 80, 1235–1245 (2020). https://doi.org/10.1007/s40265-020-01357-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01357-1