
Abstract
Background and Objective
Risankizumab is an anti-interleukin (IL)-23 monoclonal antibody being developed for treatment of moderate to severe plaque psoriasis. This study provided a comprehensive analysis of risankizumab pharmacokinetics in healthy subjects and patients with plaque psoriasis using data across phase I–III clinical trials.
Methods
Plasma pharmacokinetic data from 1899 subjects, including 13,123 observations, who received single or multiple intravenous or subcutaneous doses of risankizumab (0.01–5 mg/kg intravenous [IV], 200–1200 mg IV, 0.25–1 mg/kg subcutaneous [SC], and 18–300 mg SC) across the phase I–III clinical program were analyzed using a non-linear mixed-effects modeling approach. The developed model was qualified and the clinical relevance of covariates statistically correlated with risankizumab clearance (CL) was evaluated using simulation analyses.
Results
Risankizumab pharmacokinetics were best described using a two-compartment model with first-order absorption and elimination. Risankizumab CL, volume of distribution at steady state (Vss), and terminal-phase elimination half-life (t½) were estimated to be approximately 0.31 L/day, 11.2 L, and 28 days, respectively, for a typical 90 kg psoriatic subject, approaching steady-state plasma exposures by week 16 of dosing. Absolute SC bioavailability (F) was 89%. Bodyweight, anti-drug antibody (ADA) titers ≥ 128 (detected in only 1% of ADA-evaluable subjects in phase III studies), baseline serum albumin, high-sensitivity C-reactive protein (hs-CRP), and serum creatinine were statistically correlated with risankizumab CL; however, they had no clinically relevant impact on exposure.
Conclusion
Risankizumab is characterized by dose-proportional, bi-exponential disposition with no difference in exposure between healthy subjects and patients with psoriasis. None of the covariates identified as being statistically correlated with risankizumab CL has a clinically meaningful impact on its exposure with the proposed psoriasis clinical regimen of 150 mg administered SC at weeks 0 and 4, and every 12 weeks thereafter.
ClinicalTrials.gov Identifiers
NCT01577550, NCT02054481, NCT02596217, NCT02684370, NCT02672852, NCT02684357, NCT02694523.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Risankizumab displayed linear pharmacokinetics across the doses evaluated in the phase I–III clinical trials in psoriasis, and a two-compartment model with first-order absorption and elimination best described its pharmacokinetics. |
Body weight, anti-drug antibody, and baseline levels of albumin, high-sensitivity C-reactive protein, and serum creatinine were identified to be statistically correlated with risankizumab clearance; however, none of these covariates had a clinically meaningful impact on risankizumab exposures with the proposed psoriasis clinical regimen. |
Risankizumab pharmacokinetics were similar between healthy subjects and patients with plaque psoriasis. |
1 Introduction
Plaque psoriasis is a chronic immune-mediated skin condition with a prevalence of approximately 3% in the USA [1]. Psoriasis is characterized by marked inflammation and increased keratinocyte proliferation, leading to pruritic, often painful, demarcated, scaly, and erythematous skin lesions, which negatively impacts the quality of life of psoriatic patients. Furthermore, plaque psoriasis can be associated with other co-morbidities, including psoriatic arthritis, uveitis, and cardiometabolic diseases, among others [2].
The proinflammatory cytokine interleukin (IL)-23 has been demonstrated to play a crucial role in the pathogenesis of several autoimmune diseases, including psoriasis. IL-23 activates the T helper (Th) 17 cells, which in turn can stimulate and promote chronic tissue inflammation via various proinflammatory cytokines, including IL–17A [3]. As such, several therapies targeting IL-17 or IL-23 have been recently developed to block the IL-23/IL-17A axis for the treatment of several autoimmune disorders. Several anti-IL-23-antibodies (e.g., ustekinumab, guselkumab, and tildrakizumab) have demonstrated efficacy in treatment of plaque psoriasis [4,5,6].
Risankizumab is a humanized IgG1 monoclonal antibody that selectively binds with picomolar affinity for the p19 subunit of IL-23, preventing IL-23 from binding to its receptor [7]. Several risankizumab phase III clinical trials have now been completed and demonstrated rapid and durable clinical improvement in patients with moderate to severe plaque psoriasis, with approximately 75% of patients experiencing near-complete or complete clearance of skin lesions (i.e., Psoriasis Area Severity Index [PASI]90/PASI100 response) at week 16 with a convenient regimen of a 150 mg subcutaneous (SC) dose administered at weeks 0 and 4 and every 12 weeks thereafter [8,9,10].
Previously, we have characterized the population pharmacokinetics of risankizumab using a relatively small dataset consisting of 157 patients with moderate to severe plaque psoriasis and 115 patients with Crohn’s disease across phase I and II studies [11]. In the present analyses, pharmacokinetic data for risankizumab from healthy volunteers (n = 67) and patients with moderate to severe plaque psoriasis (n = 1844) across two phase I, one phase II, and four phase III clinical trials were analyzed simultaneously using a non-linear mixed-effects modeling approach. The objectives of the current analyses using this larger dataset were to characterize risankizumab pharmacokinetics in the moderate to severe plaque psoriasis population, evaluate the differences (if any) in risankizumab pharmacokinetics between healthy volunteers and psoriatic patients, and evaluate the impact of the relevant patient-specific covariates on risankizumab systemic exposures to inform dosing recommendations. The larger number of subjects included in the analyses and the longer duration of treatment in phase III clinical trials allows the potential impact of covariates on exposure to be more robustly assessed.
2 Methods
2.1 Study Population
Data from healthy volunteers and patients recruited in seven studies (one phase I study in healthy male volunteers, and six phase I–III studies in male and female patients with moderate to severe plaque psoriasis), who received at least a single dose of risankizumab, were included in the analyses (Table 1). Subjects recruited in these studies received single intravenous (IV) or SC dosing, or multiple SC dosing across the dose range of 0.01–5 mg/kg IV, 200–1200 mg IV, 0.25–1 mg/kg SC, and 18–300 mg SC. For SC administrations in patients, skin areas affected by psoriasis were not utilized. All studies were conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocols were approved by the ethics committee or institutional review board at each site, and all participants provided written informed consent.
2.2 Bioanalyses
Blood samples for determination of risankizumab plasma concentrations, anti-drug antibody (ADA), and neutralizing antibody (NAb) assessments were obtained by venipuncture at the sampling timepoints shown in Table 1. The actual blood sample collection times were used in the population pharmacokinetic analyses. Plasma concentrations of free risankizumab, presence and titer of ADA, and presence of NAb were measured using validated assays as previously described [11, 12].
Briefly, a validated enzyme-linked immunosorbent assay (ELISA) method was used to quantitatively determine the free risankizumab concentration in plasma within a nominal range of 5–100 ng/mL and with a lower limit of quantitation (LLOQ) of 5 ng/mL with inter-run precision (% coefficient of variation [%CV]) ≤ 5% across studies. Plasma samples above the upper limit of quantitation were diluted and re-assayed. Testing for ADA was multi-tiered, with ADA titers being determined by serial dilution for subjects confirmed to be ADA positive. A titer-based acid dissociation bridging electrochemiluminescence (ECL) immunoassay with a psoriasis-specific cut-point was developed for the detection of antibodies against risankizumab in human plasma. In addition, a cell-based assay for assessment of NAb to risankizumab was developed and a psoriasis specific cut-point with a 1% false-positive rate was established.
For subjects confirmed as ADA positive, and at the first dilution in the titer assay at which the ADAs were no longer detectable, titers were reported as < 1 and this was imputed in the analysis dataset with a value of 0.5 for testing the titer as a continuous covariate.
2.3 Population Pharmacokinetic Analyses
2.3.1 Software
The analysis utilized a non-linear mixed-effects modeling approach using NONMEM® version 7.4.1 (ICON Development Solutions, Ellicott City, MD, USA) compiled with the GNU Fortran compiler, version 4.8.3. Perl Speaks NONMEM (PsN; version 4.6.0; Uppsala University, Uppsala, Sweden [13]) and R (version 3.4.0; R Foundation for Statistical Computing, Vienna, Austria) were used to assist with model development, evaluation, and simulation analyses.
2.3.2 Model Development
Model parameters were estimated using the first-order conditional estimation (FOCE) algorithm with interaction between inter-individual variability (IIV) and residual variability (FOCE with η–ε INTERACTION) as implemented in NONMEM®. The structural, IIV, residual, and covariate models were developed in a stepwise manner. For model selection, the competing nested models were compared using the objective function value (OFV), where the difference in the OFV can serve as a likelihood ratio test approximately following a chi-squared distribution. Parameters of an alternative nested model were included if the fit improved significantly with p < 0.01.
One‐, two‐, and three‐compartment pharmacokinetic models, in addition to linear and non-linear models for elimination, were tested for the best description of risankizumab disposition.
Except for the IIV on bioavailability (F) (described below), the IIV for other pharmacokinetic parameters was assumed to follow log-normal distributions and, hence, exponential random effects models were used for their characterization (Eq. 1).
where θi,k is the estimate of the kth parameter for the ith subject, θk is the population estimate of the kth parameter, and ηi,k represents the individual deviation from θk, where ηi,k is assumed to arise from a normal distribution with a mean of 0 and a variance of ωk2: η ~ N (0, ωk2). Inclusion of IIV on F was evaluated by estimating an additive model on the logit scale to ensure Fi is bound between 0 and 1 (Eq. 2).
Additive, proportional residual error models, and a combination of both, as shown in Eqs. (3–5), respectively, were evaluated for the best description of the residual error.
where Cij is the observed plasma concentration of the ith individual at time j, \( \hat{C}_{ij} \) is the corresponding model-predicted concentration, and ε1,ij and ε2,ij represent the proportional and additive residual random error, respectively, in the residual error models. Residual random errors were assumed to arise from independent normal distributions with a mean of 0 and a variance of σn2 where n represents 1 (proportional) or 2 (additive) model structures.
Subsequently, the effects of covariates, including demographics (age, body weight, sex, race, country), laboratory values measured at baseline (serum albumin, creatinine, aspartate aminotransferase, alanine transaminase, total bilirubin, high-sensitivity C-reactive protein [hs-CRP]), estimated creatinine clearance at baseline, disease condition (healthy vs. psoriasis, baseline PASI score), immunogenicity of risankizumab (ADA titer, ADA- and NAb-positive/negative status irrespective of time, and time-dependent ADA and NAb status), and the difference in risankizumab drug supply between phase I and II studies versus phase III studies (changes related to cell bank and manufacturing batch scalability) were evaluated on model parameters as appropriate using the forward inclusion (α = 0.01) and backward elimination (α = 0.001) process implemented in PsN. Relationships between key model parameters and covariates were tested and included if significant in a multiplicative manner, as shown in Eq. (6).
where np is the number of continuous covariates, covi,p is the pth continuous covariate value in the ith subject, refp is the reference value for the pth covariate (median was used except for bodyweight where 70 kg was used as a reference), θk,p is the exponent estimate for the power model characterizing the effect of the pth continuous covariate on the kth parameter.
Similarly, nq is the number of categorical covariates, θk,q,y is the proportional difference estimate for the effect of the yth category of the qth categorical covariate on the kth parameter, where θk,q,y takes a value of 0 for the most frequent category. F was estimated separately to ascribe the difference in the risankizumab drug supplies between phase I–II studies versus phase III studies.
The effect of the time course of ADA titers (ADAeff) was evaluated on risankizumab clearance (CL) using two alternative parameterizations. First, the effect of the ADA titer was assessed using a power function (Eq. 7).
where titer is the ADA titer and θCL,ADA is the estimated exponent for the relationship between risankizumab CL and ADA titer normalized to 0.5 titer units (see Sect. 2.2). In the second parameterization, given that the ADAeff (as shown in Eq. 7) was statistically significant, various ADA titer threshold values (Titerthreshold) were evaluated using a proportional difference model to investigate if a better fit is obtained if the effect of ADA on risankizumab CL is included only when the ADA titer reaches a certain threshold value (Titerthreshold; Eq. 8)
where θCL,Titer,threshold is the estimate of the proportional difference in CL once the ADA titer reaches the Titerthreshold.
Finally, covariance between CL, central volume of distribution (Vc), F, and absorption rate constant (ka) were evaluated, and was reduced in a stepwise manner if the model was deemed unstable.
Once developed, the final model was used to predict the risankizumab plasma exposures (maximum concentration [Cmax], area under the concentration–time curve [AUC], and trough concentration [Ctrough]) in subjects with moderate to severe plaque psoriasis who received the phase III clinical regimen of 150 mg SC at weeks 0 and 4 and every 12 weeks thereafter using their actual dosing history and empirical Bayesian estimates of pharmacokinetic parameters.
2.3.3 Model Evaluation
Model evaluation was performed using goodness-of-fit plots, visual predictive checks (VPCs), and bootstrap analyses. For the VPCs, 1000 simulated replicates of the observed dataset for the same study designs were generated using NONMEM®. The median and 5th and 95th percentiles of the simulated data representing the 90% prediction intervals along with the 95% confidence intervals (CIs) of the median and 5th and 95th percentiles were calculated and compared graphically with the observed data. For the bootstrap, 1000 replicate datasets were generated from the original dataset by randomly sampling subjects with replacement. The model was fit to each of these replicated datasets, and the medians and corresponding non-parametric 95% CIs (2.5th to 97.5th percentiles) were constructed for each parameter based on the successfully converging runs and compared to the parameter estimates of the final model.
2.3.4 Simulation Analyses for Evaluating the Impact of Covariates
To evaluate the clinical relevance of the identified statistically significant covariates, simulations were carried out to compare risankizumab steady-state Cmax, and AUC over a dosing interval (AUCτ) for the risankizumab dosing regimen of 150 mg SC at weeks 0 and 4 and every 12 weeks thereafter in a subset of typical patients, or the test group, relative to the reference group. For body weight, patients weighing ≤ 100 kg were considered the reference group, while patients weighing > 100 kg were considered the test group. The 100 kg body weight was the same body weight cut-off utilized in risankizumab phase III efficacy subgroup analyses. It is also the cut-off utilized clinically for the step-dosing of ustekinumab, which was an active comparator in two of risankizumab phase III trials [4, 14]. For other continuous covariates, subjects within the 25th to 75th percentiles of the covariate distribution in the dataset served as the reference group, while subjects below the 25th percentile or above the 75th percentile were two separate test groups. For categorical covariates, subjects with the most frequent category were considered the reference group, and the other categories were the test groups.
Simulations for each of the identified covariates of interest were carried out separately while fixing other covariates to the reference value. To include parameter uncertainty in simulations, the fixed-effect parameters for each replicate were sampled from the variance–covariance matrix of the final pharmacokinetic model parameters, and the ETA distributions determined the individual subject pharmacokinetic parameters within a replicate. Cmax and AUCτ values over the interval between weeks 40 and 52 interval were calculated, and within each simulation replicate, the ratios of median Cmax and AUCτ values for each test group relative to the median Cmax and AUCτ of the reference group (normalized exposure ratio) were calculated. This process was repeated 200 times and the median of the normalized exposure ratios across the 200 replicates and the non-parametric 95% CIs (2.5th and 97.5th percentiles of the ratios) were calculated and summarized graphically using a forest plot.
3 Results
3.1 Data
A total of 15,660 risankizumab plasma concentration measurements from 1911 subjects (1844 psoriasis patients and 67 healthy subjects) were available for the analyses. After excluding measurements below the LLOQ, the majority of which were the measurements prior to the first risankizumab dose from all subjects as well as measurements during the long washout phase in single-dose studies, a total of 13,123 plasma concentration measurements from 1899 subjects were included in the analyses. Exclusion of samples below the LLOQ led to exclusion of 12 subjects from the analyses, among whom seven subjects had only risankizumab concentration measurements prior to the first risankizumab dose before dropping out from the trials.
The demographic and baseline characteristics of the subjects from whom risankizumab concentrations were available are summarized in Table 2. Overall, the population was predominantly male (71%), had a median age of 47 years (range 18–85 years), and had a median body weight of 87 kg (range 43–193 kg).
3.2 Population Pharmacokinetic Model
The two-compartment model, parameterized using the ADVAN4 TRANS4 subroutine in NONMEM® in terms of CL, Vc, peripheral volume of distribution (Vp), inter-compartmental clearance (Q), F, and ka best described risankizumab pharmacokinetics. Including IIV terms on CL, Vc, ka, and F with correlation between CL and Vc significantly improved the model fit (statistically significant decrease in OFV; p < 0.01). Residual variability was best described using a proportional error model.
Among the covariate relationships evaluated, risankizumab CL was found to have statistically significant correlation with body weight, baseline serum albumin, serum creatinine, hs-CRP, and ADA titer; risankizumab Vc and Vp were both found to have statistically significant correlation with body weight. Furthermore, different estimates for risankizumab F for the different drug supplies for phase I and II versus phase III studies significantly improved the model fit. A single variance was utilized for the IIV of SC bioavailability since the distribution of the IIV for phase I and II versus phase III was comparable after incorporation of this covariate as a fixed-effect parameter on SC bioavailability.
Presence of ADAs to risankizumab in patients with ADA titer values ≥ 128 (\( {\text{Titer}}_{\text{threshold}} = 128 \)) was found to affect risankizumab CL. The relationship was modeled in a time-varying fashion, with effect of ADAs on CL becoming detectable only when ADA titer levels are ≥ 128. Patients with ADA titer values of ≥ 128 were estimated to have an increase of risankizumab CL by 43%, on average; however, ADA titer values < 128 had no effects on risankizumab CL. Overall, a very small fraction of psoriasis subjects who received risankizumab in the phase II/III clinical trials (approximately 1% [6/598] of ADA-evaluable subjects who received the phase III clinical regimen over a 52-week time period in the UltIMMa-1 and UltIMMa-2 trials or approximately 1.5% [28/1807] of ADA-evaluable subjects who received any dose of risankizumab in all phase II/III trials) developed antibody titers ≥ 128. Other covariates such as age (18–85 years), sex, race (Asians vs. white plus others), and liver function markers (total bilirubin [3.0–57 µmol/L], alanine aminotransferase (ALT; 6.0–239 U/L], and aspartate aminotransferase [AST; 8.0–236 U/L]), and the emergence of NAbs did not correlate with risankizumab pharmacokinetic parameters.
The parameter estimates and their associated percentage relative standard errors from the final population pharmacokinetic model are provided in Table 3. For a typical 90 kg psoriasis patient, risankizumab plasma CL, Vc, Vp, volume of distribution at steady state (Vss), and elimination half-life (t½) values were estimated to be approximately 0.31 L/day, 6.52 L, 4.67 L, 11.2 L, and 28 days, respectively. The absolute SC bioavailability for the phase III clinical drug supply was 89%. The IIV for risankizumab CL, Vc, and ka was 24%, 34%, and 63%, respectively.
3.3 Model Evaluation
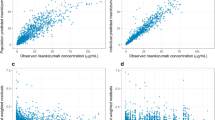
The model adequately described the data as demonstrated with the goodness-of-fit plots (Fig. 1) where population and individual predictions showed lack of any bias when plotted against observed data, and with no obvious trends in plots of conditional weighted residuals versus population predictions and time.

Goodness-of-fit plots for the final risankizumab population pharmacokinetic model. Diagnostic plots of the final risankizumab final pharmacokinetic model. a Observed versus individual predicted risankizumab plasma concentrations; b observed versus population predicted risankizumab plasma concentrations; c conditional weighted residuals versus population predicted risankizumab plasma concentrations; d conditional weighted residuals versus time since first risankizumab dose. Solid lines represent lines of identity in a and b and zero conditional residuals in c and d
Of 1000 replicates, 998 runs converged successfully in the bootstrap analyses. Based on the successful runs, the model parameters were precisely estimated as indicated by the 95% CIs, with negligible deviation from the original dataset estimates (Table 3). Furthermore, VPCs for combined data across phase III studies with similar design (studies UltIMMa-1 and UltIMMa-2) for subjects who received the clinical regimen of risankizumab 150 mg SC at weeks 0 and 4 and every 12 weeks thereafter demonstrate that the model was able to describe the central tendency and variability in the observed data well (Fig. 2).

Visual predictive checks across phase III studies in patients with psoriasis who received risankizumab 150 mg subcutaneously at weeks 0 and 4 and every 12 weeks thereafter using the final population pharmacokinetic model. The gray dots represent observed data, the lines represent observed median (solid black) and observed 5%/95% percentiles (dashed) encompassing the 90% prediction interval, and the shaded regions represent the 95% confidence intervals for the simulated median (purple) and simulated 5%/95% percentiles (blue)
3.4 Impact of Statistically Significant Covariates on Risankizumab Exposures
Based on the simulations using the developed model, subjects with body weight > 100 kg were predicted to have ~ 30% lower exposures (Cmax and AUC), on average, than subjects weighing ≤ 100 kg following administration of the clinical dosing regimen of 150 mg SC at weeks 0 and 4 and every 12 weeks thereafter. ADAs to risankizumab did not have an effect on risankizumab CL or exposure in the majority of ADA-positive subjects, with the exception of few subjects (28 subjects, representing ~ 1.5% of all ADA-evaluable subjects (n = 1807) in phase III studies or 32 subjects, representing ~ 1.7% of all ADA-evaluable subjects (n = 1911) across all risankizumab doses evaluated in phase I–III studies) who developed ADA titer of ≥ 128 titer units, in which risankizumab CL was estimated to increase by 43% and AUC was estimated to decrease by 30%, on average. None of the other statistically significant covariates identified showed a meaningful impact on risankizumab exposures. Over the evaluated covariate ranges (serum albumin 34.0–58.0 g/L, baseline hs-CRP 0.1–131 mg/L, and serum creatinine 35.4–203 µmol/L), risankizumab exposures were estimated to be well within the default equivalence boundaries of 0.8–1.25 relative to the reference groups (Fig. 3).

Impact of covariates on risankizumab exposures. Points and squares represent median values and error bars represent 95% confidence intervals of the normalized exposure ratios across 200 simulation replicates. The vertical black dashed line shows an exposure ratio of 1 relative to the reference group, and the shaded area represents the 0.8–1.25 default equivalence boundaries. ADA antidrug antibody, AUC area under the concentration–time curve between weeks 40 and 52, Cmax maximum concentration, hs–CRP high-sensitivity C-reactive protein
4 Discussion
Risankizumab is an anti-IL-23 antibody being developed for the treatment of moderate to severe plaque psoriasis and other inflammatory diseases. In four phase III studies of risankizumab in subjects with moderate to severe plaque psoriasis, almost half of the subjects achieved complete skin clearance (PASI 100) following risankizumab treatment, demonstrating a durable, superior efficacy to adalimumab [9] and ustekinumab [14]. Using data from two phase I, one phase II, and four phase III studies in healthy volunteers and moderate to severe plaque psoriasis patients (n = 1899), a population model characterizing risankizumab pharmacokinetics was developed and a variety of covariates were assessed for their impact on the pharmacokinetic variability, providing a robust comprehensive evaluation across a large pool of subjects.
A two-compartment model with first-order absorption and elimination best described the pharmacokinetics of risankizumab in healthy subjects and patients with psoriasis in these studies. Goodness-of-fit plots of the population- and individual-predicted risankizumab concentrations showed good agreement with the observed data, and no trends suggesting bias were observed in the conditional weighted residual plots. Bootstrap analyses demonstrated the robustness of the model and precision of its estimates, and VPCs demonstrated the predictive power of the model in capturing the central tendency and the variability in the observed data across the evaluated risankizumab doses.
Risankizumab displayed linear pharmacokinetics across doses evaluated in the clinical studies included in the analyses (0.01–5 mg/kg IV, 200–1200 mg IV, 0.25–1 mg/kg SC, and 18–300 mg SC). This is consistent with pharmacokinetics of other monoclonal antibodies directed against soluble targets without apparent target-mediated drug disposition [15, 16]. The t½ was estimated to be approximately 28 days, similar to the t½ of a typical IgG1 antibody [16,17,18]. Risankizumab CL and Vss for a typical 90 kg psoriasis patient were 0.31 L/day and 11.2 L, respectively. IIV (%CV) for risankizumab CL, Vc, and ka were 24%, 34%, and 63%, respectively. These pharmacokinetic parameters and their variability were consistent with our previous analyses for risankizumab based on data from phase I and II studies, confirming the previously characterized pharmacokinetic attributes of risankizumab [11, 19]. The absolute SC bioavailability of risankizumab drug supply used in phase III studies was estimated to be 89%. The estimated SC bioavailability for the drug supply used in phase I and II studies was also high (71%) and the difference in estimated SC bioavailability should be interpreted with caution because of the (1) cross-study comparison pooling IV and SC data across multiple studies; (2) availability of IV data from a relatively small sample size and at different dose levels in the phase I study; (3) relatively small number of subjects in phase I/II trials (12%) compared with phase III trials (88%); and (4) the sparse sampling for the majority of studies that assessed the SC route of administration.
Following SC dosing of the risankizumab clinical regimen (150 mg at weeks 0 and 4 and every 12 weeks thereafter), the steady-state plasma concentrations of risankizumab were approximately achieved by week 16. The mean ± standard deviation model-predicted risankizumab Ctrough were 2.48 ± 1.35 µg/mL at week 16 and 1.91 ± 1.17 µg/mL at week 52.
Based on the simulations conducted using the population pharmacokinetic model, subjects with psoriasis with body weight > 100 kg were predicted to have 30% lower exposures than subjects weighing ≤ 100 kg following administration of the clinical dosing regimen of 150 mg SC at weeks 0 and 4 and every 12 weeks thereafter. This difference in exposure was interpreted to be not clinically relevant as suggested by the comparable PASI 90 and static Physicians Global Assessment (sPGA) 0/1 responses at week 16 (co-primary endpoints) for subjects with body weight ≤ 100 kg versus > 100 kg using the pooled data across phase III studies in psoriasis patients [20]. In addition, exposure–response analyses indicated that the clinical regimen of risankizumab 150 mg SC at weeks 0 and 4 and every 12 weeks thereafter achieved the plateau of efficacy for the evaluated endpoints (PASI 90, PASI 100, and sPGA 0/1) at weeks 16 and 52 across the entire body weight range evaluated in subjects with psoriasis [20]. Detailed reports of these analyses are forthcoming.
ADAs had no effects on risankizumab CL/exposure for the majority (98.5%) of evaluated subjects with psoriasis. Only for subjects with high ADA titers ≥ 128 (approximately 1% [6/598] of ADA-evaluable subjects who received the phase III clinical regimen over a 52-week time period in the UltIMMa-1 and UltIMMa-2 trials or approximately 1.5% [28/1807] of ADA-evaluable subjects who received any dose of risankizumab in all phase II/III trials), risankizumab CL was estimated to increase by 43% (relative to ADA-negative subjects or those with ADA titers < 128), which translated to a 30% decrease in the risankizumab AUC, on average. An impact of ADAs (whether evaluated as a categorical or continuous covariate based on titer) on risankizumab CL was not detected in our previous analysis [11], which included two of the studies (a single-dose phase I study and a phase II study) analyzed herein. The present analyses had increased statistical power to detect a small effect with the much larger sample size and longer duration of the phase III trials. NAb-positive status (time varying or time independent) was not a significant predictor for risankizumab CL. Overall, the impact of ADA was interpreted to be not clinically relevant for short-term efficacy (week 16) or long-term (week 52) maintenance of efficacy (PASI 90 and sPGA 0/1), as suggested by subgroup analyses for ADA-positive versus ADA-negative subjects in phase III trials. Detailed reports of these analyses are forthcoming.
Other covariates identified to be statistically correlated with risankizumab CL (baseline levels of albumin, hs-CRP, and serum creatinine) had no clinically meaningful impact on risankizumab exposures since risankizumab exposures were predicted to be within the default 80–125% equivalence boundaries for patients with extremes of covariate values (less than 25th percentile or greater than 75th percentile) in phase III clinical trials. While a robust hypothesis for the reason of the shallow and clinically non-relevant correlation between serum creatinine and risankizumab CL has not been established, creatinine CL has been reported to have statistically significant, and also clinically irrelevant, associations with CL of other mAbs targeting IL-23 such as usetkinumab and guselkumab [21, 22].
We have previously reported a summary of population pharmacokinetic analyses of risankizumab based on data from phase I and II studies [11]. The current analyses include additional data from phase III trials in psoriasis patients as well as data from healthy volunteers and confirm the previously reported pharmacokinetic attributes of risankizumab in a larger patient pool. In the current analyses, risankizumab pharmacokinetic and immunogenicity data from phase III trials over an up to 1 year duration were included to provide a robust assessment of the impact of immunogenicity on risankizumab CL. Additionally, due to a larger sample size, the current analyses also provide a robust assessment of the impact of various patient-specific covariates on the disposition of risankizumab.
5 Conclusions
A population pharmacokinetic model was developed using phase I–III data from healthy subjects and patients with moderate to severe plaque psoriasis. Risankizumab displayed linear pharmacokinetics over the range of doses evaluated in this analysis. None of the covariates identified as being statistically significantly correlated with risankizumab pharmacokinetic parameters were predicted to be clinically relevant based on lack of meaningful impact on risankizumab exposures for the proposed clinical regimen for treatment of moderate to severe plaque psoriasis.
References
Helmick CG, Lee-Han H, Hirsch SC, Baird TL, Bartlett CL. Prevalence of psoriasis among adults in the U.S.: 2003–2006 and 2009–2010 National Health and Nutrition Examination Surveys. Am J Prev Med. 2014;47:37–45.
Conrad C, Gilliet M. Psoriasis: from pathogenesis to targeted therapies. Clin Rev Allergy Immunol. 2018;54:102–13.
Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600.
Janssen Biotech, Inc. Stelara (ustekinumab) [US prescribing information]. Horsham: Janssen Biotech, Inc; 2018.
Janssen Biotech, Inc. Tremfya (guselkumab) [US prescribing information]. Horsham: Janssen Biotech, Inc; 2017.
Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaci D, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390:276–88.
Singh S, Kroe-Barrett RR, Canada KA, Zhu X, Sepulveda E, Wu H, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7:778–91.
AbbVie. Risankizumab meets all co-primary and ranked secondary endpoints, achieving significantly greater efficacy versus standard biologic therapies in three pivotal phase 3 psoriasis studies [press release]. 2017. https://news.abbvie.com/news/risankizumab-meets-all-co-primary-and-ranked-secondary-endpoints-achieving-significantly-greater-efficacy-versus-standard-biologic-therapies-in-three-pivotal-phase-3-psoriasis-studies.htm. Accessed 4 Jun 2018.
AbbVie. Risankizumab meets all primary endpoints reporting positive results in fourth pivotal phase 3 psoriasis study [press release]. 2017. https://news.abbvie.com/news/risankizumab-meets-all-primary-endpoints-reporting-positive-results-in-fourth-pivotal-phase-3-psoriasis-study.htm. Accessed 4 Jun 2018.
Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–61. https://doi.org/10.1016/S0140-6736(18)31713-6.
Suleiman AA, Khatri A, Minocha M, Othman AA. Population pharmacokinetics of the interleukin-23 inhibitor risankizumab in subjects with psoriasis and Crohn’s disease: analyses of phase I and II trials. Clin Pharmacokinet. 2019;58:375–87.
Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551–60.
Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometr Syst Pharmacol. 2013;2:e50.
Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650–61.
Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies. BioDrugs. 2010;24:23–39.
Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493–507.
Ternant D, Paintaud G. Pharmacokinetics and concentration-effect relationships of therapeutic monoclonal antibodies and fusion proteins. Expert Opin Biol Ther. 2005;5(Suppl 1):S37–47.
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:633–59.
Krueger JG, Ferris LK, Menter A, Wagner F, White A, Visvanathan S, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136(116–24):e7.
Khatri A, Suleiman A, Polepally A, Othman A. Exposure-response relationships for efficacy and safety of risankizumab in patients with moderate to severe plaque psoriasis: integrated analyses of phase 2 and 3 clinical trials [abstract no. 9908]. In: 2019 annual meeting of American Academy of Dermatology; 1–5 Mar 2019; Washington, DC.
Yao Z, Hu C, Zhu Y, Xu Z, Randazzo B, Wasfi Y, et al. Population pharmacokinetic modeling of guselkumab, a human IgG1λ monoclonal antibody targeting IL-23, in patients with moderate to severe plaque psoriasis. J Clin Pharmacol. 2018;58:613–27.
Zhu Y, Hu C, Lu M, Liao S, Marini JC, Yohrling J, et al. Population pharmacokinetic modeling of ustekinumab, a human monoclonal antibody targeting IL-12/23p40, in patients with moderate to severe plaque psoriasis. J Clin Pharmacol. 2009;49:162–75.
AbbVie. Safety, tolerability and pharmacokinetics of BI 655066/ABBV-066 (risankizumab) in healthy Asian and caucasian male volunteers [ClinicalTrials.gov identifier NCT02596217]. National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov. Accessed 1 Apr 2019.
AbbVie, BI 655066 (risankizumab) compared to placebo and active comparator (ustekinumab) in patients with moderate to severe chronic plaque psoriasis [ClinicalTrials.gov identifier NCT02684370]. National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov. Accessed 1 Apr 2019.
AbbVie, BI 655066 compared to placebo & active comparator (ustekinumab) in patients with moderate to severe chronic plaque psoriasis [ClinicalTrials.gov identifier NCT02684357]. National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov. Accessed 1 Apr 2019.
AbbVie, BI 655066/ABBV-066 (risankizumab) compared to active comparator (adalimumab) in patients with moderate to severe chronic plaque psoriasis [ClinicalTrials.gov identifier NCT02694523]. National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov. Accessed 1 Apr 2019.
AbbVie, BI 655066/ABBV-066 (risankizumab) in moderate to severe plaque psoriasis with randomized withdrawal and re-treatment [ClinicalTrials.gov identifier NCT02672852]. National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov. Accessed 1 Apr 2019.
Acknowledgements
The authors thank the subjects, patients, and investigators who participated in the studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Compliance with Ethical Standards
Funding
This work was supported by Boehringer Ingelheim and AbbVie. Boehringer Ingelheim contributed to the study designs and data collection and AbbVie contributed to the analysis and interpretation of the data and the writing, review, and approval of the manuscript.
Conflict of interest
Ahmed A. Suleiman, Mukul Minocha, Amit Khatri, Yinuo Pang, and Ahmed A. Othman are employees of AbbVie and may hold AbbVie stock or stock options.
Ethical approval
All studies were conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocols and informed consent forms were approved by the ethics committee or institutional review board at each site.
Informed consent
All participants provided written informed consent for participation in the studies.
Data sharing
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis datasets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Suleiman, A.A., Minocha, M., Khatri, A. et al. Population Pharmacokinetics of Risankizumab in Healthy Volunteers and Subjects with Moderate to Severe Plaque Psoriasis: Integrated Analyses of Phase I–III Clinical Trials. Clin Pharmacokinet 58, 1309–1321 (2019). https://doi.org/10.1007/s40262-019-00759-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00759-z