
Abstract
With high morbidity and mortality worldwide, there is great interest in effective therapies for chronic hepatitis B (CHB) virus. There are currently several dozen investigational agents being developed for treatment of CHB. They can be broadly divided into two categories: (1) direct-acting antivirals (DAAs) that interfere with a specific step in viral replication; and (2) host-targeting agents that inhibit viral replication by modifying host cell function, with the latter group further divided into the subcategories of immune modulators and agents that target other host functions. Included among the DAAs being developed are RNA interference therapies, covalently closed circular DNA (cccDNA) formation and transcription inhibitors, core/capsid inhibitors, reverse transcriptase inhibitors, hepatitis B surface antigen (HBsAg) release inhibitors, antisense oligonucleotides, and helioxanthin analogues. Included among the host-targeting agents are entry inhibitors, cyclophilin inhibitors, and multiple immunomodulatory agents, including Toll-like receptor agonists, immune checkpoint inhibitors, therapeutic vaccines, engineered T cells, and several cytokine agents, including recombinant human interleukin-7 (CYT107) and SB 9200, a novel therapy that is believed to both have direct antiviral properties and to induce endogenous interferon. In this review we discuss agents that are currently in the clinical stage of development for CHB treatment as well as strategies and agents currently at the evaluation and discovery phase and potential future targets. Effective approaches to CHB may require suppression of viral replication combined with one or more host-targeting agents. Some of the recent research advances have led to the hope that with such a combined approach we may have a functional cure for CHB in the not distant future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Research advances with dozens of investigational agents being developed for treatment of chronic hepatitis B (CHB), including (1) direct-acting antivirals (DAAs) that interfere with a specific step in viral replication and (2) host-targeting agents that inhibit viral replication by modifying host cell function, provide hope that we may soon have a functional cure for CHB. |
DAAs being developed include RNA interference therapies, covalently closed circular DNA (cccDNA) formation and transcription inhibitors, core/capsid inhibitors, reverse transcriptase inhibitors, hepatitis B surface antigen (HBsAg) release inhibitors, antisense oligonucleotides, and helioxanthin analogues. |
Host-targeting agents being developed include entry inhibitors, cyclophilin inhibitors, and multiple immunomodulatory agents, including Toll-like receptor agonists, immune checkpoint inhibitors, therapeutic vaccines, engineered T cells, recombinant human interleukin-7 (CYT107), and SB 9200. |
1 Introduction
Hepatitis B virus (HBV) is a small, enveloped, partially double-stranded DNA virus that belongs to the Hepadnaviridae family. It is estimated that 240 million people are chronically infected with HBV worldwide [1], approximately 75% of whom reside in Asia and 12% in Africa [2]. Although the overall prevalence of chronic hepatitis B (CHB) is substantially lower in Western countries, even in the USA it is estimated that the CHB population may be as high as 2.2 million people [3]. Although not all CHB patients develop complications, it is among the leading causes of liver disease, cirrhosis, and hepatocellular carcinoma (HCC) worldwide [1], with an estimated 15–40% of CHB patients developing serious sequelae during their lifetimes [4]. More than 750,000 deaths annually worldwide are attributed to HBV-related complications, including cirrhosis of the liver, liver failure, and HCC, the second leading cause of cancer death worldwide [1, 5,6,7]. With such high morbidity and mortality worldwide, there is great interest in effective therapies for CHB.
The only approved therapies now in use for CHB are a finite course of treatment with pegylated interferon (Peg-IFN)-α and indefinite treatment with nucleos(t)ide analogue reverse transcriptase inhibitors (NUCs) [4]. Although these therapies can decrease the risks of liver decompensation and HCC and improve survival [4, 8, 9], they do not commonly yield clearance of hepatitis B surface (HBs) antigen (HBsAg). Loss of HBsAg has been referred to as a “functional cure” [10] because it is associated with reduced liver necroinflammation, increased liver fibrosis regression, normalization of alanine aminotransferase (ALT) levels, reduced risk of liver cirrhosis, decompensation and HCC, and increased survival [11,12,13,14,15,16]. Unfortunately, with Peg-IFN-α treatment HBsAg loss has only been reported in approximately 3–8% of both hepatitis B e (HBe) antigen (HBeAg)-positive and HBeAg-negative patients at 48–52 weeks [4, 17, 18]; after interferon (IFN) treatment completion, HBsAg loss may continue, with one study showing that in HBeAg-positive patients treated for a median of 16 weeks, approximately 17% had experienced HBsAg loss after a median follow-up of 8.8 years [19]. Because IFN is associated with substantial adverse effects in many patients and requires parenteral delivery, it is only used in a small percentage of CHB patients. HBsAg loss is even lower in patients receiving NUC therapy; even with long-term therapy for 5–7 years, HBsAg loss has only been seen in 0.3–5% of HBeAg-negative patients and 0–11.8% of HBeAg-positive patients [20,21,22,23]. It has been shown that even when HBV replication is well-controlled, HBsAg clearance is unlikely to occur during a patient’s lifetime [24]. With this very low rate of HBsAg loss and the high rate of viral rebound and biochemical relapse that commonly occurs with discontinuation [4, 25,26,27,28], lifelong NUC therapy is generally recommended for the majority of patients. Thus, there is strong interest in new therapies that would yield high rates of HBsAg loss, leading to functional cure in substantially more patients and allowing treatment discontinuation.
During HBV replication, relaxed circular DNA (rcDNA) is converted into covalently closed circular DNA (cccDNA), a mini-chromosome that serves as the template for viral transcription [29]. The cccDNA has a very long half-life of about 30–50 days, accumulating and persisting in the cell nuclei as a stable epigenome [30,31,32]. Long after recovery from acute HBV infection, HBV DNA is still present in very small amounts in serum, suggesting the presence of cccDNA [33]. For complete eradication of HBV infection, it will be necessary to fully eliminate cccDNA [34]. Because of the persistence of cccDNA in infected hepatocytes and the integration of the hepatitis B viral genome into the host cell, a true ‘sterilizing’ cure in which all viral DNA is eliminated, with removal of all cccDNA and integrated virus, is not currently attainable but is the ultimate goal of future therapies [35].
There are currently several dozen investigational agents being developed for treatment of CHB. They can be broadly divided into two categories: (1) direct-acting antivirals (DAAs) that interfere with a specific step in viral replication; and (2) host-targeting agents that inhibit viral replication by modifying host cell function, with the latter group further divided into the subcategories of immune modulators and agents that target other host functions [36]. In this review we discuss agents that are currently in the clinical stage of development for CHB treatment as well as strategies and agents currently at the evaluation and discovery phase and potential future targets.
2 Direct-Acting Antivirals
2.1 RNA Interference Therapies
RNA interference (RNAi) therapies have been shown to directly target HBV messenger RNA (mRNA) transcripts with high specificity. Using small, non-coding RNAs to regulate the expression of genetic information [37] they are able to profoundly reduce HBsAg production, potentially restoring effective host immunity [38]. Multiple cell culture studies have shown that RNAi successfully inhibits HBV replication [39, 40]. Although, initially, effective delivery of the RNAi to hepatocytes was considered problematic [41], there have been advances in this. Arrowhead Pharmaceuticals (Pasadena, CA, USA) has developed a polymer-based system (Dynamic PolyConjugates/DPC) for the targeted delivery of siRNA to the hepatocyte cytoplasm where RNAi occurs as a way to reduce the potential toxicity that might result from interaction with cells not intended to be targeted [38, 42]. In a transiently transgenic pHBV mouse model it was shown that a single intravenous injection of an RNAi therapy resulted in decreases in HBsAg expression of between 2 and 3 log10, along with substantial decreases in HBeAg, serum HBV DNA, and liver levels of HBV RNA [43]. In a phase IIa clinical trial, single intravenous injections of Arrowhead’s first candidate drug ARC-520 resulted in dose-dependent reductions in HBsAg; a single dose of 2 mg/kg resulted in HBsAg reduction of up to 50%, with significant reductions maintained for 43–57 days [44]. In a multidose extension of this study, there was an additional reduction in HBsAg in all patients [45]. Although development of ARC-520 has been discontinued due to toxicity in cynomolgus monkeys, an RNAi therapy that could be delivered via subcutaneous injection is now being developed by Arrowhead. In another approach aimed at effective delivery of siRNAs to hepatocytes, a mixture of three HBV siRNAs is encapsulated in a novel pH-sensitive multifunctional envelope-type nanodevice (MEND) that serves as a hepatocyte-specific drug delivery system [46]. Assessments in primary human hepatocytes and in chimeric mice with humanized liver persistently infected with HBV showed that MEND/siRNA yielded decreases in HBsAg and HBeAg both in vitro and in vivo. Table 1 shows the current RNA-based gene silencers being studied.
2.2 Covalently Closed Circular DNA (cccDNA) Formation and Transcription Inhibitors
It is theoretically possible to target cccDNA by preventing either its formation, expression, or stability [36]. By screening a small-molecule library of 85,000 drug-like compounds, two disubstituted sulfonamides (DSS), CCC-0975 and CCC-0346, were identified and, using the HepG2 cell lines, shown to inhibit the formation of cccDNA from rcDNA [47]. In duck HBV-infected hepatocytes, CCC-0975 was found to reduce cccDNA biosynthesis. This proof-of-concept study points to the possibility that therapeutics might be developed to eliminate cccDNA, and this compound is now in preclinical development. The study demonstrated that although DSS compounds neither reduced viral polymerase activity nor inhibited replication of HBV DNA directly, they blocked the conversion of HBV rcDNA into cccDNA by causing inhibition of rcDNA deproteinization, one of the possible intermediate steps in cccDNA formation.
Lymphotoxin β receptor (LTβR) activation has been shown to lead to specific and non-hepatotoxic degradation of cccDNA. LTβR agonists such as BS1 and CBE11 (a monoclonal antibody against LTβR) have been studied in oncology where they have been shown to induce degradation of cccDNA [48, 49]. In hepatitis B research, it is known that IFN-γ and tumor necrosis factor (TNF)-α can control HBV but they cause adverse effects that are too severe for them to be used as HBV treatments. As an alternative, researchers have tested the effect of activation of the LTβR, the physiological ligands for which are the TNF superfamily members LTα, LTβ, and CD258, which can induce apoptosis and activate both inflammatory and anti-inflammatory pathways [50]. In cell studies, activation of LTβR has been shown to lead to the activation of deaminases such as APOBEC3B deaminase, which acts on cccDNA leading to non-hepatocytotoxic degradation of cccDNA [50]. It is possible that these agonists combined with NUCs could be a potent anti-HBV strategy.
Site-specific nucleases that target the HBV viral genome can cleave the HBV DNA, leading to inhibition of cccDNA formation and HBV replication. Engineered nucleases bind to DNA and produce double-strand breaks at target sites and, following misrepair by pathways such as homology-directed repair (HDR) or non-homologous end joining (NHEJ) of HBV DNA, lead to inhibition of viral replication [51, 52]. The most commonly used designer endonucleases are the zinc finger nucleases (ZFNs), the transcription activator-like effector nucleases (TALENs), the meganucleases and their derivatives, and the CRISPR (clustered, regularly interspaced, short palindromic repeat)/Cas9 genome editing tool [53]. CRISPR is a series of short repeated DNA sequences in the bacterial genome. These DNA sequences are flanked by sequences of bacteriophage DNA (DNA from viruses that infect bacteria). The CRISPR locus is adjacent to the Cas gene, a type of nuclease that can degrade DNA. It has been shown that the CRISPR/Cas9 tool can be used to remove HBV cccDNA, both in cell studies [54, 55] and using a mouse model [56,57,58]. In the cell line studies, it was shown that the CRISPR/Cas9 system reduces HBV viral load by 1000-fold and the cccDNA level by 10-fold [57, 59, 60]. A complete review of current anti-HBV gene therapy is provided by Maepa et al. [61]. The concern for off-target effects leading to mutations and unwanted consequences is always present with use of these endonucleases.
2.3 Core/Capsid Inhibitors
Advances in our understanding of nucleocapsid formation has led to development of several drugs aimed at interfering with HBV capsid assembly. These drugs inhibit HBV DNA replication by both destabilizing core particle assembly and disrupting existing capsids [62]. Core inhibitors disrupt the HBV lifecycle by inducing the assembly of defective capsids. Core modifiers eliminate HBV by modulating core protein at multiple complementary points in the viral lifecycle. This class of anti-HBV drugs, variously referred to as core inhibitors, capsid inhibitors, nucleocapsid assembly inhibitors, or capsid assembly modulators, was discovered in 2003; Bay 41-4109, in the sub-class of heteroaryldihydropyrimidines (HAPs), is the prototype [63]. Other HAPs include HAP-1, morphothiadine mesilate (GLS-4), HAP-18, and NVR-010-001-E2 [64]. In rodent models, Bay 41-4109 was shown to inhibit the virus replication cycle but was hepatotoxic in high doses. GLS-4 is a derivative of Bay 41-4109 that has been shown in preclinical studies to be equally effective but much less toxic to primary human hepatocytes [62]. It is now in phase II clinical studies.
AL-3778 (formerly NVR 3-778) is a first-in-class HBV core inhibitor that is thought to inhibit several core-mediated HBV life cycle functions [65]. In a humanized mouse model study that compared monotherapy with either AL-3778 (then called NVR 3-778), entecavir, or Peg-IFN, to combination therapy with NVR 3-778 + entecavir and or NVR 3-778 + Peg-IFN it was shown that serum HBV viral load reduction with NVR 3-778 monotherapy was similar to that seen with entecavir monotherapy and larger than that obtained with Peg-IFN monotherapy; the largest reduction was seen with NVR 3-778 + Peg-IFN combination therapy [66]. HBsAg serum levels were reduced most in the Peg-IFN groups, with only a minimal effect seen with NVR 3-778 alone. Levels of cccDNA were similar across treatment groups. In a later study, the nucleotide and corresponding amino acid differences in serum HBV DNA from humanized mice treated with either AL-3778 (then called NVR 3-778), Peg-IFN, or vehicle were compared [67]. NVR 3-778 was again shown to more effectively suppress serum HBV DNA than Peg-IFN; there were no nucleotide sequence changes in the core protein coding region of any of the mice treated with NVR 3-778. In a study that assessed efficacy and safety of AL-3778 (then called NVR 3-778), given alone and in combination with Peg-IFN for 28 days, in six dosing cohorts of treatment-naïve CHB patients, it was shown that there were dose-related HBV DNA reductions and early HBeAg reductions, effects that were increased when NVR 3-778 was given with Peg-IFN. An ongoing study is assessing the relative oral bioavailability of a tablet formulation of AL-3778 (formerly NVR 3-778) given under either fasted or fed (high-fat meal) conditions as well as the drug–drug interaction between AL-3778 and entecavir or tenofovir disoproxil fumarate (TDF) (ClinicalTrials.gov identifier NCT03032536) [193].
Phenylpropenamides, including AT-61 and AT-130, are also capsid inhibitors [64]. They behave as assembly accelerators, leading to formation of defective capsids without viral genome [68]. NZ-4 is an isothiafludine compound derived from leucamide A that has been shown to inhibit HBV replication in cell lines by increasing the deficient capsids [69]. Compound 3711 is a biaryl derivative shown to inhibit HBV replication by inducing genome-free capsid formation [70]. BCM 599 (N-(2, 6-dichloropyridin-3-yl)-2-[(4-fluorophenyl)formamido]acetamide) is in a novel class of capsid inhibitor that has been shown to have synergistic action with lamivudine in cell culture lines [71]. Table 2 shows the capsid inhibitors or core protein allosteric modifiers (CpAMs) currently undergoing trials [194].
2.4 Reverse Transcriptase Inhibitors
NUCs do not directly suppress cccDNA, viral transcription, or translation. They interfere with the synthesis of viral DNA from pregenomic RNA by targeting the RT function of the HBV polymerase [35]. Tenofovir alafenamide fumarate (TAF; Vemlidy®, Gilead Sciences, Foster City, CA, USA) is a recently US Food and Drug Administration (FDA)-approved NUC that is a novel prodrug of tenofovir, which is phosphorylated by host nucleotide kinases to the pharmacologically active form tenofovir diphosphate [72]. Highly bioavailable and efficiently delivered to lymphoid tissue and hepatocytes in its active form, TAF has been shown to be non-inferior to TDF in both HBeAg-positive and -negative patients with a better safety profile [73, 74]. After 1 year of treatment in the two major trials that compared TAF with TDF, patients treated with TAF had significantly smaller decreases in bone mineral density at both the hip (−0.10 vs. −1.72% in HBeAg-positive patients, and −0.29 vs. −2.16% in HBeAg-negative patients) and spine (−0.42 vs. −2.29% in HBeAg-positive patients and −0.88 vs. −2.51% in HBeAg-negative patients) [73, 74]. There were also smaller mean increases in serum creatinine in patients treated with TAF compared with TDF, although the difference was only statistically significant in HBeAg-positive patients [73, 74]. As has generally been the case with the other NUCs, HBsAg loss was low even at the end of 96 weeks of therapy, occurring in 7 of 576 TAF-treated HBeAg-positive patients (1%) and 4 of 288 TDF-treated HBeAg-positive patients (1%) [75]. Only one HBeAg-negative patient treated with TAF achieved HBsAg loss, with the loss seen at week 80; no TDF-treated HBeAg-negative patients achieved this [76].
CMX157 is a lipid conjugate of TDF that has been shown in vitro to be 4.5-fold more active against HBV than TDF and has been evaluated in phase I and II trials [36, 77, 78]. Clevudine is a NUC that has been approved for HBV treatment in South Korea and the Philippines [79]. Individual use of clevudine has been discontinued because of the development of drug resistance and myopathy, but it is used in lower doses in combination with adefovir [80,81,82]. Besifovir (LB80380) is a NUC that has been shown to have antiviral activity against both wild-type virus and virus with drug resistance mutations [83]. Studies have shown it to be well-tolerated and effective at reducing viral load in CHB patients with lamivudine-resistant virus [84] and non-inferior to entecavir 0.5 mg daily in treatment-naive CHB patients [85]; the only significant adverse effect of besifovir was l-carnitine depletion that required carnitine supplementation [85]. AGX-1009, a prodrug of TDF that is activated by a different molecular side chain, has demonstrated good efficacy for inhibition of viral replication in preclinical studies and is currently being studied in a phase I trial in China [86].
It has been observed that although RT inhibitors can effectively reduce viral load, they only have limited ability to reduce cccDNA levels, total intrahepatic HBV DNA, and serum HBsAg [87]. One of the reasons is that HBV reverse transcription occurs after infection has been developed with formation of HBV cccDNA in the infected nuclei of hepatocytes, in contrast to HIV where reverse transcription occurs after the infection but before the integration of DNA into the host genome [88]. It is encouraging that it has recently been shown that with many years of NUC treatment (median period 126 months) there is marked depletion of cccDNA in the majority of patients, but it was also shown that although serum HBsAg levels were reduced, they remained detectable in 42 of 43 patients [89]. Long-term treatment with NUCs can be associated with drug toxicity and drug resistance [90, 91]. Therefore, there is a need to identify compounds that can lead to eradication of the virus.
2.5 Hepatitis B Surface Antigen (HBsAg) Release Inhibitors
Nucleic acid polymers (NAPs) are broadly active against a wide range of enveloped viruses with type 1 entry mechanisms through their use of phosphorothioate oligonucleotides (PS-ONs) to inhibit protein interactions that are involved in viral replication. In a duck model, NAPs have been shown to have both entry and post-entry antiviral activity, preventing HBsAg secretion by blocking the formation of HBV subviral particles, which are mainly composed of HBsAg protein [92, 93]. REP 9 AC is an amphipathic DNA polymer that has been shown to inhibit HBsAg release from infected hepatocytes in CHB patients. The agent is currently in phase I and II clinical trials. Interim data have shown rapid clearance of HBsAg, HBV DNA suppression, and formation of hepatitis B surface antibody (anti-HBs), allowing patients to achieve durable immunity with REP 9 AC and its modified form [94, 95]. In a study of 12 HBeAg-positive CHB patients, the NAP HBsAg release inhibitor REP2139-Ca was given for 20–38 weeks, with response defined as decline in serum HBsAg; responders were then treated with add-on pegylated interferon alpha 2a (pegIFN-α-2a) and/or thymosin α-1 [96]. At week 0–24, mean HBV RNA, HBV DNA, and HBsAg levels had declined significantly and HBV RNA was undetectable in eight of 12 patients and remained undetectable during the treatment-free follow-up period in seven of these eight patients (mean 21.9 weeks, range 7–27 weeks). HBsAg loss and anti-HBs seroconversion were achieved in four of eight patients during treatment-free follow-up. REP2139-Ca is now being studied in phase I and II clinical trials in combination with Peg-IFN and TDF.
2.6 Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are short, synthetic, nucleic acid fragments that bind to target RNA sequences in order to form either an RNA:RNA duplex (antisense RNA) or a DNA:RNA hybrid (antisense DNA). Their structure can easily be modified to increase their affinity, stability, and specificity for the targets [97, 98]. The most commonly targeted area is the phosphate, which is the backbone of these oligonucleotides. A significant decrease in viremia along with a decrease in intrahepatic HBV DNA, HBV RNA, and surface and core proteins has been observed in polyethylenimine-based ASOs in a duck HBV model [99]. ASOs have been used to decrease the level of asialoglycoprotein receptor (ASGPR) 1, which is present exclusively in hepatocytes. HBV can upregulate ASGPR levels, which are responsible for HBV infection in hepatocytes by mediating hepatic endocytosis of HBV particles [100,101,102]. Thus, downregulation of ASGPR1 can block HBV replication by inhibiting hepatic endocytosis of HBV, which is the target for ASOs. AS2 is one of the ASOs that could inhibit viral replication through this mechanism [101]. Another mechanism of action of ASOs is to utilize intracellular enzyme RNase H, which degrades RNA strand into RNA–DNA hetero-duplex [103]. Gapmer ASOs are created through insertion of chemically modified nucleic acid analogs at each end of the oligonucleotides, which increases affinity and protects oligonucleotides from exonucleases and mediates efficient induction of RNase H degradation against mutated RNA [104]. Numerous therapies in this class are currently under development.
2.7 Helioxanthin Analogues
Helioxanthin analogues are another class of small molecules that inhibit HBV DNA, HBV RNA, and viral protein expression. Helioxanthin and its derivative 5-4-2 inhibited HBV mRNA levels as well as HBV transcripts in HepG2 2.2.15 cells in one study [105]. The helioxanthin analogues target multiple steps of the viral life cycle including inhibition of preS/S promotor activity and pregenomic/PreC activity using a gene reporter system [106]. In more recent research, additional helioxanthin analogues have been identified and research is ongoing [107].
3 Host-Targeting Agents
Included in this category are a broad variety of agents that modify various aspects of host cell function in ways that inhibit viral replication. These agents can be broadly subdivided into immune modulators and agents that target other host functions [36]. It is hoped that host-targeting agents could stimulate both innate and adaptive immunity and play a critical role in the clearance of HBV-infected hepatocytes.
3.1 Entry Inhibitors
Entry inhibitors are designed to block HBV’s entry into hepatocytes by either targeting cellular components in ways that interfere with HBV binding to the cell receptor(s) or by targeting HBV viral particles in ways that block HBV attachment to hepatocytes [108]. Entry inhibitors are of particular interest since inhibiting viral entry could block virus replication before cccDNA, the persistent viral reservoir, is formed [109]. Sodium taurocholate co-transporting polypeptide (NTCP), a key bile acid transporter whose primary role is transport of bile salts from the portal blood into the liver, is mainly expressed in hepatocytes at the basolateral membrane [110]. It has been shown that NTCP is a functional receptor for HBV and hepatitis D virus (HDV) that helps the viruses gain entry into the hepatocytes [111, 112]. The pre-S1 region of the HBV large surface protein is used to attach to the NTCP receptor.
The first-in-class agent myrcludex B is a synthetic N-myristoylated peptide composed of the residues from 2 to 48 of preS1 that competitively attaches to the NTCP receptor, blocking HBV’s entry into the hepatocyte [113]. In an animal model it was shown that myrcludex B both inhibited HBV transmission from cell to cell and inhibited increase of the intrahepatic cccDNA pool by impairing the conversion of rcDNA to cccDNA [113]. In a phase I study, myrcludex B was well-tolerated in doses of up to 20 mg given intravenously and 10 mg administered subcutaneously, with no serious adverse effects [114]. Interim results of a phase Ib/IIa trial in patients co-infected with CHB and chronic hepatitis delta (HDV) who were treated with myrcludex B, Peg-IFN-α-2a, or the combination of both showed that HDV RNA significantly decreased in all three treatment groups, becoming negative in two of eight patients in both the myrcludex B and Peg-IFN-α-2a monotherapy cohorts, and in five patients in the combination treatment group [115]. ALT decreased significantly only in the myrcludex B cohort. There was a significant decrease in HBV DNA only in the combination treatment group. HBsAg levels were unchanged in all three treatment groups.
Ciclosporin (cyclosporine A; CsA), a cyclic peptide composed of 11 amino acids that is used as an immunosuppressive agent, has been found to have affinity for and to bind to the NTCP receptor in a way that is independent of the cyclophilin (CYP) pathway responsible for its immunosuppressive effects. Using the human hepatocyte-derived cell line HepaRG and NTCP-based infection systems, it was shown that ciclosporin inhibits HBV and HDV entry into the hepatocyte by direct NTCP inhibition [116]. Ciclosporin disrupted the binding of NTCP to HBVpreS. Several ciclosporin derivatives, including SCYX618806, SCYX827830, and SCYX1454139, have been shown to have more potent anti-HBV activity than CsA53 [117]. The non-immunosuppressive cyclosporine alisporivir (DEBIO-025) has been shown to have anti-HBV activity equivalent to ciclosporin [117].
In a study that combined in vitro testing with computational approaches, 31 FDA-approved drugs were found to inhibit NTCP, most of which are classified as antifungals, antihyperlipidemics, antihypertensives, anti-inflammatories, or glucocorticoids [110]. Of these, it was found that nine agents reduced taurocholate uptake into NTCP-transfected cells by more than 50%: bendroflumethiazide, ciclosporin, ezetimibe, irbesartan, losartan, nefazodone, nifedipine, ritonavir, and simvastatin. Irbesartan, ezetimibe, and ritonavir have all been shown to have an inhibitory effect on HBV receptors in cell lines. In a HepG2-NTCP cell line that supports HBV infection, irbesartan was shown to effectively inhibit HBV entry [118]. Using differentiated HepaRG cells as a cell-culture infection model it was shown that altering hepatic cholesterol uptake with ezetimibe reduces early HBV infection through NTCP inhibition [119]. Other compounds that inhibit HBV entry through NTCP include epigallocatechin-3-gallate, a catechin flavonoid present in green tea extract, which has been shown to induce endocytosis of NTCP from the plasma membrane leading to protein degradation [120], and Ro41-5253, which has been shown to reduce the expression of both NTCP mRNA and protein by dysregulating the retinoic acid receptor [121]. The fungal metabolite vanitaracin A, a novel tricyclic polyketide, has been shown to inhibit entry of both HBV and HDV by inhibition of NTCP’s transporter activity [109]. Some NAPs have been shown to use the properties of PS-ONs as amphipathic polymers to block the amphipathic interactions used in viral entry, inhibiting attachment of the virion to the hepatocyte [92]. In vitro, several NAPs have been shown to inhibit duck HBV entry into hepatocytes. Although some of the NAPs tested caused significant toxicity in ducks, others such as REP 2005 were both effective against duck HBV infection and were well-tolerated [92].
3.2 Cyclophilin Inhibitors
In humans, there are seven main CYPs, cellular proteins with peptidyl-prolyl isomerase enzymatic activity [122, 123]. Included are CYPA, CYPB, CYPC, CYPD, CYPE, CYP40, and CYP natural killer (CYPNK) [122]. CYPs are host factors that are needed for the replication of many different viruses, including both hepatitis B and C [124]. CYPA has been shown to play an important role in both HBV replication and in HBV envelope protein secretion from hepatocytes [124]. Ciclosporin is the prototype CYP inhibitor but through modification of the ciclosporin molecule, inhibitors without immunosuppressive qualities have been developed, including alisporivir, NIM811, and SCY-635 [125]. In cell studies, when CYP inhibitors (either alisporivir or NIM811) were used to block CYP enzymatic activity, both HBV DNA replication and HBsAg production and secretion were substantially reduced [124]. Using alisporivir in combination with the DAA telbivudine yielded increased antiviral effects compared with the use of either agent alone. NVP018 is an orally available, sangamide-based second-generation CYP inhibitor, which has shown in vitro dual anti-HBV effects, causing immune modulation through IFN regulatory factors and inhibiting HBV replication [126]. However, since the dissolution in October 2016 of a development agreement between NVP018 developer NeuroVive Pharmaceutical AB (Lund, Sweden) and another company, development of the drug as an HBV therapy has been on hold.
3.3 Immunomodulatory Agents
With CHB patients, the goal of immunotherapy is to target or manipulate the immune system in ways that restore efficient antiviral immune responses. Multiple immunomodulatory agents are now being studied to treat CHB, including Toll-like receptor (TLR) agonists, immune checkpoint inhibitors, therapeutic vaccines, engineered T cells, and others.
3.3.1 Toll-Like Receptor Agonists
TLRs play a key role in innate immune defenses [127]. They are a type of pattern recognition receptor that can sense the presence of foreign pathogens and stimulate the release of inflammatory cytokines and subsequent adaptive immune responses [128]. It has been shown that HBV can suppress the TLR-induced antiviral activity of liver cells [129]. TLR7 stimulation in hepatocytes induces an endogenous type I IFN response leading to development of broad protective immunity against hepatitis viruses [130]. In animal models, it has been shown that agonists of several different TLRs (including TLRs 3, 7, 8, and 9) have anti-HBV effects [131,132,133,134,135], including substantial reductions in HBV viral load, serum HBsAg, and HBeAg as seen with the TLR7 agonist GS-9620 given in multiple doses to chimpanzees [136]. With phase I trials showing that although GS-9620 induced peripheral IFN-stimulated gene 15 (ISG15) expression it did not reduce HBsAg levels or HBV DNA [137], it is now being studied in combination with TDF in phase II trials [128].
3.3.2 Engineered T Cells
To fully address HBV infection, a strong T cell response is needed both to induce cellular immune responses to kill infected hepatocytes and to signal B cells to produce antibodies against HBeAg and HBsAg. This occurs in patients with acute HBV infection that does not become chronic [138,139,140,141] but does not occur in CHB patients. The goal of engineering T cells is to improve the affinity of T cell receptors for specific antigens. This is done by harvesting T cells from the patient, modifying them to express chimeric antigen receptors, which will allow them to recognize specific antigens on target cells, and then reintroducing the cells into the patient. In a mouse model, it was shown that T cells that had been engineered with a chimeric antigen receptor specific for HBV envelope proteins localized to the liver and rapidly reduced HBV replication, yielding a profound reduction in viral load [142]. There was immune-mediated damage to the liver but it was not severe and was only transient. To date, there has been no research to show that this approach can be safely and effectively used in humans. One factor that is likely to limit the use of engineered T cells is that this is a very costly individualized therapy, which would preclude its use in the developing world where a substantial portion of the CHB population resides.
3.3.3 Immune Checkpoint Inhibitors
As in many other chronic infections and cancers, in CHB there is exhaustion of T cells with weak virus-specific T cell responses that impede the clearance of virus, characterized by progressive loss of T cell effector functions and increased expression of several inhibitory receptors called checkpoint proteins, including cluster of differentiation (CD) 244, CD160, cytotoxic T lymphocyte (CTL) antigen-4 (CTLA-4), programmed death (PD)-1, cell immunoglobulin mucin-3 (Tim-3), and others [143,144,145,146,147,148,149]. Although the original focus was on CD8+ exhausted T cells, there is increasing evidence that CD4+ exhausted T cells also play important roles in this problem [150]. It is thought that several mechanisms may contribute to the T cell exhaustion, including high viral load and antigen levels, loss of CD4+ T cell help, and suppressive cytokines such as interleukin (IL)-10 and transforming growth factor (TGF)-β [151, 152]. As our understanding of T cell exhaustion has improved, there has been increasing interest in the possibility that by blocking inhibitory molecules with immune checkpoint inhibitors, the function of exhausted T cells could be restored in CHB patients [150]. It has been shown in a study that used liver biopsies from CHB patients that blocking the interaction of PD-1 with its ligand PD-L1 by incubating T cells with HBV peptides in the presence of anti-PD-L1 resulted in increased intrahepatic CD8+ T cell proliferation and production of IFN-γ and IL-2 [146]. In another cell study, the combination of CD137 stimulation with CD137L and blockade of PD-1 interaction with PD-L1 using anti-PD-L1 resulted in increased responses of intrahepatic HBV-specific T cells [147]. Results with nivolumab, an anti-PD-1 monoclonal antibody, in which 15% of chronic HCV patients had a significant reduction in HCV RNA following a single dose [153], has increased interest in the possibility that checkpoint inhibitors might also have efficacy in CHB patients. Experts in the field have theorized that there is a moderate to high probability that checkpoint inhibitors could become an effective therapy for CHB, most likely as one component of a combined approach [128]. Because there is a substantial risk of adverse events with these agents, cautious assessment of their effects in CHB patients with careful monitoring will be required.
3.3.4 Therapeutic Vaccines
Therapeutic vaccines are currently of considerable interest because it is thought that they could help counter HBV-specific T cell exhaustion, restoring the level of the HBV-specific T cell population, boosting CD4+ T cell responses, and activating humoral and cytolytic immune responses against HBV antigens [154, 155]. There is considerable genetic diversity in HBV, with eight distinct genotypes and multiple subtypes, giving complexity to the development of effective vaccines. There are several types of HBV therapeutic vaccines currently in development.
3.3.4.1 Protein- or Peptide-Based Vaccines
Protein- or peptide-based vaccines induce HBV-specific antibodies in high titers but yield only weak cellular immune responses and thus require repeated doses and adjuvant therapy for the best effects [156]. Combining HBsAg and highly immunogenic hepatitis B core antigen (HBcAg) has led to significant improvement in protein-based vaccination [157]. HBcAg activates B cells, which enhances T cell priming, enabling them to act as potent antigen-presenting cells (APCs), increasing the immunogenic effects of vaccine and causing highly potent HBs-specific T cell responses [158,159,160,161]. It is thought that this type of protein-based vaccination that combines HBsAg and HBcAg may work well to reduce viral load and induce seroconversion in CHB patients [157].
3.3.4.2 DNA-Based Vaccines
DNA-based vaccines provide genes encoding the antigen rather than consisting of the antigen itself. They induce strong humoral and cellular immune responses, including T helper (Th) 1 and cytotoxic responses, in contrast to protein-based vaccinations, which mainly mount humoral responses [162]. Multiple trials have been conducted to assess the efficacy of DNA-based vaccination in CHB patients. In one small study of patients who had not responded to standard treatment with IFN-α and/or lamivudine, DNA vaccine (encoding for S and preS2 domains of HBV envelope proteins) yielded a transient decrease in HBV DNA levels in 50% of patients, HBV-specific IFN-γ-secreting T cells significantly increased, and most patients showed transient proliferative responses to HBcAg [163]. It has been observed in multiple studies that the immunogenicity of these vaccinations can be enhanced with DNA prime and viral vector boost vaccinations [164]. In another study, a DNA vaccine encoding HBV proteins and modified human IL-12 was given for 12 months to CHB patients being treated with lamivudine [165]. Detectable HBV-specific IFN-γ-secreting T cell responses were observed at the end of treatment and during follow-up, with the type 1 T cell responses, particularly CD4+ memory T cell responses, maintained for at least 40 weeks after completion of therapy.
It has been observed in multiple studies that the immunogenicity of these vaccinations can be enhanced with DNA prime and viral vector booster vaccinations [164]. A large number of multifunctional T cells can result with this type of combined DNA vaccine and viral vector booster (poxviruses or adenoviruses), as observed in trials of vaccines for tuberculosis and malaria [166].
3.3.4.3 Viral Vector-Based Vaccines
Sustained immune responses can be generated with live attenuated viral vaccines, and with other diseases multiple vaccines have been developed with recombinant viral vectors such as poxviruses and adenoviruses [167, 168]. In a chimpanzee study, treatment with a recombinant retroviral vector vaccine expressing HBcAg resulted in increased anti-HBe antibodies and restoration of HBV-specific cytotoxic T cell responses; in only one of the three chimpanzees, HBeAg seroconversion and HBV clearance occurred [169]. In a study of a DNA/MVA (recombinant modified vaccinia virus Ankara) prime/boost vaccine given with or without lamivudine treatment to 12 HBeAg-negative and 12 HBeAg-positive CHB patients, no added beneficial effects were seen beyond those that occurred with antiviral therapy alone [170]. It is possible that the effects of these types of vaccines could be improved by lowering HBV viral load prior to vaccination, a strategy that in the woodchuck model resulted in strong woodchuck hepatitis surface antigen (WHsAg)- and woodchuck hepatitis core antigen (WHcAg)-specific CD4+ and CD8+ T cell responses in animals pre-treated with entecavir and then given a DNA prime-adenovirus boost vaccine regimen; these T cell responses were not detectable in the animals treated only with entecavir [171]. The TG1050 vaccine (Transgene, Strasbourg, France) based on a viral vector expressing three HBV antigens (core, polymerase, and envelope) is now being studied in a randomized, multicenter, double-blind, placebo-controlled phase I study (ClinicalTrials.gov identifier NCT02428400) [195].
3.3.4.4 Cell-Based Vaccines
Dendritic cells (DCs) are APCs that are present throughout the body and are important for antigen presentation to CD4+ and CD8+ T cells [172]. It has been shown that in CHB patients there are functional defects in APCs, particularly the DCs, which may be responsible for a decreased ability for HBV antigens to be presented to the host immune system for clearance of HBV [173]. Antigen-presenting autologous DCs (ADCs), primed with antigen, have been studied as immunotherapy in multiple diseases [172]. In one large study of CHB patients not currently receiving any other anti-HBV therapy, ADCs were pulsed with HBcAg18-27 peptide (FLPSDFFPSV) and the HBV Pre-S244-53 peptide (SILSKTGDPV) and reinfused intravenously, twice monthly during the first 3 months and once monthly during the following 3 months [172]. HBV DNA became undetectable in 46.36% of HBeAg-negative patients and 3.13% of HBeAg-positive patients. Of 195 patients with ALT levels above 40 IU/mL at baseline, ALT levels in 50.26% had returned to normal by 48 weeks, with a significant effect on the normalization of ALT observed in both HBeAg-positive and HBeAg-negative patients. The researchers noted that this DC vaccine appeared to trigger a strong CD8+ CTL response in both HBeAg-negative and -positive patients. Because in HBeAg-positive patients strong viral replication reduced viral load clearance by the DC vaccine, they suggested that it might be preferable to combine the vaccine with antiviral drugs.
Loss of HBsAg and production of anti-HBs has been observed in a study with transgenic mice where endogenous DCs were pulsed with HBsAg [174]. Based on this, 5 million HBsAg-pulsed DCs were administered intradermally one to three times in five CHB patients, resulting in anti-HBs in two patients and HBsAg-specific cellular immunity in one patient [175]. The HBsAg-pulsed DCs resulted in induction of higher levels of IFN-γ and IL-12 than did un-pulsed DCs. The HBsAg-pulsed DCs did not result in generalized inflammation, exacerbation of liver damage, abnormal kidney function, or features of autoimmunity and were considered safe in all patients. Based on these results, additional clinical trials are planned.
3.3.5 Other Immune Modulators and Associated Therapies
There are multiple other therapies in this category currently being tested, among which the farthest along in development are the cytokine agents. Researchers are looking at the immune restoration and vaccine adjuvant effects of certain cytokines and the possibility that cytokines and cytokine receptor agonists could lead to HBV cccDNA elimination. IL-7 is necessary for the correct development of T cells, DC subsets, and B cells. A combination of NUC therapy (entecavir or tenofovir) with recombinant human IL-7 (CYT107) or with both CYT107 and HBV vaccine (GenHevac B®, Pasteur, Merieux, France) is now being studied (ClinicalTrials.gov identifier NCT01027065) [196]. Because pre-clinical data had shown that IL-12 inhibits HBV replication through the stimulation of IFN-γ production, one study investigated the results of adding recombinant human IL-12 therapy to lamivudine treatment [176]. In CHB patients given the combination there was enhanced T cell reactivity to HBV and IFN-γ production but HBV replication inhibition was not maintained after discontinuation of lamivudine.
SB 9200, an oral prodrug of the dinucleotide SB 9000, is a novel therapy that is believed to have both direct antiviral properties and induce endogenous IFN by activating two viral sensor proteins: retinoic acid-inducible gene 1 (RIG-I) and nucleotide-binding oligomerization domain-containing protein 2 (NOD2). It is thought that this activation results in IFN-mediated antiviral immune responses in HBV-infected cells. In the woodchuck model, two oral doses of SB 9200 (15 and 30 mg/kg) were studied [177]. After 12 weeks of treatment, there were reductions in serum woodchuck hepatitis virus (WHV) DNA of 2.2 and 3.7 log10 and in WHV surface antigen of 0.5 and 1.6 log10 with the lower and higher doses, respectively. The drug was well-tolerated with no adverse effects noted. The reduction in surface antigen is thought to be due to inhibition of cccDNA synthesis or transcription. Because it has been shown in vitro that SB 9200 has potent activity against both wild-type and mutant HBV and has synergistic effects with NUCs, it is now being studied in a combination approach. In an ongoing phase IIa trial, patients are assigned to one of four dosing cohorts (25, 50, 100, or 200 mg) of SB 9200 or placebo, with the SB 9200 given once daily for 12 weeks; all patients will then receive tenofovir 300 mg once daily for an additional 12 weeks of treatment (ClinicalTrials.gov identifier NCT02751996) [197]. In a planned phase IIb trial the concomitant use of SB 9200 and tenofovir given for 12 weeks will be assessed, followed by tenofovir given alone.
Because the efficacy of some of the immune modulator therapies discussed here, including therapeutic vaccines, IFN, and checkpoint inhibitors, can be compromised by high antigen levels [178,179,180], the CRISPR/Cas9 genome editing tool, discussed in Sect. 2.2 as a cccDNA inhibitor, is of interest. Although this tool may not be available outside the laboratory for some time, the hope is that it could someday be used to directly remove cccDNA, which would, in turn, lower HBsAg levels and improve the efficacy of some of the immune modulators [128].
4 Novel Therapies
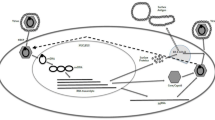
Current evidence suggests that existing HBV therapies reduce progression to chronic liver disease and its sequelae, including HCC [181, 182]. In the present treatment paradigm, new-generation drugs such as tenofovir alafenamide and besifovir may have similar efficacy with fewer adverse effects [73, 85, 182]. However, the ultimate goal of chronic HBV treatment would be the elimination of cccDNA [181, 182]. To achieve this goal, multiple therapies with a variety of targets are under investigation [183]. The approaches can be divided into virologic approaches and host immune approaches. With the exceptions of tenofovir alafenamide and besifovir, all other compounds are in pre-clinical or phase I or II trials [184]. Novel therapies with virologic approaches are shown in Fig. 1 and those with immune approaches in Fig. 2 [47, 50, 183,184,185,186,187,188]. The main features of the HBV life cycle and potential antiviral targets are shown in Fig. 3.


Novel therapies targeting hepatitis B virus with immunological approaches [46, 49, 66, 115, 116, 175,176,177,178,179]. Among the therapies currently being assessed as possible agents to counter hepatitis B virus with host-targeting approaches, including (a) entry inhibitors: myrcludex B, ciclosporin (cyclosporine A) and several ciclosporin derivatives (including SCYX618806, SCYX827830, and SCYX1454139), alisporivir, bendroflumethiazide, ezetimibe, irbesartan, losartan, nefazodone, nifedipine, ritonavir, simvastatin, epigallocatechin-3-gallate, Ro41-5253, vanitaracin A, and the nucleic acid polymer REP 2005; (b) cyclophilin inhibitors: ciclosporin, alisporivir, NIM811, SCY-635, and NVP018; (c) Toll-like receptor agonists: GS-9620; (d) engineered T cells: T cells engineered with a chimeric antigen receptor specific for HBV envelope proteins; (e) immune checkpoint inhibitors: nivolumab; (f) therapeutic vaccines: (1) protein/peptide-based: vaccination that combines HBsAg and HBcAg, (2) DNA vaccines: DNA vaccine encoding for S and preS2 domains of HBV envelope proteins, DNA prime and viral vector boost vaccines, and DNA vaccine encoding HBV proteins and modified human IL-12, (3) viral vector-based vaccines: TG1050 vaccine, recombinant retroviral vector vaccine expressing HBcAg, DNA/MVA prime/boost vaccine, and DNA prime-adenovirus boost vaccine, (4) cell-based vaccines: dendritic cell vaccines; and (g) other immune modulators: recombinant human IL-7 (CYT107), recombinant human IL-12, and SB 9200. HBcAg hepatitis B core antigen, HBsAg hepatitis B surface antigen, HBV hepatitis B virus, IL interleukin, MVA modified vaccinia virus Ankara

The main features of the HBV life cycle and potential antiviral targets [186, 189,190,191,192]. (1) HBV entry inhibitors. Lipopeptides mimicking pre-S1 domain competing with Dane particle for binding to NTCP (e.g., myrcludex B™). Other small molecules are under evaluation. (2) Targeting cccDNA. Damage and destruction of cccDNA via cytokines or cccDNA sequence-specific nucleases. Functional silencing via modulation of host cellular epigenetic-modifying enzymes by cytokines or inhibition of viral protein function. (3) HBV mRNAs. Small-interfering RNA approaches or antisense oligonucleotides to block viral replication and viral protein expression. (4) HBV Pol inhibitors. Reverse transcriptase inhibitors of the nucleos(t)ide analog family are part of the standard of care. RNAse H inhibitors are in preclinical evaluation. (5) Core modulators. Nucleocapsid assembly and pgRNA packaging. Capsid assembly modulators can affect nucleocapsid assembly, pgRNA encapsidation, and the nuclear functions of HBc (cccDNA regulation and interferon stimulated gene expression). (6) Egress inhibitors. Phosphorothioate oligonucleotides inhibiting HBsAg release and monoclonal antibodies to decrease circulating HBsAg load are under evaluation. Reproduced with permission from Revill et al. [189]. cccDNA covalently closed circular DNA, dslDNA double-stranded linear DNA, HBc hepatitis B core protein, HBe hepatitis e antigen, HBs hepatitis B surface, HBsAg hepatitis B surface antigen, HBV hepatitis B virus, HBx hepatitis B x protein, HSC hepatic stellate cell, IFNβ interferon-β, IL6 interleukin-6, KC Kuppfer cell, LSEC liver sinusoidal endothelial cells, mRNA messenger RNA, NTCP sodium taurocholate co-transporting polypeptide, pgRNA pregenomic RNA, Pol polymerase, RC DNA relaxed circular DNA, TGFβ transforming growth factor-β, MVB multivesicular bodies, SVP subviral envelope particles
5 Conclusion
In recent years there has been remarkable progress in our understanding of HBV virology and the body’s immune response to it, and significant research that has led to possible new treatments. Until the day comes that we have a true sterilizing cure in which all viral DNA is eliminated, with removal of all cccDNA and integrated virus, effective approaches to CHB may require suppression of viral replication combined with one or more host-targeting agents. Some of the recent research advances have led to the hope that with such an approach we may have a functional cure for CHB in the not distant future.
References
Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30(12):2212–9.
Gust ID. Epidemiology of hepatitis B infection in the Western Pacific and South East Asia. Gut. 1996;38(Suppl 2):S18–23.
Kowdley KV, Wang CC, Welch S, Roberts H, Brosgart CL. Prevalence of chronic hepatitis B among foreign-born persons living in the United States by country of origin. Hepatology. 2012;56(2):422–33.
Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology. 2009;50:661–2.
Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127(5 Suppl 1):S35–50.
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–128.
Marcellin P, Castelnau C, Martinot-Peignoux M, Boyer N. Natural history of hepatitis B. Minerva Gastroenterol Dietol. 2005;51(1):63–75.
Rijckborst V, ter Borg MJ, Cakaloglu Y, Ferenci P, Tabak F, Akdogan M, et al. A randomized trial of peginterferon alpha-2a with or without ribavirin for HBeAg-negative chronic hepatitis B. Am J Gastroenterol. 2010;105(8):1762–9.
Sung JJ, Tsoi KK, Wong VW, Li KC, Chan HL. Meta-analysis: treatment of hepatitis B infection reduces risk of hepatocellular carcinoma. Aliment Pharmacol Ther. 2008;28(9):1067–77.
Ruan P, Xu SY, Zhou BP, Huang J, Gong ZJ. Hepatitis B surface antigen seroclearance in patients with chronic hepatitis B infection: a clinical study. J Int Med Res. 2013;41(5):1732–9.
Chu CM, Liaw YF. Hepatitis B surface antigen seroclearance during chronic HBV infection. Antivir Ther. 2010;15(2):133–43.
Arase Y, Ikeda K, Suzuki F, Suzuki Y, Saitoh S, Kobayashi M, et al. Long-term outcome after hepatitis B surface antigen seroclearance in patients with chronic hepatitis B. Am J Med. 2006;119(1):71 e9–16.
Liu J, Yang HI, Lee MH, Lu SN, Jen CL, Batrla-Utermann R, et al. Spontaneous seroclearance of hepatitis B seromarkers and subsequent risk of hepatocellular carcinoma. Gut. 2014;63(10):1648–57.
Moucari R, Marcellin P. HBsAg seroclearance: prognostic value for the response to treatment and the long-term outcome. Gastroenterol Clin Biol. 2010;34(Suppl. 2):S119–25.
Yuen MF, Wong DK, Fung J, Ip P, But D, Hung I, et al. HBsAg seroclearance in chronic hepatitis B in Asian patients: replicative level and risk of hepatocellular carcinoma. Gastroenterology. 2008;135(4):1192–9.
Yuen MF, Wong DK, Sablon E, Tse E, Ng IO, Yuan HJ, et al. HBsAg seroclearance in chronic hepatitis B in the Chinese: virological, histological, and clinical aspects. Hepatology. 2004;39(6):1694–701.
Buster EHCJ, Janssen HLA. Antiviral treatment for chronic hepatitis B virus infection—immune modulation or viral suppression. Neth J Med. 2006;64:175–85.
Lampertico P. The royal wedding in chronic hepatitis B: the haves and the have-nots for the combination of pegylated interferon and nucleos(t)ide therapy. Hepatology. 2015;61(5):1459–61.
van Zonneveld M, Honkoop P, Hansen BE, Niesters HG, Darwish Murad S, de Man RA, et al. Long-term follow-up of alpha-interferon treatment of patients with chronic hepatitis B. Hepatology. 2004;39(3):804–10.
Chang TT, Lai CL, Kew Yoon S, Lee SS, Coelho HS, Carrilho FJ, et al. Entecavir treatment for up to 5 years in patients with hepatitis B e antigen-positive chronic hepatitis B. Hepatology. 2010;51(2):422–30.
Buti M, Tsai N, Petersen J, Flisiak R, Gurel S, Krastev Z, et al. Seven-year efficacy and safety of treatment with tenofovir disoproxil fumarate for chronic hepatitis B virus infection. Dig Dis Sci. 2015;60(5):1457–64.
Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131(6):1743–51.
Pan CQ, Tong M, Kowdley KV, Hu KQ, Chang TT, Lai CL, et al. High rates of viral suppression after long-term entecavir treatment of Asian patients with hepatitis B e antigen-positive chronic hepatitis B. Clin Gastroenterol Hepatol. 2012;10(9):1047–50.e1.
Chevaliez S, Hezode C, Bahrami S, Grare M, Pawlotsky JM. Long-term hepatitis B surface antigen (HBsAg) kinetics during nucleoside/nucleotide analogue therapy: finite treatment duration unlikely. J Hepatol. 2013;58(4):676–83.
Seto WK, Hui AJ, Wong VW, Wong GL, Liu KS, Lai CL, et al. Treatment cessation of entecavir in Asian patients with hepatitis B e antigen negative chronic hepatitis B: a multicentre prospective study. Gut. 2015;64(4):667–72.
Jafri SM, Lok AS. Antiviral therapy for chronic hepatitis B. Clin Liver Dis. 2010;14(3):425–38.
Frenette CT, Gish RG. To “be” or not to “be”: that is the question. Am J Gastroenterol. 2009;104(8):1948–52.
Fung J, Lai CL, Tanaka Y, Mizokami M, Yuen J, Wong DK, et al. The duration of lamivudine therapy for chronic hepatitis B: cessation vs. continuation of treatment after HBeAg seroconversion. Am J Gastroenterol. 2009;104(8):1940–6.
Warner N, Locarnini S. Replication of hepatitis B virus. In: Boyer TD, Manns MP, Sanyal AJ, editors. Zakim and Boyer’s hepatology: a textbook of liver disease. 6th ed. Philadelphia: Elsevier; 2012.
Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA. 2009;106(47):19975–9.
Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51(3):581–92.
Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, Miller DS, et al. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75(1):311–22.
Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat Med. 1996;2(10):1104–8.
Ohno M, Otsuka M, Kishikawa T, Yoshikawa T, Takata A, Koike K. Novel therapeutic approaches for hepatitis B virus covalently closed circular DNA. World J Gastroenterol. 2015;21(23):7084–8.
Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL, et al. Chronic hepatitis B: virology, natural history, current management and a glimpse at future opportunities. Antivir Res. 2015;121:47–58.
Block TM, Rawat S, Brosgart CL. Chronic hepatitis B: a wave of new therapies on the horizon. Antivir Res. 2015;121:69–81.
Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–55.
Schluep T, Lickliter J, Hamilton J, Lewis DL, Lai CL, Lau JY, et al. Safety, tolerability and pharmacokinetics of ARC-520 Injection, an RNA interference-based therapeutic for the treatment of chronic hepatitis B virus infection, in healthy volunteers. Clin Pharmacol Drug Dev. 2016. doi:10.1002/cpdd.318 (Epub 2016 Oct 14).
Konishi M, Wu CH, Wu GY. Inhibition of HBV replication by siRNA in a stable HBV-producing cell line. Hepatology. 2003;38(4):842–50.
Hamasaki K, Nakao K, Matsumoto K, Ichikawa T, Ishikawa H, Eguchi K. Short interfering RNA-directed inhibition of hepatitis B virus replication. FEBS Lett. 2003;543(1–3):51–4.
Chen Y, Cheng G, Mahato RI. RNAi for treating hepatitis B viral infection. Pharm Res. 2008;25(1):72–86.
Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, Roesch PL, et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci USA. 2007;104(32):12982–7.
Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21(5):973–85.
Yuen M, Chan H, Given B, Hamilton H, Schluep T, Lewis DL, et al. Phase II, dose ranging study of ARC-520, a siRNA-based therapeutic, in patients with chronic hepatitis B virus infection. Hepatology. 2014;60(Suppl 1):LB-21.
Yuen MF, Liu K, Chan HL, Given BD, Schluep T, Hamilton J, et al. Prolonged RNA interference therapy with ARC-520 injection in treatment naïve, HBeAg positive and negative patients with chronic HBV results in significant reductions of HBs antigen. J Hepatol. 2017;66(Suppl 1):S27.
Yamamoto N, Sato Y, Munakata T, Kakuni M, Tateno C, Sanada T, et al. Novel pH-sensitive multifunctional envelope-type nanodevice for siRNA-based treatments for chronic HBV infection. J Hepatol. 2016;64(3):547–55.
Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother. 2012;56(8):4277–88.
Lukashev M, LePage D, Wilson C, Bailly V, Garber E, Lukashin A, et al. Targeting the lymphotoxin-beta receptor with agonist antibodies as a potential cancer therapy. Cancer Res. 2006;66(19):9617–24.
Hu X, Zimmerman MA, Bardhan K, Yang D, Waller JL, Liles GB, et al. Lymphotoxin beta receptor mediates caspase-dependent tumor cell apoptosis in vitro and tumor suppression in vivo despite induction of NF-kappaB activation. Carcinogenesis. 2013;34(5):1105–14.
Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343(6176):1221–8.
Gupta RM, Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124(10):4154–61.
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–46.
Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21(2):121–31.
Seeger C, Sohn JA. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids. 2014;3:e216.
Karimova M, Beschorner N, Dammermann W, Chemnitz J, Indenbirken D, Bockmann JH, et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci Rep. 2015;5:13734.
Dong C, Qu L, Wang H, Wei L, Dong Y, Xiong S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antivir Res. 2015;118:110–7.
Lin SR, Yang HC, Kuo YT, Liu CJ, Yang TY, Sung KC, et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Nucleic Acids. 2014;19(3):e186.
White MK, Hu W, Khalili K. The CRISPR/Cas9 genome editing methodology as a weapon against human viruses. Discov Med. 2015;19(105):255–62.
Kennedy EM, Bassit LC, Mueller H, Kornepati AV, Bogerd HP, Nie T, et al. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology. 2015;476:196–205.
Weber ND, Stone D, Sedlak RH, De Silva Feelixge HS, Roychoudhury P, Schiffer JT, et al. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS One. 2014;9(5):e97579.
Maepa MB, Roelofse I, Ely A, Arbuthnot P. Progress and prospects of anti-HBV gene therapy development. Int J Mol Sci. 2015;16(8):17589–610.
Wu G, Liu B, Zhang Y, Li J, Arzumanyan A, Clayton MM, et al. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob Agents Chemother. 2013;57(11):5344–54.
Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299(5608):893–6.
Cole AG. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr Opin Pharmacol. 2016;30:131–7.
Yuen MF, Kim DJ, Weilert F, Chang HLY, Lalezari JP, Hwang SG, et al. NVR 3-778, a first-in-class HBV core inhibitor, alone and in combination with peg-interferon (PegIFN), in treatment naive HBeAg-positive patients: early reductions in HBV DNA and HBeAg. J Hepatol. 2016;64(Suppl 1):S210–1.
Klumpp K, Shimada T, Allweiss L, Volz T, Luetgehetman M, Flores O, et al. High antiviral activity of the HBV core inhibitor NVR 3-778 in the humanized uPA/SCID mouse model. J Hepatol. 2015;62(Suppl 2):S250.
Lam AM, Shimada T, Dandri M, Flores L, Klumpp K. No changes in the coding sequence for HBV core protein after 6 weeks of treatment of HBV infected humanized mice with NVR 3-778. J Hepatol. 2017;66(suppl 1):S477–8.
Katen SP, Chirapu SR, Finn MG, Zlotnick A. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem Biol. 2010;5(12):1125–36.
Yang L, Shi LP, Chen HJ, Tong XK, Wang GF, Zhang YM, et al. Isothiafludine, a novel non-nucleoside compound, inhibits hepatitis B virus replication through blocking pregenomic RNA encapsidation. Acta Pharmacol Sin. 2014;35(3):410–8.
Wang YJ, Lu D, Xu YB, Xing WQ, Tong XK, Wang GF, et al. A novel pyridazinone derivative inhibits hepatitis B virus replication by inducing genome-free capsid formation. Antimicrob Agents Chemother. 2015;59(11):7061–72.
Cho MH, Jeong H, Kim YS, Kim JW, Jung G. 2-amino-N-(2,6-dichloropyridin-3-yl)acetamide derivatives as a novel class of HBV capsid assembly inhibitor. J Viral Hepat. 2014;21(12):843–52.
Murakami E, Wang T, Park Y, Hao J, Lepist EI, Babusis D, et al. Implications of efficient hepatic delivery by tenofovir alafenamide (GS-7340) for hepatitis B virus therapy. Antimicrob Agents Chemother. 2015;59(6):3563–9.
Buti M, Gane E, Seto WK, Chan HLY, Chuang WL, Stepanova T, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol. 2016;1(3):196–206.
Chan HLY, Fung S, Seto WK, Chuang WL, Chen CY, Kim HJ, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol. 2016;1(3):185–95.
Agarwal K, Fung S, Seto WK, Lim YS, Gane E, Janssen HL, et al. A phase 3 study comparing tenofovir alafenamide (TAF) to tenofovir disoproxil fumarate (TDF) in patients with HBeAg-positive, chronic hepatitis B (CHB): efficacy and safety results at week 96 [abstract no. ILC2017-RS-244]. In: International Liver Congress 2017, European Association for the study of the liver, 19–23 Apr 2017, Amsterdam.
Brunetto M, Lim YS, Gane E, Seto WK, Osipenko M, Ahn SH, et al. A phase 3 study comparing tenofovir alafenamide (TAF) to tenofovir disoproxil fumarate (TDF) in patients with HBeAg-negative, chronic hepatitis B (CHB): efficacy and safety results at week 96 [abstract no. ILC2017-RS-246]. In: International Liver Congress 2017, European Association for the study of the liver, 19–23 Apr 2017, Amsterdam.
Menendez-Arias L, Alvarez M, Pacheco B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: mechanism of action and resistance. Curr Opin Virol. 2014;8:1–9.
Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, et al. Present and future therapies of hepatitis B: from discovery to cure. Hepatology. 2015;62(6):1893–908.
Jang JH, Kim JW, Jeong SH, Myung HJ, Kim HS, Park YS, et al. Clevudine for chronic hepatitis B: antiviral response, predictors of response, and development of myopathy. J Viral Hepat. 2011;18(2):84–90.
Seok JI, Lee DK, Lee CH, Park MS, Kim SY, Kim HS, et al. Long-term therapy with clevudine for chronic hepatitis B can be associated with myopathy characterized by depletion of mitochondrial DNA. Hepatology. 2009;49(6):2080–6.
Kim SB, Song IH, Kim YM, Noh R, Kang HY, Lee H, et al. Long-term treatment outcomes of clevudine in antiviral-naive patients with chronic hepatitis B. World J Gastroenterol. 2012;18(47):6943–50.
Tak WY, Yang JM, Kim BI, Baik SK, Cheon GJ, Byun KS, et al. A randomized, open-label study comparing low-dose clevudine plus adefovir combination therapy with clevudine monotherapy in naive chronic hepatitis B patients. Hepatol Int. 2014;8(3):375–81.
Fung J, Lai CL, Yuen MF. LB80380: a promising new drug for the treatment of chronic hepatitis B. Expert Opin Investig Drugs. 2008;17(10):1581–8.
Yuen MF, Han KH, Um SH, Yoon SK, Kim HR, Kim J, et al. Antiviral activity and safety of LB80380 in hepatitis B e antigen-positive chronic hepatitis B patients with lamivudine-resistant disease. Hepatology. 2010;51(3):767–76.
Lai CL, Ahn SH, Lee KS, Um SH, Cho M, Yoon SK, et al. Phase IIb multicentred randomised trial of besifovir (LB80380) versus entecavir in Asian patients with chronic hepatitis B. Gut. 2014;63(6):996–1004.
Wang XY, Chen HS. Emerging antivirals for the treatment of hepatitis B. World J Gastroenterol. 2014;20(24):7707–17.
Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, et al. Chronic hepatitis B: what should be the goal for new therapies? Antivir Res. 2013;98(1):27–34.
Glebe D, Konig A. Molecular virology of hepatitis B virus and targets for antiviral intervention. Intervirology. 2014;57(3–4):134–40.
Lai CL, Wong D, Ip P, Kopaniszen M, Seto WK, Fung J, et al. Reduction of covalently closed circular DNA with long-term nucleos(t)ide analogue treatment in chronic hepatitis B. J Hepatol. 2017;66(2):275–81.
Scaglione SJ, Lok AS. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology. 2012;142(6):1360–8 e1.
Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology. 2009;137(5):1593–608 e1–2.
Noordeen F, Vaillant A, Jilbert AR. Nucleic acid polymers prevent the establishment of duck hepatitis B virus infection in vivo. Antimicrob Agents Chemother. 2013;57(11):5299–306.
Noordeen F, Vaillant A, Jilbert AR. Nucleic acid polymers inhibit duck hepatitis B virus infection in vitro. Antimicrob Agents Chemother. 2013;57(11):5291–8.
Mahtab MA, Bazinet M, Vaillant A. REP 9 AC: a potent HBsAg release inhibitor that elicits durable immunological control of chronic HBV infection. Hepatology. 2011;54(Suppl S1):478A.
Mahtab MA, Bazinet M, Patient R, Roingeard P, Vaillant A. Nucleic acid polymers REP 9 AC/REP 9 AC’ elicit sustained immunologic control of chronic HBV infection. Glob Antivir J. 2011;7(Suppl 1):64A.
Jansen L, Vaillant A, Stelma F, Kootstra NA, Bazinet M, Al-Mahtab M, et al. Serum HBV-RNA levels decline significantly in chronic hepatitis B patients dosed with the nucleic-acid polymer REP2139-CA. J Hepatol. 2015;62(Suppl 2):S250.
Jensen KD, Kopeckova P, Kopecek J. Antisense oligonucleotides delivered to the lysosome escape and actively inhibit the hepatitis B virus. Bioconjug Chem. 2002;13(5):975–84.
Prakash TP, Siwkowski A, Allerson CR, Migawa MT, Lee S, Gaus HJ, et al. Antisense oligonucleotides containing conformationally constrained 2’,4’-(N-methoxy)aminomethylene and 2’,4’-aminooxymethylene and 2’-O,4’-C-aminomethylene bridged nucleoside analogues show improved potency in animal models. J Med Chem. 2010;53(4):1636–50.
Robaczewska M, Guerret S, Remy JS, Chemin I, Offensperger WB, Chevallier M, et al. Inhibition of hepadnaviral replication by polyethylenimine-based intravenous delivery of antisense phosphodiester oligodeoxynucleotides to the liver. Gene Ther. 2001;8(11):874–81.
Treichel U, Meyer zum Buschenfelde KH, Dienes HP, Gerken G. Receptor-mediated entry of hepatitis B virus particles into liver cells. Arch Virol. 1997;142(3):493–8.
Yang J, Bo XC, Yao J, Yang NM, Wang SQ. Differentially expressed cellular genes following HBV: potential targets of anti-HBV drugs? J Viral Hepat. 2005;12(4):357–63.
Treichel U, Meyer zum Buschenfelde KH, Stockert RJ, Poralla T, Gerken G. The asialoglycoprotein receptor mediates hepatic binding and uptake of natural hepatitis B virus particles derived from viraemic carriers. J Gen Virol. 1994;75(Pt 11):3021–9.
Campbell JM, Bacon TA, Wickstrom E. Oligodeoxynucleoside phosphorothioate stability in subcellular extracts, culture media, sera and cerebrospinal fluid. J Biochem Biophys Methods. 1990;20(3):259–67.
Fluiter K, Mook OR, Vreijling J, Langkjaer N, Hojland T, Wengel J, et al. Filling the gap in LNA antisense oligo gapmers: the effects of unlocked nucleic acid (UNA) and 4’-C-hydroxymethyl-DNA modifications on RNase H recruitment and efficacy of an LNA gapmer. Mol Biosyst. 2009;5(8):838–43.
Cheng YC, Ying CX, Leung CH, Li Y. New targets and inhibitors of HBV replication to combat drug resistance. J Clin Virol. 2005;34(Suppl 1):S147–50.
Wu GY, Chen HS. Novel approaches towards conquering hepatitis B virus infection. World J Gastroenterol. 2007;13(6):830–6.
Janmanchi D, Lin CH, Hsieh JY, Tseng YP, Chen TA, Jhuang HJ, et al. Synthesis and biological evaluation of helioxanthin analogues. Bioorg Med Chem. 2013;21(7):2163–76.
Ye X, Zhou M, He Y, Wan Y, Bai W, Tao S, et al. Efficient inhibition of hepatitis B virus infection by a preS1-binding peptide. Sci Rep. 2016;07(6):29391.
Kaneko M, Watashi K, Kamisuki S, Matsunaga H, Iwamoto M, Kawai F, et al. A novel tricyclic polyketide, vanitaracin A, specifically inhibits the entry of hepatitis B and D viruses by targeting sodium taurocholate cotransporting polypeptide. J Virol. 2015;89(23):11945–53.
Dong Z, Ekins S, Polli JE. Structure-activity relationship for FDA approved drugs as inhibitors of the human sodium taurocholate cotransporting polypeptide (NTCP). Mol Pharm. 2013;10(3):1008–19.
Yan H, Liu Y, Sui J, Li W. NTCP opens the door for hepatitis B virus infection. Antivir Res. 2015;121:24–30.
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;13(1):e00049.
Volz T, Allweiss L, Ben MM, Warlich M, Lohse AW, Pollok JM, et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol. 2013;58(5):861–7.
Blank A, Markert C, Hohmann N, Carls A, Mikus G, Lehr T, et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J Hepatol. 2016;65(3):483–9.
Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol. 2016;65(3):490–8.
Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, et al. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol. 2014;60(4):723–31.
Watashi K, Sluder A, Daito T, Matsunaga S, Ryo A, Nagamori S, et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology. 2014;59(5):1726–37.
Ko C, Park WJ, Park S, Kim S, Windisch MP, Ryu WS. The FDA-approved drug irbesartan inhibits HBV-infection in HepG2 cells stably expressing sodium taurocholate co-transporting polypeptide. Antivir Ther. 2015;20(8):835–42.
Lucifora J, Esser K, Protzer U. Ezetimibe blocks hepatitis B virus infection after virus uptake into hepatocytes. Antivir Res. 2013;97(2):195–7.
Huang HC, Tao MH, Hung TM, Chen JC, Lin ZJ, Huang C. (−)-Epigallocatechin-3-gallate inhibits entry of hepatitis B virus into hepatocytes. Antivir Res. 2014;111:100–11.
Tsukuda S, Watashi K, Iwamoto M, Suzuki R, Aizaki H, Okada M, et al. Dysregulation of retinoic acid receptor diminishes hepatocyte permissiveness to hepatitis B virus infection through modulation of sodium taurocholate cotransporting polypeptide (NTCP) expression. J Biol Chem. 2015;290(9):5673–84.
Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6(7):226.
Lin K, Gallay P. Curing a viral infection by targeting the host: the example of cyclophilin inhibitors. Antivir Res. 2013;99(1):68–77.
Phillips S, Chokshi S, Chatterji U, Riva A, Bobardt M, Williams R, et al. Alisporivir inhibition of hepatocyte cyclophilins reduces HBV replication and hepatitis B surface antigen production. Gastroenterology. 2015;148(2):403–14.e7.
Naoumov NV. Cyclophilin inhibition as potential therapy for liver diseases. J Hepatol. 2014;61(5):1166–74.
Nilsson J, Moss S, Coates N, Bobardt M, Gallay P, Gregory M. NVP018, a cyclophilin inhibitor for treatment of chronic HBV infection. J Hepatol. 2014;60(Suppl):S423.
Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406(6797):782–7.
Pham EA, Perumpail RB, Fram BJ, Glenn JS, Ahmed A, Gish RG. Future therapy for hepatitis B virus: role of immunomodulators. Curr Hepatol Rep. 2016;15(4):237–44.
Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology. 2009;49(4):1132–40.
Funk E, Kottilil S, Gilliam B, Talwani R. Tickling the TLR7 to cure viral hepatitis. J Transl Med. 2014;14(12):129.
Chang J, Guo JT. Treatment of chronic hepatitis B with pattern recognition receptor agonists: current status and potential for a cure. Antivir Res. 2015;121:152–9.
Menne S, Tumas DB, Liu KH, Thampi L, AlDeghaither D, Baldwin BH, et al. Sustained efficacy and seroconversion with the Toll-like receptor 7 agonist GS-9620 in the Woodchuck model of chronic hepatitis B. J Hepatol. 2015;62(6):1237–45.
Wu J, Huang S, Zhao X, Chen M, Lin Y, Xia Y, et al. Poly(I:C) treatment leads to interferon-dependent clearance of hepatitis B virus in a hydrodynamic injection mouse model. J Virol. 2014;88(18):10421–31.
Jo J, Tan AT, Ussher JE, Sandalova E, Tang XZ, Tan-Garcia A, et al. Toll-like receptor 8 agonist and bacteria trigger potent activation of innate immune cells in human liver. PLoS Pathog. 2014;10(6):e1004210.
Huang LR, Wohlleber D, Reisinger F, Jenne CN, Cheng RL, Abdullah Z, et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol. 2013;14(6):574–83.
Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL, Brasky KM, et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology. 2013;144(7):1508–17, 1517.e1–10.
Gane EJ, Lim YS, Gordon SC, Visvanathan K, Sicard E, Fedorak RN, et al. The oral toll-like receptor-7 agonist GS-9620 in patients with chronic hepatitis B virus infection. J Hepatol. 2015;63(2):320–8.
Boettler T, Panther E, Bengsch B, Nazarova N, Spangenberg HC, Blum HE, et al. Expression of the interleukin-7 receptor alpha chain (CD127) on virus-specific CD8+ T cells identifies functionally and phenotypically defined memory T cells during acute resolving hepatitis B virus infection. J Virol. 2006;80(7):3532–40.
Maini MK, Boni C, Ogg GS, King AS, Reignat S, Lee CK, et al. Direct ex vivo analysis of hepatitis B virus-specific CD8(+) T cells associated with the control of infection. Gastroenterology. 1999;117(6):1386–96.
Nayersina R, Fowler P, Guilhot S, Missale G, Cerny A, Schlicht HJ, et al. HLA A2 restricted cytotoxic T lymphocyte responses to multiple hepatitis B surface antigen epitopes during hepatitis B virus infection. J Immunol. 1993;150(10):4659–71.
Rehermann B, Fowler P, Sidney J, Person J, Redeker A, Brown M, et al. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J Exp Med. 1995;181(3):1047–58.
Krebs K, Bottinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145(2):456–65.
Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–76.
Park JJ, Wong DK, Wahed AS, Lee WM, Feld JJ, Terrault N, et al. Hepatitis B virus-specific and global T-cell dysfunction in chronic hepatitis B. Gastroenterology. 2016;150(3):684–95 e5.
Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One. 2012;7(10):e47648.
Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology. 2010;138(2):682–93, 93 e1–4.
Fisicaro P, Valdatta C, Massari M, Loggi E, Ravanetti L, Urbani S, et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver T cells against HBV, but not HCV. Gastroenterology. 2012;143(6):1576–85 e4.
Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229(1):114–25.
Schurich A, Khanna P, Lopes AR, Han KJ, Peppa D, Micco L, et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology. 2011;53(5):1494–503.
Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. 2015;19(6):e1694.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9.
Bertoletti A, Gehring AJ. The immune response during hepatitis B virus infection. J Gen Virol. 2006;87(Pt 6):1439–49.
Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One. 2013;8(5):e63818.
Wilson EM, Tang L, Kottilil S. Eradication strategies for chronic hepatitis B infection. Clin Infect Dis. 2016;1(62 Suppl 4):S318–25.
Shih C, Chou SF, Yang CC, Huang JY, Choijilsuren G, Jhou RS. Control and eradication strategies of hepatitis B virus. Trends Microbiol. 2016;24(9):739–49.
Wen Y, Wang X, Wang B, Yuan Z. Vaccine therapies for chronic hepatitis B: can we go further? Front Med. 2014;8(1):17–23.
Kutscher S, Bauer T, Dembek C, Sprinzl M, Protzer U. Design of therapeutic vaccines: hepatitis B as an example. Microb Biotechnol. 2012;5(2):270–82.
Milich DR, Chen M, Schodel F, Peterson DL, Jones JE, Hughes JL. Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci USA. 1997;94(26):14648–53.
Lazdina U, Cao T, Steinbergs J, Alheim M, Pumpens P, Peterson DL, et al. Molecular basis for the interaction of the hepatitis B virus core antigen with the surface immunoglobulin receptor on naive B cells. J Virol. 2001;75(14):6367–74.
Wen YM. Antigen-antibody immunogenic complex: promising novel vaccines for microbial persistent infections. Expert Opin Biol Ther. 2009;9(3):285–91.
Wen YM, Wu XH, Hu DC, Zhang QP, Guo SQ. Hepatitis B vaccine and anti-HBs complex as approach for vaccine therapy. Lancet. 1995;345(8964):1575–6.
Donnelly JJ, Wahren B, Liu MA. DNA vaccines: progress and challenges. J Immunol. 2005;175(2):633–9.
Mancini-Bourgine M, Fontaine H, Scott-Algara D, Pol S, Brechot C, Michel ML. Induction or expansion of T-cell responses by a hepatitis B DNA vaccine administered to chronic HBV carriers. Hepatology. 2004;40(4):874–82.
Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25(2):98–104.
Yang SH, Lee CG, Park SH, Im SJ, Kim YM, Son JM, et al. Correlation of antiviral T-cell responses with suppression of viral rebound in chronic hepatitis B carriers: a proof-of-concept study. Gene Ther. 2006;13(14):1110–7.
Gilbert SC, Moorthy VS, Andrews L, Pathan AA, McConkey SJ, Vuola JM, et al. Synergistic DNA-MVA prime-boost vaccination regimes for malaria and tuberculosis. Vaccine. 2006;24(21):4554–61.
Paoletti E. Applications of pox virus vectors to vaccination: an update. Proc Natl Acad Sci USA. 1996;93(21):11349–53.
Tatsis N, Ertl HC. Adenoviruses as vaccine vectors. Mol Ther. 2004;10(4):616–29.
Sallberg M, Hughes J, Javadian A, Ronlov G, Hultgren C, Townsend K, et al. Genetic immunization of chimpanzees chronically infected with the hepatitis B virus, using a recombinant retroviral vector encoding the hepatitis B virus core antigen. Hum Gene Ther. 1998;9(12):1719–29.
Cavenaugh JS, Awi D, Mendy M, Hill AV, Whittle H, McConkey SJ. Partially randomized, non-blinded trial of DNA and MVA therapeutic vaccines based on hepatitis B virus surface protein for chronic HBV infection. PLoS One. 2011;6(2):e14626.
Kosinska AD, Zhang E, Johrden L, Liu J, Seiz PL, Zhang X, et al. Combination of DNA prime—adenovirus boost immunization with entecavir elicits sustained control of chronic hepatitis B in the woodchuck model. PLoS Pathog. 2013;9(6):e1003391.
Luo J, Li J, Chen RL, Nie L, Huang J, Liu ZW, et al. Autologus dendritic cell vaccine for chronic hepatitis B carriers: a pilot, open label, clinical trial in human volunteers. Vaccine. 2010;28(13):2497–504.
Wang FS, Xing LH, Liu MX, Zhu CL, Liu HG, Wang HF, et al. Dysfunction of peripheral blood dendritic cells from patients with chronic hepatitis B virus infection. World J Gastroenterol. 2001;7(4):537–41.
Akbar SM, Furukawa S, Hasebe A, Horiike N, Michitaka K, Onji M. Production and efficacy of a dendritic cell-based therapeutic vaccine for murine chronic hepatitis B virus carrierer. Int J Mol Med. 2004;14(2):295–9.
Akbar SM, Furukawa S, Horiike N, Abe M, Hiasa Y, Onji M. Safety and immunogenicity of hepatitis B surface antigen-pulsed dendritic cells in patients with chronic hepatitis B. J Viral Hepat. 2011;18(6):408–14.
Rigopoulou EI, Suri D, Chokshi S, Mullerova I, Rice S, Tedder RS, et al. Lamivudine plus interleukin-12 combination therapy in chronic hepatitis B: antiviral and immunological activity. Hepatology. 2005;42(5):1028–36.
Korolowicz KE, Iyer RP, Czerwinski S, Suresh M, Yang J, Padmanabhan S, et al. Antiviral efficacy and host innate immunity associated with SB 9200 treatment in the woodchuck model of chronic hepatitis B. PLoS One. 2016;11(8):e0161313.
Barnes E. Therapeutic vaccines in HBV: lessons from HCV. Med Microbiol Immunol. 2015;204(1):79–86.
Weng M, Zeng WZ, Wu XL, Zhang Y, Jiang MD, Wang Z, et al. Quantification of serum hepatitis B surface antigen in predicting the response of pegylated interferon alfa-2a in HBeAg-positive chronic hepatitis B with prior lamivudine exposure. Virol J. 2013;10:277.
Reignat S, Webster GJ, Brown D, Ogg GS, King A, Seneviratne SL, et al. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J Exp Med. 2002;195(9):1089–101.
Lin CL, Yang HC, Kao JH. Hepatitis B virus: new therapeutic perspectives. Liver Int. 2016;36(Suppl 1):85–92.
Coffin CS, Lee SS. New paradigms in hepatitis B management: only diamonds are forever. Br Med Bull. 2015;116:79–91.
Petersen J, Thompson AJ, Levrero M. Aiming for cure in HBV and HDV infection. J Hepatol. 2016;65(4):835–48.
Lin CL, Kao JH. Review article: novel therapies for hepatitis B virus cure—advances and perspectives. Aliment Pharmacol Ther. 2016;44(3):213–22.
Durantel D, Zoulim F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J Hepatol. 2016;64(1 Suppl):S117–31.
Urban S, Bartenschlager R, Kubitz R, Zoulim F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology. 2014;147(1):48–64.
Palumbo GA, Scisciani C, Pediconi N, Lupacchini L, Alfalate D, Guerrieri F, et al. IL6 inhibits HBV transcription by targeting the epigenetic control of the nuclear cccDNA minichromosome. PLoS One. 2015;10(11):e0142599.
Chen J, Zhang W, Lin J, Wang F, Wu M, Chen C, et al. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol Ther. 2014;22(2):303–11.
Revill P, Testoni B, Locarnini S, Zoulim F. Global strategies are required to cure and eliminate HBV infection. Nat Rev Gastroenterol Hepatol. 2016;13(4):239–48.
Koniger C, Wingert I, Marsmann M, Rosler C, Beck J, Nassal M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci USA. 2014;111(40):E4244–53.
Gunsar F. Treatment of delta hepatitis. Expert Rev Anti Infect Ther. 2013;11(5):489–98.
Heidrich B, Yurdaydin C, Kabacam G, Ratsch BA, Zachou K, Bremer B, et al. Late HDV RNA relapse after peginterferon alpha-based therapy of chronic hepatitis delta. Hepatology. 2014;60(1):87–97.
Alios Biopharma Inc. Study of the relative oral bioavailability of AL-3778 tablets and drug interaction with entecavir or tenofovir disoproxil fumarate in healthy volunteers [ClinicalTrials.gov identifier NCT03032536]. US National Institutes of Health, ClinicalTrials.gov. https://www.clinicaltrials.gov. Accessed 10 May 2017.
Hepatitis B Foundation. Drug Watch. Compounds in development for chronic hepatitis B. Updated May 2017. http://www.hepb.org/treatment-and-management/drug-watch. Accessed 10 May 2017.
Transgene. Safety and tolerability of TG1050: a dose-finding study [ClinicalTrials.gov identifier NCT02428400]. US National Institutes of Health, ClinicalTrials.gov. https://www.clinicaltrials.gov. Accessed 10 May 2017.
Cytheris SA. Dose escalation of interleukin-1 (IL-7) added on antiviral treatment and vaccination in HBeAg-negative chronic hepatitis B virus (HBV) infected patients (CONVERT) [ClinicalTrials.gov identifier NCT01027065]. US National Institutes of Health, ClinicalTrials.gov. https://www.clinicaltrials.gov. Accessed 10 May 2017.
Spring Bank Pharmaceuticals, Inc. A study evaluating the safety, pharmacokinetics, and antiviral efficacy of SB 9200 in subjects infected with chronic HBV (ACHIEVE) [ClinicalTrials.gov identifier NCT02751996]. US National Institutes of Health, ClinicalTrials.gov. https://www.clinicaltrials.gov. Accessed 10 May 2017.
Acknowledgements
The authors would like to express their gratitude to independent medical editor Dr. Lark Lands for her invaluable assistance in preparing the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was provided for this paper.
Conflicts of interest
Drs. Dawood, Abdul Basit, and Jayaraj report no conflicts of interest. Dr. Gish reports grants/research support from Gilead, Merck, Benitec, AbbVie; consultancy/advising for Abbott, AbbVie, Akshaya, Alexion, Arrowhead, Astra-Zeneca, Bayer AG, Bristol-Myers Squibb Company, ContraVir Pharmaceuticals, Eiger, Enyo Pharma, Genentech, Gilead Sciences, Hoffmann-LaRoche Ltd., HumAbs, Intellia, Intercept, Ionis Pharmaceuticals, Isis, Janssen, MedImmune, Merck & Co., Nanogen, Novira, and Quest; speaker’s contracts with Alexion, Bayer, BMS, Gilead Sciences Inc., Salix/Valeant, AbbVie, and Merck; and stock in Kinex, Synageva, RiboSciences, CoCrystal, and Arrowhead Pharmaceuticals. Dr. Gish has the following relationships with companies engaged in research to develop pharmaceutical agents for the treatment of chronic hepatitis B: grants/research support from Gilead Sciences, and Merck & Co.; performed as consultant and/or advisor to Akshaya Pharmaceuticals, Arbutus Biopharma Corporation, Arrowhead Research Corporation, Bristol-Myers Squibb, ContraVir Pharmaceuticals, Enyo Pharma, Gilead Sciences, HumAbs BioMed, Ionis Pharmaceuticals, Merck & Co., Nanogen Biopharmaceutical, and Novira Therapeutics; current activity with the scientific or clinical advisory boards of Arrowhead Research Corporation, Merck & Co., ContraVir Pharmaceuticals, Gilead Sciences, Isis Pharmaceuticals, Enyo Pharma, HumAbs BioMed, and Nanogen Biopharmaceutical; member of the Speakers Bureau for Bristol-Myers Squibb, Gilead Sciences, and Merck & Co.; and stock options with Arrowhead Research Corporation.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dawood, A., Abdul Basit, S., Jayaraj, M. et al. Drugs in Development for Hepatitis B. Drugs 77, 1263–1280 (2017). https://doi.org/10.1007/s40265-017-0769-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0769-2