
Abstract
Neuropsychiatric systemic lupus erythematosus (NPSLE) is a generic definition referring to a series of neurological and psychiatric symptoms directly related to systemic lupus erythematosus (SLE). NPSLE includes heterogeneous and rare neuropsychiatric (NP) manifestations involving both the central and peripheral nervous system. Due to the lack of a gold standard, the attribution of NP symptoms to SLE represents a clinical challenge that obligates the strict exclusion of any other potential cause. In the acute setting, management of these patients does not differ from other non-SLE subjects presenting with the same NP manifestation. Afterwards, an individualized therapeutic strategy, depending on the presenting manifestation and severity of symptoms, must be started. Clinical trials in NPSLE are scarce and most of the data are extracted from case series and case reports. High-dose glucocorticoids and intravenous cyclophosphamide remain the cornerstone for patients with severe symptoms that are thought to reflect inflammation or an underlying autoimmune process. Rituximab, intravenous immunoglobulins, or plasmapheresis may be used if response is not achieved. When patients present with mild to moderate NP manifestations, or when maintenance therapy is warranted, azathioprine and mycophenolate may be considered. When symptoms are thought to reflect a thrombotic underlying process, anticoagulation and antiplatelet agents are the mainstay of therapy, especially if antiphospholipid antibodies or antiphospholipid syndrome are present. Recent trials on SLE using new biologicals, based on newly understood SLE mechanisms, have shown promising results. Based on what we currently know about its pathogenesis, it is tempting to speculate how these new therapies may affect the management of NPSLE patients. This article provides a comprehensive and critical review of the literature on the epidemiology, pathophysiology, diagnosis, and management of NPSLE. We describe the most common pharmacological treatments used in NPSLE, based on both a literature search and our expert opinion. The extent to which new drugs in the advanced development of SLE, or the blockade of new targets, may impact future treatment of NPSLE will also be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A strict differential diagnosis and individualization of treatment, depending on the neuropsychiatric presentation and severity of symptoms, are crucial in neuropsychiatric systemic lupus erythematosus (NPSLE). |
In clinical practice, therapies are directed to a presumptive pathophysiologic process (inflammatory, thrombotic, or both). |
New biological drugs against different novel targets are currently being studied in systemic lupus erythematosus (SLE). Some of these targets are thought to contribute to NPSLE pathogenesis and may offer potential therapeutic options. |
Increased understanding of the pathogenesis of NPSLE may lead to the development of novel immunomodulatory therapies that may replace the current therapies. |
1 Introduction
Systemic lupus erythematosus (SLE) is a chronic multisystem inflammatory autoimmune disease with a waxing and waning course and a broad spectrum of clinical presentations [1]. The involvement of the nervous system in SLE patients leads to a nonspecific and heterogeneous group of neuropsychiatric (NP) manifestations [2]. A major issue in clinical evaluation is the attribution of NP symptoms to SLE. No laboratory or radiological biomarker nor other formal system exists for establishing a diagnosis and guiding therapy decisions in neuropsychiatric SLE (NPSLE). In clinical practice, an individual multidisciplinary diagnostic and therapeutic approach based on the suspected cause and severity of symptoms is recommended [3].
To date, only one randomized controlled treatment trial in NPSLE has been undertaken. In addition to the scarcity of trials, several therapeutic strategies report benefits in different aspects of NPSLE management: primary prevention, resolution and stabilization of acute symptoms, maintenance therapy, and secondary prevention.
We provide a comprehensive and critical review of the literature on the epidemiology, classification, pathophysiology, and diagnostic approach of NPSLE, and discuss the current pharmacological armamentarium and potential future therapies to treat patients with NPSLE.
2 Epidemiology
Epidemiological studies have suggested differences in the prevalence of SLE and NPSLE according to age, sex, and ethnicity. It is well-established that SLE is substantially increased in females of child-bearing age (female:male ratio is 8–15:1) [4]. Although a greater incidence of neurological involvement in females and an increased risk of seizures in males have been reported, there is limited evidence to support an association between gender and NPSLE since most of the studies do not provide correction for important confounders such as comorbidities [5–7]. NP manifestations are more frequently seen in African descendants, Hispanics, and Asians than in White individuals;[8–10] however, NP damage occurs more frequently among White patients, as described in the LUMINA and Maryland cohorts [11, 12].
NP manifestations usually occur early in the course of SLE, and in 39–50 % of patients it is the presenting symptom of SLE [13]. An important variability in NPSLE prevalence (range 4–91 %), incidence (range 8–40 %), and frequency of specific NPSLE events has been reported [13–15]. Unterman et al. reported a meta-analysis pooling all available studies, resulting in a population of 5057 SLE patients. The prevalence of NPSLE was 44.5 % in prospective studies versus 17.6 % in retrospective studies [16]. Disparities in frequency have been mainly attributed to differences within the definition used and stringency in attributing the events to SLE. Most of these studies included minor, nonspecific symptoms (e.g. mild depression or anxiety) that, to some degree, are known to be investigator-dependent [17]. After exclusion of these minor events and peripheral nervous system (PNS) syndromes, Kampylafka et al. reported an NPSLE prevalence of 4.3 % and an incidence rate of 7.8/100 person years [15]. Subsequently, the proportion of major central nervous system (CNS) increased significantly when compared with data reported previously (i.e. myelopathy with a prevalence of 22 % instead of 1–1.5 %) [14, 15].
NPSLE is a severe complication of SLE that contributes considerably to quality of life, morbidity and mortality. NP events are associated with a lower quality of life over time, with poor prognosis in SLE patients [18]. We recently reported a tenfold increase in mortality rate in NPSLE compared with the general population [19].
3 Classification of Neuropsychiatric Systemic Lupus Erythematosus (NPSLE)
In 1999, the American College of Rheumatology (ACR) published a set of NPSLE case definitions, including 12 CNS and 7 PNS manifestations. The CNS manifestations can be divided into four psychiatric syndromes and eight neurological syndromes, and were also divided into focal (events presenting as focal neurologic deficits) and diffuse (including cognitive disorder, mood disorder, psychosis, acute confusional state, and anxiety disorder) (Table 1) [20]. Although this standardized approach to categorize NP events in SLE patients has improved the description and classification of NPSLE in clinical studies, its usefulness in clinical practice is limited. Several CNS syndromes (headache, anxiety, mood disorder, and mild cognitive disorder) included in these definitions are nonspecific. Although frequently seen in SLE patients, these syndromes may only be attributed to an SLE in selected cases within an appropriate clinical context [21, 22]. Other CNS manifestations not included in the ACR definitions, such as neuromyelitis optica or posterior reversible encephalopathy syndrome, have been increasingly recognized to be linked to SLE [23, 24].
In a clinical setting, NPSLE can also be classified according to the suspected underlying pathophysiologic process. Two different pathophysiologic processes are recognized to contribute to NP symptoms in NPSLE: (1) inflammatory, related to a pro-inflammatory and/or autoimmune-mediated cause; and (2) thrombotic/ischemic, associated with vascular occlusion, microangiopathy, and hemorrhage (see below). In a considerable proportion of NPSLE patients, both pathophysiologic changes have been reported to coexist and to manifest as a wide heterogeneous group of NP features. Both ischemic and inflammatory NPSLE have been included in the term primary NPSLE. In addition, secondary NPSLE refers to SLE patients whose NP symptoms are due to the medication for SLE or to SLE-related organ damage [3].
4 Pathogenesis
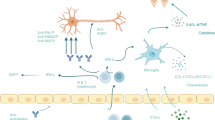
SLE is characterized by the loss of tolerance to nuclear antigens, the formation of autoantibodies, and immune complexes, resulting in complement activation, cell destruction and tissue inflammation. Prominent features are alterations in B- and T-cell activation, aberrant clearance of apoptotic material, and an activated type I interferon (IFN) system [1]. The pathogenic processes that lead to damage or dysfunction in the nervous system of SLE patients, and results in pathophysiological changes and ultimately in clinical manifestations, remains poorly understood.
Dozens of risk factors indicative of putative mechanisms have been proposed as candidates in the genesis of nervous system involvement in SLE (Table 2) [2, 25]. Two separate main pathogenic mechanisms are hypothesized to lead to NPSLE [3, 26].
-
1.
Autoimmune or inflammation characterized by brain dysfunction due to autoantibodies or inflammatory mediators with either a disrupted blood–brain barrier (BBB) or intrathecal formation of immune complexes and the presence of inflammatory mediators. Neuronal dysfunction may be induced directly by these mediators or indirectly through activation of other neural cells.
-
2.
Vascular injury and occlusion characterized by a thrombotic process of the large and small (microangiopathy) intracranial vessels due to autoantibody-mediated vascular injury, immune complexes, complement deposition, leukoagglutination, and accelerated atherosclerosis.
Several genetic studies implicate the possible role of mutations in TREX1, a gene encoding the three-prime repair exonuclease 1 (DNase III), in NPSLE [27]. Polymorphisms in this gene have been associated with neurological involvement in European SLE patients, especially with seizures [28]. Loss-of-function mutations in TREX1 have been shown to augment production of type I IFNs in mice and also in a patient with early-onset cerebral NPSLE [29, 30]. Furthermore, the HLA-DRB1*04 and rs10181656(G) alleles have been associated with ischemic cardiovascular disease (CVD) in White SLE patients, independent of the cardiovascular traditional risk factors and anticardiolipin antibody (aCL) status [31, 32].
Autoantibodies are thought to play a crucial role in the NPSLE pathogenesis. Brain tissue-reactive antibodies in NPSLE can be synthesized in the CNS or in peripheral organs (lymph nodes and bone marrow). In the latter, these autoantibodies must pass through the BBB of SLE patients to exert an effect upon neurons. Why and how the integrity of the BBB is compromised is not fully understood. The majority of studies simply report the higher presence of a certain antibody in serum and/or cerebrospinal fluid (CSF) of NPSLE patients, and none of these findings have remained as reproducible as to become a specific biomarker for NPSLE or any individual NP manifestation. Although no specific autoantibodies have been identified, several studies have confirmed an important association between antiphospholipid antibodies (aPL), especially lupus anticoagulant (LAC), and antiribosomal P antibodies, and cerebrovascular disease and psychosis, respectively [33].
There is also evidence that accelerated atherosclerotic disease occurring in SLE patients can lead to early cerebrovascular disease. Factors contributing to premature atherosclerotic vascular disease include traditional factors such as smoking, dyslipidemia, increasing age, obesity, hypertension, and longer disease duration, in addition to other factors such as homocysteinemia, higher damage score, duration and cumulative doses of corticosteroids, and low levels of vitamin D [34–37].
A substantial need exists for developing and validating a range of laboratory and radiological biomarkers to reliably capture all the different aspects of NPSLE heterogeneity and to further improve its prevention, diagnosis, and therapy.
5 Diagnostic Approach in NPSLE
In SLE, <40 % of the NP symptoms will be attributed to SLE-induced nervous system damage. In the remaining cases, other causes (i.e. therapy, primary NP disorders) will better explain these symptoms. Given the absence of a gold standard in the diagnostic approach, NPSLE remains a diagnosis per exclusionem and is mainly based on expert opinion. In all patients, there is an obligation to first exclude other causes such as infection, coincidental disease processes, metabolic abnormalities, or drug side effects. In SLE patients presenting with unexplained NP symptoms or signs suggestive of NP disease, the first step would be to evaluate and characterize the NP symptoms, similarly to patients without SLE [38]. Neurological assessment should be focused on headache, signs of seizures, alertness, and motor and sensory deficits, while psychiatric evaluation should assess behavior, cognition, perception, and thinking, as well as mood and affect. Formal neuropsychological testing must be carried out when there is a clinical suspicion of impaired cognitive ability. General SLE activity must be assessed by a rheumatologist, which may contribute to the attribution of NP events to SLE, especially diffuse manifestations. Symptoms of atherosclerotic disease, thrombotic events, and cardiovascular risk factors should be assessed [3, 26].
The diagnostic work-up of these patients should consider all investigations that should be carried out in non-SLE patients presenting with the same manifestations [26, 38]. In clinical practice, NPSLE diagnosis is achieved case-by-case using different clinical, laboratory, electrophysiological, and neuroimaging data, depending on the clinical presentation (Table 3). Among all circulating autoantibodies, aPL, including aCL, LAC, and β2-glycoprotein antibodies, provide the greatest diagnostic information in NPSLE, especially in patients with focal NP events such as cerebrovascular disease and seizures [39]. It has been suggested serum antiribosomal P antibodies are specifically related to lupus psychosis. An association between NPSLE and anti-NMDA (N-methyl-d-aspartate antibodies) has also been suggested but studies are contradictory and its presence is not associated with a specific NPSLE syndrome [33]. Autoantibodies to aquaporin 4 can help in the diagnostic process of a patient presenting with myelopathy and optic neuritis [23]. The measurement of autoantibodies or cytokines in CSF is not recommended in clinical practice at this time due to the lack of specificity [26].
Magnetic resonance imaging (MRI) is the neuroimaging technique of choice in NPSLE and remains the neuroimaging test used in clinical practice. This technique is able to localize abnormalities in the brain and spine, allowing the identification of lesions associated with NPSLE (e.g. infarcts or myelopathy) and many differential disorders (e.g. tumors or infections). However, MRI is nonspecific and in a significant number of patients no abnormalities or only aspecific white matter hyperintensities are found, independent of the NPSLE syndrome and severity [40].
Advanced neuroimaging methods based on MRI (diffusion-weighted imaging, proton magnetic resonance spectroscopy, and magnetic transfer imaging) and nuclear imaging testing (positron emission tomography and single photon emission computed tomography) have been proposed to be useful in the identification of NP involvement in SLE patients; however, the clinical relevance of these tests in individual patients requires further evidence and validation. Since no single imaging test covers all the different mechanisms leading to brain injury occurring in these patients, multimodal imaging has been proposed as a future potential standard of practice in NPSLE [41].
Electrophysiological studies are used when seizures and neuropathies are suspected clinically. These tests will help in the diagnostic process of the neurological syndrome; however, there are no specific signs for NPSLE. Electroencephalography alterations may be somehow useful in guiding therapy. Focal epileptiform discharges would be more suggestive of an ischemic nidus compared with diffuse disorganized activity, which is more suggestive of inflammation. Typical epileptiform patterns are present in less than half of the SLE patients with seizure, which may be predictive of seizure recurrence [42, 43]. Florica et al. described the electrophysiological abnormalities in a large SLE cohort. In total, 8 % of these patients had a neuropathy associated with SLE. Axonal neuropathy was seen in 70 % of patients, and signs of demyelination was seen in 20 % of patients [44].
Due to the lack of specificity of all the tests used in the diagnostic process, the attribution of NP to SLE remains a challenge. Bortoluzzi et al. proposed an algorithm that may assist the rheumatologist in the attribution of NP events to SLE [45]. We have previously recommended multidisciplinary expert consensus after a standardized assessment as the best strategy for diagnosing and classifying NPSLE. In some cases, the diagnosis will be inevitably presumptive; therefore, patients must be prospectively followed and reanalyzed to avoid misclassification [3].
6 Therapeutic Strategies
Once other potential causes are ruled out and NP symptoms of the patient are attributed to SLE, an individualized therapeutic strategy, depending on the presenting manifestation and severity, should be started. In 2010, the European League Against Rheumatism (EULAR) published a series of recommendations for the management of NPSLE. The management of these patients always includes a general therapeutic approach that does not differ from other non-SLE individuals presenting with the same NP manifestation. Correction of aggravating factors, nonpharmacological interventions, and symptomatic therapy when appropriate may initially be considered as general treatment [38].
The potential role of some therapies (e.g. hydroxychloroquine) in the primary prevention of major NPSLE, especially cerebrovascular disease, has also been suggested [46].
Specific NPSLE therapy remains relatively empirical due to the scarcity of controlled trials. In clinical practice, depending on the suspected underlying pathophysiological process, therapy will be directed at inflammation or at prevention of ischemic events [3]. Manifestations that are thought to reflect an immune-inflammatory state or, in the presence of generalized lupus activity, initiation of immunosuppressive therapy is warranted (corticosteroids alone or in combination with another immunosuppressant), with the main objective of resolving/stabilizing symptoms. On the other hand, anticoagulation and antiplatelet agents are the mainstay of secondary prevention after ischemic NPSLE, especially in the presence of aPL (Fig. 1) [38]. Some authors have suggested that when ischemic and inflammatory NPSLE syndromes coexist, a broad therapy approach with both immunosuppressive and anticoagulation and/or antiplatelet therapy should be considered. In the Leiden NPSLE clinic, a treatment algorithm was developed based on available evidence and our own expertise, although the treatment of NPSLE patients must always be individually tailor made (Fig. 2; Table 4) [38, 47].

Therapeutic options in NPSLE. aCL anticardiolipin antibodies, β2GP I β2-glycoprotein I, LAC lupus anticoagulant, NPSLE neuropsychiatric systemic lupus erythematosus

6.1 General Treatment
6.1.1 Symptomatic Therapy
The management of patients with NPSLE is multimodal. In several NPSLE syndromes, symptomatic therapy is needed. In SLE patients with mood disorders, psychosis, seizure disorders, movement disorders, and headaches, current drugs used to treat these symptoms are often the first treatment step, without taking into account NPSLE as the potential underlying cause. In mild NP manifestations, this symptomatic therapy can be sufficient; however, more severe NPSLE manifestations or inadequate response to symptomatic treatment warrants additional therapy with immunosuppressive and/or antithrombotic medication.
Antidepressive and antipsychotic agents, as well as anxiolytics, are prescribed according to the standard indications in psychiatric disorders. Antiepileptic therapy is started when high-risk features are present: second seizure (>24 h after the first event), serious brain injury, brain structural abnormalities (MRI), focal neurological signs, partial seizure as the first seizure, and epileptiform EEG discharges. Generalized seizures are usually managed with phenytoin or barbiturates, and partial complex seizures are usually managed with carbamazepine, clonazepam, valproic acid, or gabapentin. Symptomatic therapy in movement disorders consists of dopamine agonists. Nonsteroidal anti-inflammatory drugs can be added for symptomatic pain relief and migraine treatment for specific headaches [38, 48].
6.1.2 Nonpharmacological Intervention
Cognitive complaints in SLE patients are common, with most studies reporting a prevalence ranging from 17 to 66 % [26]; however, most cases of cognitive dysfunction are mild and immunosuppressive therapy is not indicated in these patients. Cognitive dysfunction has been associated with psychosocial factors such as fatigue, sleep deprivation, pain, depression, and anxiety. No consistent evidence-based symptomatic therapy exists for cognitive dysfunction in SLE. It has been reported that some patients with depression and cognitive dysfunction may benefit from antidepressants [49]. In patients who reported cognitive dysfunction but were not globally impaired on neuropsychological testing, the positive effects of an 8-week psychoeducational group intervention were reported to improve memory self-efficacy, memory function, and the ability to perform daily activities [50]. A study evaluating a 6-month psychoeducational group intervention in a group of 34 SLE patients also resulted in a significant and sustained improvement in coping skills of SLE patients and hence in their quality of life [51].
6.2 Primary Prevention
6.2.1 Antimalarial Drugs
Hydroxychloroquine and chloroquine are 4-aminoquinolines, and quinacrine is an acridine derivative. The use of antimalarials as a treatment for SLE became popular after reports were first published in the 1950s [52]. Nowadays, the prescription of hydroxychloroquine (200 or 400 mg/day) is considered mandatory in the treatment of SLE, and is recommended during the whole course of the disease, independently of severity, and even continued during pregnancy. This therapy is extensively used for cutaneous and musculoskeletal involvement [38, 53]. Widespread use of chloroquine and hydroxychloroquine led to recognition of retinal toxicity as the most frequent complication. Quinacrine differs from hydroxychloroquine and chloroquine in that it has no recognized retinal toxicity. Other known but uncommon antimalarial side effects are dermatitis, gastrointestinal symptoms, leukopenia, thrombocytopenia, aplastic anemia and cardiotoxicity [54]. Moreover, these drugs are also known as a rare cause of psychosis, and chloroquine has been associated with the occurrence of epileptic seizures in patients with a history of epilepsy [55, 56]. Although no studies specifically address its effect on NP symptoms, a preventive role of these drugs in CNS lupus has been suggested, especially in cerebrovascular disease [46, 57].
A reduction of mortality in multiple SLE cohorts, and reduced accrual of damage, was shown in SLE patients treated with antimalarials [58]. Moreover, several studies using antimalarials have demonstrated that immunologic effects can reduce the risk of CVD, such as improvement of dyslipidemias, prevention of diabetes, and reducing aPL titers [59–62]. In an SLE cohort study of 1150 patients, antimalarials had an independent protective effect on the development of metabolic syndrome over time, showing its potential as an atheroprotective drug in these patients [63].
Antithrombotic effect is another known additional benefit of antimalarials. Jung et al. demonstrated how antimalarial drugs were associated with a 68 % reduction in the risk of all thrombovascular events in SLE [64]. Other prospective SLE studies have also found a positive effect of hydroxychloroquine on reducing the risk of future thrombotic events [60, 65].
Another potential benefit attributed to antimalarial drugs in SLE is the protective effect against seizures, such as was reported in the LUMINA cohort [66]. Recently, another cohort study on 1631 SLE patients has confirmed this association. The mechanism of how the protection occurs is unknown [67].
6.2.2 Statins
Statins act by competitive inhibition of the hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase enzyme, blocking the transformation of HMG-CoA into mevalonate and subsequently inhibiting cholesterol synthesis [68]. This therapy is used extensively to treat hypercholesterolemia. A reduction in levels of low-density lipoprotein (LDL)-cholesterol have demonstrated a marked benefit in the prevention of CVD and a dramatic reduction of morbidity and mortality in established CVD [68, 69].
Some immunomodulating effects, such as reduction of inflammatory cytokines and adhesion molecules, prevention of endothelial cell activation induced by aPL, inhibition of T-cell function, and reduction of the number and activity of inflammatory cells in atherosclerotic plaque have been attributed to statins [68, 70, 71]. Statins may improve the regulation of the inflammatory processes involved in atherosclerosis [72].
All these properties have been reported to be of interest in SLE since this disease is associated with an increased risk of accelerated atherosclerosis not fully explained by traditional risk factors [35]. For some authors, SLE is considered a coronary heart disease risk factor equivalent to diabetes. It has been proposed that as the 10-year cardiovascular risk charts do not take lupus into account as a risk factor, it results in undertreatment of these patients. Based on this, Elliot and Manzi, as well as other authors, have proposed an LDL goal of <100 mg/dl in all SLE patients and <70 mg/dl for those SLE patients with subclinical CVD, known stroke/transient ischemic attack (TIA) or other cardiovascular event, peripheral vascular disease, or diabetes [73, 74]. However, to date no studies have demonstrated the efficacy of statin therapy as primary or secondary cardiovascular prevention in SLE patients. The Lupus Atherosclerosis Prevention Study, a 2-year trial of atorvastatin in 200 adult patients with SLE without clinical CVD, showed no benefit in the primary (coronary artery calcium) or secondary (carotid intima media thickness and carotid plaque) atherosclerosis outcomes [75]. The APPLE trial, a recently published 3-year prospective study on pediatric SLE, reported no benefit of atorvastatin use on preclinical CVD markers [76].
From our point of view, and as other authors have pointed out, the lack of evidence does not support the widespread use of statins in all SLE patients as primary prevention. Subsequently, they recommend the use of statins in SLE patients with hyperlipidemia who meet criteria for their use based on standard CVD guidelines such as the National Cholesterol Education Program [77]. Furthermore, SLE patients with a stroke or TIA presumed to be of atherosclerotic origin must be treated according to the use of the American Stroke Association Guidelines. The use of statins is recommended in these patients when the LDL cholesterol level is ≥100 mg/dL. Statin therapy may also be considered when the LDL cholesterol level is <100 mg/dL, but with less levels of evidence [78].
6.3 Management of Inflammatory NPSLE
6.3.1 Corticosteroids
Glucocorticoids are 21-carbon steroid hormones. The biological effects of glucocorticoids are mediated via the glucocorticoid receptor. These hormones are involved in the regulation of the immune response and inflammation, and also play an important role in various metabolic processes. Glucocorticoids, especially prednisone, have been the hallmark medication for SLE. This therapy offers the most immediate anti-inflammatory effect of all immunosuppressive therapies and is subsequently the more widely used therapy to control mild–severe flares of SLE. The basis for the use of different dosages of glucocorticoids in a specific clinical setting in SLE is essentially empirical [79]. Glucocorticoids are also known to induce an important number of undesirable side effects, including, among others, hypertension, dyslipidemia, osteoporosis, cataracts, glaucoma, electrolyte alteration, diabetes, Cushing’s syndrome, peptic ulcer, risk of infection, and reactivation of latent viruses. Overall, prolonged application is a high-risk factor for these side effects, whereas total dose is of secondary importance [80]. Furthermore, several NP disturbances have been related to the use of glucocorticoids. For example, approximately 10 % of patients treated with 1 mg/kg/day or more of prednisone will have a glucocorticoid-induced psychiatric disease. Depression, hypomania, and overt psychosis are known glucocorticoid-induced psychiatric manifestations. Symptoms usually occur within 8 weeks of initiation or augmentation of glucocorticoids and resolve completely in most cases through dose reduction, which may help in the differentiation with NPSLE [81].
In NPSLE patients, although corticosteroids are commonly used when NPSLE is thought to reflect an inflammatory process, this practice can only be substantiated by clinical experience [38]. Despite the lack of solid evidence, in severe NPSLE manifestations an accepted practice consists of 1 g of methylprednisolone intravenously for 3 consecutive days followed by prednisolone orally (starting dose 1 mg/kg/day), with a tapering scheme of 3–12 months. In less severe manifestations, the starting dose is usually 0.5–1 mg/kg/day, and tapering schemes are very diverse.
Although the benefits of glucocorticoids in an acute setting are unquestionable, some studies have addressed the important influence of corticosteroids, especially prolonged use, on future damage accrual in SLE patients [82–84]. Glucocorticoids may aggravate underlying metabolic abnormalities and other factors contributing to the risk of clinical accelerated atherosclerosis, which may lead to early cerebrovascular disease. Zonana-Nacach et al. showed how each 2-month exposure to high-dose prednisone was associated with a 1.2-fold increase in the risk of stroke in SLE [82]. Other authors have suggested that doses <7.5 mg/day, as well as methylprednisolone pulses, may not be associated with damage accrual [83].
6.3.2 Cyclophosphamide
As an alkylating agent, cyclophosphamide adds an alkyl group to DNA, thereby interfering with DNA replication. Cyclophosphamide is a prodrug that is converted by liver cytochrome 450 enzymes to its metabolite 4-hydroxy cyclophosphamide. Both the dose and duration of therapy will influence the degree of inhibition of immune function. Cyclophosphamide is used for the most serious SLE involvements [85]. In SLE patients, cyclophosphamide is mostly administered in an intermittent intravenous schedule instead of oral treatment. This was established by two trials performed at the National Institutes of Health (NIH) [86, 87]. The so-called NIH regimen has been used for decades in severe SLE flares. This regimen consists of monthly (for 6 months) and then quarterly (for up to 1 year after complete remission) doses of 0.75–1.5 g/m2 intravenous cyclophosphamide, combined with intravenous methylprednisolone. In order to minimize the side effects of this drug, low-dose intravenous regimens (six fortnightly pulses of a fixed dose of 500 mg) were later introduced in patients with lupus nephritis. Ten-year follow-up data of the Euro-Lupus Nephritis Trial comparing both doses of cyclophosphamide showed that this low-dose protocol achieved results comparable to the NIH regimen [88].
Cyclophosphamide has significant side effects. Common complications are alopecia, nausea and vomiting, and bone marrow depression. Although severe myelotoxicity is rare, myelosuppression consisting primarily of leukopenia is probably the most significant of the cyclophosphamide-induced toxicities. Leukopenia and granulocytopenia increase the patient’s risk of infection [89, 90]. Other organ-specific toxicities of cyclophosphamide are hemorrhagic cystitis, cardiotoxicity, interstitial pneumonitis, pulmonary fibrosis, and cervical intraepithelial neoplasia [91]. Azoospermia and ovarian failure are also well-documented consequences of long-term therapy. The latter association is largely dependent on its cumulative dose, and the effect is stronger with the patient’s increasing age [92, 93].
Severe NPSLE manifestations, mainly CNS involvement, have been treated with cyclophosphamide. Several case series have described positive effects of treatment with cyclophosphamide in severe NPSLE manifestations in both adults and children [94, 95]. A report from Mok et al. demonstrated a favorable response of 13 patients with lupus psychosis to 6 months of treatment with oral cyclophosphamide (1–2 mg/kg/day) and oral prednisone (1 mg/kg/day), with the dose tapered gradually, followed by azathioprine (1–2 mg/kg/day) [96]. A retrospective study of 31 NPSLE patients suggested patients benefited from concomitant glucocorticoids and monthly intravenous cyclophosphamide (250–1000 mg/m2) [97]. Ramos et al. retrospectively analyzed 25 NPSLE patients with CNS involvement. All patients were treated with weekly low-dose cyclophosphamide pulses (500 mg/m2) and all but one patient achieved a good response after a mean of 11 days without major side effects [98]. Stojanovich et al. conducted a study on 60 NPSLE patients and compared the outcomes of two groups, one receiving monthly low-dose intravenous cyclophosphamide (200–400 mg/m2) plus prednisone versus prednisone alone. They concluded that patients treated with cyclophosphamide showed more clinical and electrophysiological improvement on cerebral function [99].
Cyclophosphamide is the only therapy tested in a small, randomized controlled clinical trial in NPSLE. This trial compared intravenous methylprednisolone with intravenous cyclophosphamide. All patients received methylprednisolone 1 g/day for 3 days as induction treatment. This was followed by methylprednisolone 1 g/month for 4 months, then bimonthly for 6 months and subsequently every 3 months for 1 year, or cyclophosphamide 750 mg/m2 monthly for 1 year and then every 3 months for another year. Furthermore, oral prednisone (1 mg/kg/day) was started on the fourth day of treatment (1 mg/kg/day), for no more than 3 months, and tapered according to disease activity/remission. Treatment response, defined as 20 % improvement from basal conditions by clinical, serological, and specific neurological measures at 24 months, was observed in 94.7 % (18/19) of patients using intravenous cyclophosphamide compared with 46.2 % (6/13) of patients in the methylprednisolone group. Other benefits of cyclophosphamide were a reduction in prednisone requirements and electroencephalographic improvement. No statistically significant differences in adverse effects were reported between the groups [100].
A recent Cochrane systematic review remarked that this study was the only very-low-quality evidence that cyclophosphamide is more effective in reducing NPSLE symptoms compared with methylprednisolone [101]. Besides the lack of good evidence, in clinical practice it is common to treat severe manifestations of NPSLE thought to reflect an inflammatory/neurotoxic process with an induction regimen of cyclophosphamide (500–750 mg/m2/month) in combination with glucocorticoids (prednisolone 1 mg/kg/day, with a tapering schedule of 3–6 months with or without a previous 3 days of intravenous methylprednisolone 500–1000 mg/day). After 6 months of therapy, cyclophosphamide can be continued in the same dose every 3 months for 18 months, or maintenance therapy during 1 year with other immunosuppressive therapy, for example azathioprine, may be considered to prevent recurrence [38].
From our point of view, since there is no evidence to support one or the other regimen, azathioprine may be a better option due to its better side effect profile; however, this decision must be taken based on expert opinion after evaluation of an adequate response to induction therapy with cyclophosphamide, taking into account the type of NPSLE syndrome, other concomitant organ SLE involvements, age of the patient, and the desire to become pregnant in the future.
6.3.3 Azathioprine
Azathioprine is a prodrug that is rapidly converted to mercaptopurine and methylnitroimidazole by thiopurine methyltransferase (TPMT). Mercaptopurine is known to inhibit several enzymatic process involved in purine synthesis, which affect both cellular and humoral immune functions [102].
The positive effects of this drug in SLE patients, especially in the prevention of flares, have been previously described in the 1970s [103]. Nowadays, azathioprine (2–3 mg/kg/day) is mainly used in SLE patients presenting with arthritis, mucocutaneous manifestations, and serositis, and as maintenance therapy in lupus nephritis [57].
The side effects of azathioprine include bone marrow suppression, hepatotoxicity, gastrointestinal intolerance, and a mild increased risk for infections [104]. Furthermore, in the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression; therefore, determination of TPMT activity before initiating therapy with azathioprine is recommended by some authors [102].
The effect of azathioprine on NP manifestations in SLE has been scarcely studied. Due to its relatively mild side effect profile, azathioprine is frequently used for maintenance or as a glucocorticoid-sparing agent; however, controlled trials are lacking. A study including 68 SLE patients with a poor prognosis due to renal or NP manifestations showed that the 54 patients treated with azathioprine had a significantly improved long-term survival and fewer hospitalizations compared with those who did not receive this drug. The authors remarked on the importance of this therapy in the maintenance of clinical remission and inhibition of further deterioration of CNS function [105]. However, in a randomized trial of azathioprine (3–4 mg/kg/day plus prednisone initially) versus prednisone (60 mg/day initially) in 24 patients with life-threatening SLE, no significant short-term benefit was observed for azathioprine on NP and other SLE manifestations [106].
A single-center cohort study by Lim et al. reported the clinical course and response to immunosuppressive treatment of 53 children with psychiatric manifestations attributed to SLE (all with cognitive dysfunction and 40 with psychosis). Eighteen of 32 patients treated with azathioprine required a change to cyclophosphamide due to poor response; however, none of the patients receiving cyclophosphamide required a change [107].
One open-label trial has been conducted using azathioprine as maintenance therapy after 6 months of oral cyclophosphamide and oral prednisone in a very small case series of 13 patients with lupus psychosis. After a follow-up period of 86 ± 51 months, only one patient (8 %) had a relapse of psychosis and three patients developed other NP symptoms [96]. In patients with psychiatric disorders in the context of generalized SLE activity, induction therapy with glucocorticoids and cyclophosphamide followed by maintenance with azathioprine may result in a significant improvement (60–80 % response), although relapses may occur (up to 50 %) [38].
In addition to these results, and although the use of this therapy in NPSLE is not supported by current evidence, azathioprine is widely used in clinical practice as maintenance therapy after cyclophosphamide induction in severe NPSLE symptoms, and as the first option in mild NPSLE symptoms as a glucocorticoid-sparing agent. Further studies are needed.
6.3.4 Mycophenolate Mofetil
Mycophenolate mofetil is a prodrug of mycophenolic acid, an inhibitor of lymphocyte proliferation via reversible inhibition of inosine-5′-monophosphate dehydrogenase, which is critical for the de novo synthesis of guanosine nucleotides. Mycophenolic acid suppresses antibody formation, cell-mediated immune response, the expression of adhesion molecules, and recruitment of lymphocytes and monocytes to sites of inflammation [108].
Historically, this drug has been used to reduce acute and chronic rejection in allograft recipients. In SLE patients, mycophenolate mofetil is widely used as the first-line option for both induction and maintenance therapy of lupus nephritis [110, 111]. Several observational studies have suggested the potential benefit of mycophenolate mofetil in nonrenal manifestations of SLE, especially in hematological and dermatological manifestations; however, none focused on NP manifestations [112].
Mycophenolate mofetil and mycophenolic acid are administered orally at a dose of 1000–3000 mg/day and 1080–1440 mg/day, respectively. The main adverse effects of mycophenolate mofetil are gastrointestinal intolerance (nausea, abdominal pain, mild to moderate diarrhea), bone marrow suppression, and infections [109].
Ginzler et al. analyzed, as a secondary endpoint, the nonrenal flares of the Aspreva Lupus Management Study, which compared the effect of mycophenolate mofetil with intravenous cyclophosphamide in 370 patients with lupus nephritis. Three NPSLE patients were treated with mycophenolate mofetil and prednisolone after 3-day methylprednisolone pulses; two patients responded partially, whereas one had complete recovery. Moreover, two patients in the cyclophosphamide group developed new neurologic manifestations versus none in the mycophenolate mofetil group [113]. In a SLE cohort study of 75 patients who were all treated with mycophenolate mofetil, neurologic manifestations improved in two of two patients, but new neurologic manifestations occurred in six patients [114].
Other authors have described individual cases or small case series of NPSLE patients treated with mycophenolate mofetil, mainly as maintenance therapy after initial treatment with cyclophosphamide [109, 115–118]. Three patients with myelopathy were treated with mycophenolate mofetil as the first immunosuppressive agent. One patient did not respond, one patient achieved partial recovery, and the other patient achieved complete recovery [109, 115].
Saison et al. published a series of 20 SLE patients with myelopathy. In nine patients, mycophenolate mofetil was chosen as maintenance therapy. Four of these patients relapsed at least once in the first 2 years [119].
In general, the efficacy of this drug in NPSLE patients is very modest. Furthermore, since most patients were simultaneously or previously treated with high-dose glucocorticoids, intravenous immunoglobulins (IVIGs), or another immunosuppressant, it is difficult to draw any conclusions.
6.3.5 Methotrexate
Methotrexate is a folic acid antagonist. The polyglutamated derivatives of methotrexate are potent inhibitors of various enzymes, including dihydrofolate reductase and 5-aminoimidazole-4-carboxamide ribonucleotide transformylase. Among the multiple immunosuppressive effects, the inhibition of interleukin (IL)-1, as well as an effect on proteolytic enzymes, has been described. Methotrexate is usually administered orally, subcutaneously, or intramuscularly. Methotrexate is routinely used in SLE patients with musculoskeletal and skin manifestations. It has also been administered intrathecally or into the cerebral ventricles, particularly in patients with CNS tumors. In these cases, 10 mg methotrexate and 10 mg dexamethasone is diluted by 3 ml normal saline and injected by conventional lumbar puncture. This route of administration may solve the problem that this drug may have to get through the BBB and increase the methotrexate drug concentration in CSF. The more common side effects of methotrexate include gastrointestinal symptoms, stomatitis, increased levels of liver enzymes and mild cytopenia. Furthermore, severe complications such as liver fibrosis, interstitial pneumonitis, and severe pancytopenia have been reported [120, 121].
This drug is used very rarely in NPSLE and evidence is limited to several case series reporting the effect of intrathecal methotrexate. Valesini et al. reported the improvement of three NPSLE patients after a combination of intrathecal methotrexate and dexamethasone [121]. A Chinese group published several reports on the same cohort. A total of 109 patients received a combination of intrathecal methotrexate and dexamethasone (one to five injections), and showed an association with a positive outcome [122]. Wang et al. reported an effectiveness rate of 89 % of methylprednisolone treatment combined with intrathecal methotrexate in 36 NPSLE patients [123]. In addition to these results, intrathecal administration of methotrexate is not considered common practice and is limited to a few centers [38].
6.3.6 Cyclosporin A
Cyclosporin A is a calcineurin inhibitor initially derived from a fungus; it depresses T-cell activity by inhibiting transcription of IL-2 and other cytokines. Cyclosporin A has immunosuppressive properties and has been used in patients with SLE at a daily dose of 2.5–3 mg/kg [124]. The place of cyclosporin A in the treatment of SLE patients is limited and most data regarding these agents come from experience with lupus nephritis. This therapy is known to cause side effects such as hypertension, deterioration of renal function, and hypertrichosis [125].
No studies explicitly describe the effect of cyclosporin A on NP symptoms in SLE. Cyclosporin A, together with therapeutic plasma exchange (TPE), has been used in 18 NPSLE patients with organic brain syndrome and psychosis. When this combination was added to the previous standard therapy including corticosteroids and azathioprine or cyclophosphamide during a flare, earlier improvement in NP symptoms was achieved. However, the real efficacy of plasma exchange or cyclosporin A remains unknown due to the concomitant use of both therapies [126].
6.3.7 Rituximab/Anti-CD20
Rituximab is a chimeric monoclonal antibody directed against the B-cell-specific antigen CD20. B-cell depletion is achieved by rituximab through different mechanisms such as antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and induction of apoptosis. The use of B-cell depletion therapy in SLE is based on the aspect that B cells play a central role in the pathogenesis of SLE, as antigen-presenting cells and in the production of autoantibodies, cytokines, and chemokines. Currently, rituximab is widely used as an alternative therapy in patients with active SLE who are nonresponsive to standard immunosuppressive therapy [127].
Two randomized controlled trials (RCTs) on rituximab in SLE patients, one with renal involvement [the Lupus Nephritis Assessment with Rituximab (LUNAR) trial] and the other without renal involvement [the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial] failed to find superiority of rituximab over standard immunosuppressive regimens (glucocorticoids, cyclophosphamide, and mycophenolate mofetil) in mild to moderately active SLE. In these studies, severe and/or refractory patients were not included [128, 129]. On the other hand, the efficacy and safety of rituximab in the treatment of nonrenal SLE has recently been analyzed in a systematic review including one RCT, two open-label studies and 22 cohort studies, with a total of 1231 patients. Rituximab was shown to be safe and effective in the treatment of nonrenal SLE, especially in terms of disease activity, immunologic parameters, and corticosteroid-sparing effect [130]. The efficacy of rituximab in NPSLE was not assessed in the previous studies; however, in a substantial number of case reports and some open-label studies, good efficacy of rituximab was shown in refractory cases of NPSLE. Tokunaga and colleagues reported a response rate of 100 % in a series of ten severe refractory NPSLE patients treated with rituximab [131]. Narvaez and colleagues summarized all published data concerning adult patients with refractory NPSLE. A clinical response was observed in 85 % (29/34) of patients, classified as complete response in 50 % (17/34) and partial response in 35 % (12/34) of patients. However, 45 % of these patients relapsed after a median of 17 months despite maintenance therapy. Different therapeutic regimens of rituximab were used. The most frequently used regimen was 1000 mg doses separated by 15 days. Different dosing schedules appeared to show no difference in response, tolerability, or side effects. In all cases, rituximab was administered together with corticosteroids [132]. A recent case series on 18 pediatric NPSLE patients showed promising effect of rituximab. The authors divided the benefit into definite (five patients), probable (seven patients), possible (five patients) and no effect (one patient) [133]. Long-term information regarding rituximab therapy in NPSLE is scarce. Current data support the use of rituximab as a second-line therapy in patients with severe refractory NPSLE but additional controlled studies are needed to define the exact place of rituximab in the therapeutic regimen for NPSLE.
6.3.8 Intravenous Immunoglobulins
IVIGs constitute a mixture of natural antibodies of IgG subclass derived from the blood of healthy donors [134]. In SLE patients, the beneficial effects of IVIGs are thought to be due to (1) suppression of the function of autoreactive B lymphocytes and neutralization of pathogenic autoantibodies produced by these cells; and (2) inhibition of type I IFN-mediated differentiation of dendritic cells and suppression of endocytosis of nucleosomes [135].
Available evidence for the efficacy of IVIGs in SLE is limited and is derived from case reports and case series. Current indications proposed for IVIGs in SLE are severe cases nonrespondent to conventional immunosuppressive drugs, patients with active SLE and concomitant infection, and use as a corticosteroid-sparing agent when SLE can be controlled only with high-dose glucocorticoids. Furthermore, IVIGs can be safely used during pregnancy, without teratogenic risk, and might be a good choice in severe and life-threatening cases. In SLE patients, IVIGs have been administered as 2 g/kg, with divided doses over 2–5 days as the more commonly practiced regimen [136, 137]. The occurrence of adverse effects attributed to IVIGs is reported in up to 20 % of patients. The majority of side effects described are mild and self-limited (i.e. headaches, fever, flushing, chills, arthralgia, back pain, and myalgia) and do not require discontinuation of therapy. Other serious anecdotal systemic reactions, such as thromboembolic complications, have also been described [136, 138].
Based on case reports/series and retrospective studies, IVIGs have been successfully used to treat NPSLE patients with a broad spectrum of symptoms, including CNS manifestations (acute confusional syndrome, seizures, headache, chorea, psychosis), PNS manifestations (chronic inflammatory demyelinating polyneuropathy, Guillain–Barré, peripheral neuropathy associated with vasculitis), and psychiatric symptoms (psychosis, depression, mood disorder) [137, 139]. Camara and colleagues recently reported a series of 52 SLE patients, including 11 patients with nervous system involvement treated with IVIGs. Among these patients, six had CNS involvement and five had peripheral neuropathy. After IVIG treatment, eight of these patients experienced improvement (four total remission and four partial responses), and three (CNS vasculitis, peripheral neuropathy, and other with a nonspecified NPSLE) did not respond to this therapy [139]. In general, in the majority of the NPSLE cases reported to date, IVIGs were administered because of no response after corticosteroid pulses, failure of or contraindication for standard immunosuppressive therapy, or the presence of a concomitant infection. As seen in other SLE manifestations, the beneficial effects of IVIGs on NP symptoms were rapidly achieved and patients did not experience any relapse after the last infusion [137, 139, 140]. IVIGs may be used in severe refractory NPSLE not responding to conventional immunosuppression, or even as the first-choice agent when patients are pregnant or if symptoms are life-threatening and a concomitant infection is present. Future RCTs are needed for identifying the efficacy, safety, optimal dose, and duration of therapy, to support the evidence-based use of IVIGs in NPSLE patients.
6.3.9 Therapeutic Plasma Exchange
TPE is an extracorporeal blood purification technique designed for the removal of large molecular weight substances from the plasma. Either fresh frozen or stored plasma are used to replace large quantities of plasma from a patient by passing venous blood through an extracorporeal continuous flow centrifugation device. The rationale of this technique is that removal of substances will reduce further damage and may permit reversal of the pathologic process [141]. How this therapeutic procedure works exactly in SLE patients remains a matter of debate [142].
TPE is an expensive complex technique that requires specific equipment and experience. On the other hand, the technique is relatively safe. Complications are minor and include mild allergic reactions, fever, and hypocalcemic symptoms such as paresthesias, nausea, and cramps. Potentially life-threatening adverse reactions are rare (<0.15 %) [142, 143].
Although TPE has been used to treat several SLE manifestations with beneficial effects, its use in clinical practice is limited. Apheresis is accepted by the American Society for Apheresis (ASFA) as second-line therapy, either as standalone treatment or in conjunction with other modes of treatment, although the evidence is low [144].
TPE has been used alone or in combination with other immunosuppressants as adjuvant therapy in patients with NPSLE. It has been suggested that TPE modulates humoral components of the immune response removing autoantibodies and/or immune complexes, while immunosuppressive therapy suppresses de novo antibody production [144, 145].
Neuwelt reviewed 26 SLE patients with CNS involvement treated with TPE alone or in combination with cyclophosphamide. After this therapy, 74 % of patients improved, 13 % stabilized, and 13 % progressed, highlighting a potential benefit of TPE in refractory or critically ill patients not responding to cyclophosphamide [145]. A retrospective study on 13 patients with NPSLE flares undergoing TPE showed complete remissions in 54 % of patients and partial remissions in 46 % of patients. All but one patient had been administered cyclophosphamide and adjuvant plasma exchange during the same week [146].
TPE and cyclosporine were used in a prospective trial of 28 SLE patients (18 patients with organic brain syndrome and psychosis). This combination was added to the previous therapy (glucocorticoids and azathioprine or cyclophosphamide) during severe SLE flares. Earlier improvement in organ involvement, including rapid resolution of NP symptoms, was achieved; however, the real efficacy of TPE is not known due to the concomitant use of cyclosporine [126].
The ASFA guidelines suggest that a daily or every other day course (1–1.5 total plasma volumes) for three to six times is sufficient to see response in patients with lupus cerebritis. The guidelines also set out the treatment of acute inflammatory demyelinating disease, but there are no recommendations regarding myelopathy in SLE. In this particular case, TPE has been successfully used in severe cases; however, its real efficacy in these patients remains to be clarified since most of the successful cases reported used concomitant immunosuppressive therapy [146, 147]. TPE may also have an indication in other neurological entities related to SLE, such myasthenia gravis and Guillain–Barré syndrome. Although there are no specific recommendations for these NP symptoms, good functional outcomes have been reported when TPE has been added to immunosuppressive therapy in SLE patients with these complications [144, 148–150].
In conclusion, TPE in combination with other immunosuppressive therapy has acceptable toxicity and may be a good alternative in severe NPSLE; however, controlled trials are needed to better define the role of this therapy in these patients.
6.3.10 Hematopoietic Stem Cell Transplantation
Autologous hematopoietic stem cell transplantation (AHSCT) has emerged as an effective treatment modality in intractable SLE patients. AHSCT can achieve sustained clinical and immunologic remissions (50–70 % disease-free survival [DFS] at 5 years) even in patients with severe disease who are not responding to any other conventional immunosuppressive therapy [151]. The aim of this therapy is resetting the patient’s immune system by (1) depletion of autoreactive memory cells; and (2) reconstitution of the immune system by a normalized repertoire of graft-derived younger T cells [152]. Peripheral blood is the preferred source of hematopoietic stem cells, and the combination of cyclophosphamide (2–4 g/m2), antithymocyte globulin (30 mg/kg), and granulocyte colony-stimulating factor (G-CSF; 5 µg/kg) represents the most used ablation/mobilization strategy [151, 152].
AHSCT is associated with an increased risk of short-term mortality, attributable in most cases to infectious complications or disease flares. Transplant-related mortality ranges from 0 to 25 % [153]. Furthermore, it has been reported that SLE patients have the highest 100-day transplant-related mortality and highest 5-year post-transplant mortality compared with other autoimmune diseases; therefore, it is important that the risk:benefit ratio is optimized in these patients [154].
According to recent literature, more than 200 patients have received autologous stem cell transplants as therapy for lupus refractory to aggressive standard immunosuppressive therapy [151]. The largest number of cases published come from the European Group for Blood and Marrow Transplantation (EBMT) registry (n = 85), and a North American single-center experience (n = 50). The probability of 5-year DFS in both studies was approximately 50 %. Furthermore, patients not achieving sustained remission had better response to therapies that had previously failed [151, 154, 155].
Among the 50 SLE patients undergoing AHSCT reported by Burt and colleagues, 32 patients presented with any CNS involvement (seizures, psychosis, headache, aseptic meningitis, focal deficits, TIA, or myelopathy). In 18 cases, CNS involvement was the primary indication for AHSCT. No specific data regarding the outcome of NPSLE patients were reported; however, in general the disease activity significantly improved and remained significantly lower for up to 5 years after AHSCT [155]. Data from the EBMT registry have been reported in two articles, depending on the period of recruitment [156, 157]. Jayne and colleagues reported that 46 % of SLE patients recruited from 1995 to 2002 had CNS involvement. The frequency of CNS lupus activity declined to <5 % after AHSCT and only one patient developed new CNS disease post-AHSCT [156]. Among the 28 SLE patients reported to the EBMT registry between 2001 and 2008, 12 had neurologic disorder. The 3-year DFS, which was defined as the time interval from ASCT to either relapse or death, was 38 ± 15 % in NPSLE patients. Overall survival and DFS were not associated with the presence of neurologic or any other involvement before AHSCT [156]. Other authors have also reported long-standing clinical remission in NPSLE treated with AHSCT but the results are limited to case reports or small series [152, 157]. In addition to the improvement in overall lupus activity and serological status, AHSCT has been reported to reverse antiphospholipid syndrome (APS). Levels of aCL and LAC markedly improved or normalized after transplantation in a high proportion of patients [158]. Burt and colleagues reported that of 22 patients receiving anticoagulation, 18 were able to discontinue this therapy a mean of 4 months after AHSCT, and 78 % remained free of subsequent thrombotic events [155].
AHSCT has been used as a salvage therapy for patients with severe NPSLE refractory to intense standard immunosuppressive therapy. In spite of the promising rates of success, more RCTs are needed in SLE patients in order to establish criteria for patient selection and the exact role of this therapy.
6.4 Management of Ischemic NPSLE and Secondary Prevention
6.4.1 Antiplatelet Treatment
The disruption of atherosclerotic plaques leads to enhanced platelet deposition and, subsequently, to the formation of arterial thrombus, which can precipitate in an acute cerebrovascular event. Since platelets have been described as key cellular components of arterial occlusive thrombi, acetylsalicylic acid, a drug with a potent effect to diminish platelet aggregation, is intended to have a significant impact on morbidity and mortality [159]. Atherosclerosis has been described as occurring prematurely in SLE patients and being independent of traditional risk factors for CVD [35]. Platelet activation is increased in SLE patients compared with controls and may have a role in accelerated atherosclerosis [160]. Furthermore, the simultaneous presence of SLE and aPL antibodies, namely LAC and isolated persistently positive aCL at medium–high titers, has been associated with an increased risk of thrombosis. Tektonidou and colleagues reported that low-dose acetylsalicylic acid had a mild protective role against thrombosis in aPL-positive SLE patients [161]. Experts in APS recommend the use of low-dose acetylsalicylic acid in SLE patients with a positive LAC or isolated persistent aCL at medium–high titers as primary thromboprophylaxis when no major contraindications are present [162].
As secondary thromboprophylaxis, therapy depends on the presence of definite APS. In patients without aPL or with aPL who do not fulfill the criteria for APS, acetylsalicylic acid (50–325 mg/day) monotherapy, the combination of acetylsalicylic acid and extended-release dipyridamole, and clopidogrel 75 mg monotherapy are acceptable options for initial therapy. These options are recommended as the first choice over double therapy (acetylsalicylic acid/clopidogrel) or anticoagulants [162, 163]. In patients who have had a second cerebrovascular event while receiving acetylsalicylic acid, alternative antiplatelet agents such as clopidogrel are often considered; however, no single agent or combination has been well-studied in the general population or in SLE patients [163].
In SLE patients with stroke fulfilling the criteria for APS, optimal treatment is a matter of ongoing debate. Sole antiplatelet treatment has been proposed as an option, while other authors support the use of combined antiplatelet treatment and lower intensity anticoagulation [international normalized ratio (INR) 2.0–3.0] or high-intensity oral anticoagulation (INR >3.0) [162].
6.4.2 Anticoagulation
Anticoagulant drugs have demonstrated its effectiveness for the prevention and treatment of venous and arterial thromboembolism; however, their use is also associated with an increased risk for bleeding, and in some administration routes, especially oral, management can be challenging. The use and benefits of anticoagulant therapy in both primary and secondary prevention of ischemic cerebrovascular disease in the subset of SLE patients with aPL or APS remains controversial [164]. Cuadrado and colleagues recently reported a 5-year prospective, randomized, open, controlled trial on 166 SLE patients with aPL randomly receiving treatment with low-dose acetylsalicylic acid or low-dose acetylsalicylic acid and low-intensity warfarin. The authors concluded that there were no differences in thrombosis rate between both groups. Furthermore, warfarin was less safe and worse tolerated [165].
Secondary prevention of stroke in patients with aPL has also been addressed in several studies. A study focusing on arterial cerebral events showed similar rates of recurrent thrombotic events over 2 years and risk of major bleeding in patients treated with low-dose aspirin or warfarin therapy [166]. Two prospective, randomized studies on APS reported that treatment with high-intensity warfarin (INR range 3–4) was not superior to moderate-intensity warfarin (INR 2.0–3.0) in preventing recurrent thrombosis. The rate of minor hemorrhagic complications was slightly increased in patients treated with high-intensity anticoagulation [167, 168]. However, other authors have emphasized the limitations of these trials and recommend the use of prolonged warfarin therapy at a target INR >3.0 for all APS patients with a first arterial thrombotic event [169]. The EULAR recommendations for the management of NPSLE suggest that anticoagulation may be superior to antiplatelet therapy for secondary prevention of arterial events (including stroke/TIA) in APS [38]. Furthermore, recent international guidelines for the prevention and long-term management of thrombosis in aPL-positive patients recommend the use of warfarin therapy with a target INR >3.0, or an anti-aggregant combined with anticoagulant therapy (INR 2.0–3.0) in patients with definite APS and arterial thrombosis. The lack of consensus over the optimal treatment in these cases exemplifies the debate generated on this topic in the last few years, and stresses the need to start new trials [38, 47, 162].
In SLE patients with cerebral venous and sinus thrombosis, treatment with oral anticoagulation is recommended. The optimal duration of oral anticoagulant therapy after the acute phase is unclear. Guidelines recommend oral anticoagulation (INR range 2.0–3.0) for 3–6 months, and in patients with definite APS, long-term anticoagulation should be considered [170].
Oral anticoagulation has also been used in NPSLE other than cerebrovascular disease. In some SLE aPL-positive patients with myelopathy, good functional outcomes have been reported when added to immunosuppressive therapy [38, 171]; however, other authors found no additional benefit over standard immunosuppression alone and claim that there is not enough evidence for anticoagulation in these patients [172]. In SLE patients with chorea or seizure, anticoagulation may be considered when associated with aPL or when an ischemic subjacent process is suspected. This therapy has also been successfully used in aPL-related ischemic optic neuropathy and in SLE patients with positive aPL and cranial neuropathy not responding to immunosuppressive therapy [38]. A single uncontrolled study has suggested that APS patients with cognitive dysfunction may benefit from anticoagulation therapy [173].
6.5 Potential Future Therapies
6.5.1 Drugs in Development
Advances in the understanding of immunopathogenesis of SLE have led to the development of immunotherapies targeting B cells, T cells, the costimulatory modulation, and cytokines. Pathogenic mechanisms in NPSLE are still poorly understood. Furthermore, experimental models using these new therapies are lacking; however, according to what we already know about NPSLE pathogenesis, we could speculate about the potential role of some of these drugs in the future treatment of these manifestations.
The promising effect of rituximab may suggest an important contribution of B cells to NPSLE pathogenesis. Belimumab, a humanized monoclonal antibody targeted against B lymphocyte stimulator (BLyS), is now licensed in the US and Europe for the management of SLE. The BLISS trials were neither designed nor powered to definitively demonstrate the efficacy of belimumab in specific organ systems. Other trials on therapies targeting BLyS, such as tabalumab (phase II) and blisibimod (phase III) are ongoing. Atacicept, a humanized fusion protein that binds BLyS and APRIL (a proliferation-inducing ligand) has also been tested in SLE patients [174]. Both BLyS and APRIL were shown to be elevated in the CSF of SLE patients. Furthermore, APRIL was increased in the CSF of NPSLE patients compared with SLE patients without NP symptoms and other neurological diseases. It has been suggested that BLyS and APRIL are produced locally in the astrocytes of the brain and that they may play a role in NPSLE etiology. Hence, antagonists of these cytokines could have beneficial effect in these patients [175, 176]. To date, results in SLE patients are promising and further studies with these therapies are awaited; however, patients with severe CNS manifestations were excluded from all these trials, which will limit any conclusion in this respect [177].
Epratuzumab, a humanized monoclonal antibody that targets CD22 on B cells and results in modulation of B-cell function and migration, has also been studied in SLE patients. Although the EMBLEM and ALLEVIATE trials showed promising results, the EMBODY I and EMBODY II phase III clinical studies for epratuzumab in SLE did not meet their primary clinical efficacy endpoints [178, 179].
Several drugs targeting cytokines that are thought to contribute to the pathogenesis of both SLE and NPSLE are currently being tested. For example, IFN-α is considered one of the most promising therapeutic targets in SLE. Sifalimumab, a human anti-IFN-α monoclonal antibody, and rontalizumab, a humanized monoclonal antibody IgG1, have shown promising results in reducing SLE disease activity across multiple clinical measures [180]. Although not confirmed in all studies, IFN-α is one of the inflammatory mediators related to NPSLE pathogenesis. Type I IFNs are found in glia and neurons. Among their functions, IFNs induce other inflammatory mediators such as IL-6, alter brain neurotransmitters such as serotonin, and generate brain toxic metabolites. Subsequently, IFN-α has been hypothesized as a potential target in NPSLE [181–183].
A common characteristic of these new therapies is that the impact on disease activity seems promising but must still be assessed in phase III trials. Moreover, in most of these trials, CNS involvement was an exclusion criterion, and the potential to treat NPSLE will remain unknown [177].
Several studies have confirmed the intrathecal presence of higher levels of other cytokines in NPSLE. High levels of tumor necrosis factor (TNF)-α, IL-6, and IFN-γ are found in CSF of NPSLE patients. The overproduction of these cytokines is thought to play a role in the pathogenesis and severity of NP symptoms, and they have been proposed as candidate targets for future treatment [175, 184–186]. Two phase I trials with tocilizumab, an anti-IL-6 blocker, and AMG 811, a monoclonal antibody against IFN-γ, have shown acceptable results and more studies are awaited [187, 188].
Although antibodies are administered intravenously and may have a therapeutic effect on the brain, taking into account the BBB disruption in NPSLE, these drugs may require transport across the BBB using an endogenous BBB peptide receptor transporter [189]. Ischemic NPSLE, especially in the presence of aPL or APS, may benefit from new-generation direct oral anticoagulants in the future, including dabigatran etexilate, a direct thrombin inhibitor, and rivaroxaban, apixaban and edoxaban, which are direct anti-Xa inhibitors [190]. Although not currently recommended in APS, these therapies may represent a potential alternative for long-term anticoagulation in APS. Rivaroxaban has shown good results in both arterial and venous thrombosis; however, information is controversial [191, 192]. More data will be drawn from ongoing studies.
6.5.2 Potential Future Targets
The BBB is a network of endothelial cells and pericyte and astrocyte projections that regulates the entry of soluble molecules and cells into the brain parenchyma. It has been proposed that a disruption of the integrity of the BBB may have a potential pathogenic role in NPSLE since this may permit the influx of neuropathic antibodies across the BBB. Many modulators of the integrity of the BBB have been proposed. Among them, anti-endothelial cell antibodies, complement components, cytokines and chemokines, and environmental mediators have an essential role [193].
It has been speculated that ameliorating the disruption of the BBB may have an important effect in the control of NPSLE. Studies in MRL/lpr mice, accurately reflecting human NPSLE, have shown the importance of TWEAK, a pro-inflammatory cytokine member of the TNF superfamily, and the alternative complement cascade in BBB disruption.
TWEAK variably induces cellular proliferation, angiogenesis, apoptosis, and the production of metalloproteinase, cytokines, and chemokines [194]. TWEAK has been found to be increased in the cerebral cortices of MRL/lpr mice. Furthermore, in a murine knockout model for its receptor Fn14, mice were found to improve in cognitive function and to have less depression and anhedonia [195].
Complement component C5 has been reported to play a role in the maintenance of the BBB in mice [196]. Selective inhibition of C5aR alleviated CNS lupus [197]. Also, inhibition of the classical and alternative complement cascade with the complement inhibitor Crry was demonstrated to alleviate experimental CNS lupus in mice [198]. Furthermore, complement plays a role in microvascular injury. Mice deficient in C3 and C5 components are resistant to enhanced thrombosis and endothelial cell activation induced by aPL antibodies, indicating the important role of alternative pathway complement activation on aPL antibody-mediated thrombogenesis [199, 200]. Based on this information, eculizumab, a humanized monoclonal antibody blocking the generation of terminal complement components C5a and C5b-9, may be a potential drug to be used in the future in NPSLE [201].
7 Conclusions
NP symptoms constitute an uncommon and poorly understood event in SLE patients, and pose a diagnostic and therapeutic challenge to the physician. Management of NPSLE patients has not evolved substantially in the last decades and is characterized by the lack of good evidence and the use of empirical therapies to date. It seems reasonable that increased understanding of the pathogenesis of NPSLE and any of its manifestations will promote the possibility of finding targeted therapies and an evidence-based approach to management.
References
Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–21.
Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol. 2014;10(10):579–96.
Zirkzee EJ, Steup-Beekman GM, van der Mast RC, Bollen EL, van der Wee NJ, Baptist E, et al. Prospective study of clinical phenotypes in neuropsychiatric systemic lupus erythematosus; multidisciplinary approach to diagnosis and therapy. J Rheumatol. 2012;39(11):2118–26.
Amur S, Parekh A, Mummaneni P. Sex differences and genomics in autoimmune diseases. J Autoimmun. 2012;38(2–3):J254–65.
Miller MH, Urowitz MB, Gladman DD, Killinger DW. Systemic lupus erythematosus in males. Medicine (Baltimore). 1983;62(5):327–34.
Ward MM, Studenski S. Systemic lupus erythematosus in men: a multivariate analysis of gender differences in clinical manifestations. J Rheumatol. 1990;17(2):220–4.
Murphy G, Isenberg D. Effect of gender on clinical presentation in systemic lupus erythematosus. Rheumatology (Oxford). 2013;52(12):2108–15.
Fernandez M, Alarcon GS, Calvo-Alen J, Andrade R, McGwin G Jr, Vila LM, et al. A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum. 2007;57(4):576–84.
Samanta A, Feehally J, Roy S, Nichol FE, Sheldon PJ, Walls J. High prevalence of systemic disease and mortality in Asian subjects with systemic lupus erythematosus. Ann Rheum Dis. 1991;50(7):490–2.
Gonzalez LA, Toloza SM, Alarcon GS. Impact of race and ethnicity in the course and outcome of systemic lupus erythematosus. Rheum Dis Clin North Am. 2014;40(3):433–54 (vii–viii).
Mikdashi J, Handwerger B. Predictors of neuropsychiatric damage in systemic lupus erythematosus: data from the Maryland lupus cohort. Rheumatology (Oxford). 2004;43(12):1555–60.
Gonzalez LA, Pons-Estel GJ, Zhang J, Vila LM, Reveille JD, Alarcon GS. Time to neuropsychiatric damage occurrence in LUMINA (LXVI): a multi-ethnic lupus cohort. Lupus. 2009;18(9):822–30.
Hanly JG, Urowitz MB, Sanchez-Guerrero J, Bae SC, Gordon C, Wallace DJ, et al. Neuropsychiatric events at the time of diagnosis of systemic lupus erythematosus: an international inception cohort study. Arthritis Rheum. 2007;56(1):265–73.
Bertsias GK, Boumpas DT. Pathogenesis, diagnosis and management of neuropsychiatric SLE manifestations. Nat Rev Rheumatol. 2010;6(6):358–67.
Kampylafka EI, Alexopoulos H, Kosmidis ML, Panagiotakos DB, Vlachoyiannopoulos PG, Dalakas MC, et al. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. Plos One. 2013;8(2):e55843.
Unterman A, Nolte JE, Boaz M, Abady M, Shoenfeld Y, Zandman-Goddard G. Neuropsychiatric syndromes in systemic lupus erythematosus: a meta-analysis. Semin Arthritis Rheum. 2011;41(1):1–11.
Borowoy AM, Pope JE, Silverman E, Fortin PR, Pineau C, Smith CD, et al. Neuropsychiatric lupus: the prevalence and autoantibody associations depend on the definition: results from the 1000 faces of lupus cohort. Semin Arthritis Rheum. 2012;42(2):179–85.
Hanly JG, Urowitz MB, Su L, Bae SC, Gordon C, Wallace DJ, et al. Prospective analysis of neuropsychiatric events in an international disease inception cohort of patients with systemic lupus erythematosus. Ann Rheum Dis. 2010;69(3):529–35.
Zirkzee EJ, Huizinga TW, Bollen EL, van Buchem MA, Middelkoop HA, van der Wee NJ, et al. Mortality in neuropsychiatric systemic lupus erythematosus (NPSLE). Lupus. 2014;23(1):31–8.
The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42(4):599–608.
Ainiala H, Hietaharju A, Loukkola J, Peltola J, Korpela M, Metsanoja R, et al. Validity of the new American College of Rheumatology criteria for neuropsychiatric lupus syndromes: a population-based evaluation. Arthritis Rheum. 2001;45(5):419–23.
Hanly JG. ACR classification criteria for systemic lupus erythematosus: limitations and revisions to neuropsychiatric variables. Lupus. 2004;13(11):861–4.
Birnbaum J, Petri M, Thompson R, Izbudak I, Kerr D. Distinct subtypes of myelitis in systemic lupus erythematosus. Arthritis Rheum. 2009;60(11):3378–87.
Barber CE, Leclerc R, Gladman DD, Urowitz MB, Fortin PR. Posterior reversible encephalopathy syndrome: an emerging disease manifestation in systemic lupus erythematosus. Semin Arthritis Rheum. 2011;41(3):353–63.
Zandman-Goddard G, Chapman J, Shoenfeld Y. Autoantibodies involved in neuropsychiatric SLE and antiphospholipid syndrome. Semin Arthritis Rheum. 2007;36(5):297–315.
Hanly JG. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol. 2014;10(6):338–47.
de Vries B, Steup-Beekman GM, Haan J, Bollen EL, Luyendijk J, Frants RR, et al. TREX1 gene variant in neuropsychiatric systemic lupus erythematosus. Ann Rheum Dis. 2010;69(10):1886–7.
Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12(4):270–9.
Ellyard JI, Jerjen R, Martin JL, Lee AY, Field MA, Jiang SH, et al. Identification of a pathogenic variant in TREX1 in early-onset cerebral systemic lupus erythematosus by Whole-exome sequencing. Arthritis Rheumatol. 2014;66(12):3382–6.
Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134(4):587–98.
Lundstrom E, Gustafsson JT, Jonsen A, Leonard D, Zickert A, Elvin K, et al. HLA-DRB1*04/*13 alleles are associated with vascular disease and antiphospholipid antibodies in systemic lupus erythematosus. Ann Rheum Dis. 2013;72(6):1018–25.
Svenungsson E, Gustafsson J, Leonard D, Sandling J, Gunnarsson I, Nordmark G, et al. A STAT4 risk allele is associated with ischaemic cerebrovascular events and anti-phospholipid antibodies in systemic lupus erythematosus. Ann Rheum Dis. 2010;69(5):834–40.
Sciascia S, Bertolaccini ML, Roccatello D, Khamashta MA, Sanna G. Autoantibodies involved in neuropsychiatric manifestations associated with systemic lupus erythematosus: a systematic review. J Neurol. 2014;261(9):1706–14.
Petri M, Perez-Gutthann S, Spence D, Hochberg MC. Risk factors for coronary artery disease in patients with systemic lupus erythematosus. Am J Med. 1992;93(5):513–9.
Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349(25):2399–406.
Schanberg LE, Sandborg C, Barnhart HX, Ardoin SP, Yow E, Evans GW, et al. Premature atherosclerosis in pediatric systemic lupus erythematosus: risk factors for increased carotid intima-media thickness in the atherosclerosis prevention in pediatric lupus erythematosus cohort. Arthritis Rheum. 2009;60(5):1496–507.
Toloza SM, Uribe AG, McGwin G Jr, Alarcon GS, Fessler BJ, Bastian HM, et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA). XXIII. Baseline predictors of vascular events. Arthritis Rheum. 2004;50(12):3947–57.
Bertsias GK, Ioannidis JP, Aringer M, Bollen E, Bombardieri S, Bruce IN, et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann Rheum Dis. 2010;69(12):2074–82.
Zirkzee EJ, Magro Checa C, Sohrabian A, Steup-Beekman GM. Cluster analysis of an array of autoantibodies in neuropsychiatric systemic lupus erythematosus. J Rheumatol. 2014;41(8):1720–1.
Luyendijk J, Steens SC, Ouwendijk WJ, Steup-Beekman GM, Bollen EL, van der Grond J, et al. Neuropsychiatric systemic lupus erythematosus: lessons learned from magnetic resonance imaging. Arthritis Rheum. 2011;63(3):722–32.
Ercan E, Ingo C, Tritanon O, Magro-Checa C, Smith A, Smith S, et al. A multimodal MRI approach to identify and characterize microstructural brain changes in neuropsychiatric systemic lupus erythematosus. Neuroimage Clin. 2015;8:337–44.
Gonzalez-Duarte A, Cantu-Brito CG, Ruano-Calderon L, Garcia-Ramos G. Clinical description of seizures in patients with systemic lupus erythematosus. Eur Neurol. 2008;59(6):320–3.
Appenzeller S, Cendes F, Costallat LT. Epileptic seizures in systemic lupus erythematosus. Neurology. 2004;63(10):1808–12.
Florica B, Aghdassi E, Su J, Gladman DD, Urowitz MB, Fortin PR. Peripheral neuropathy in patients with systemic lupus erythematosus. Semin Arthritis Rheum. 2011;41(2):203–11.
Bortoluzzi A, Scire CA, Bombardieri S, Caniatti L, Conti F, De Vita S, et al. Development and validation of a new algorithm for attribution of neuropsychiatric events in systemic lupus erythematosus. Rheumatology (Oxford). 2015;54(5):891–8.
Petri M. Use of hydroxychloroquine to prevent thrombosis in systemic lupus erythematosus and in antiphospholipid antibody-positive patients. Curr Rheumatol Rep. 2011;13(1):77–80.
Fanouriakis A, Boumpas DT, Bertsias GK. Pathogenesis and treatment of CNS lupus. Curr Opin Rheumatol. 2013;25(5):577–83.
Hermosillo-Romo D, Brey RL. Diagnosis and management of patients with neuropsychiatric systemic lupus erythematosus (NPSLE). Best Pract Res Clin Rheumatol. 2002;16(2):229–44.
Petri M, Naqibuddin M, Carson KA, Wallace DJ, Weisman MH, Holliday SL, et al. Depression and cognitive impairment in newly diagnosed systemic lupus erythematosus. J Rheumatol. 2010;37(10):2032–8.
Harrison MJ, Morris KA, Horton R, Toglia J, Barsky J, Chait S, et al. Results of intervention for lupus patients with self-perceived cognitive difficulties. Neurology. 2005;65(8):1325–7.
Haupt M, Millen S, Janner M, Falagan D, Fischer-Betz R, Schneider M. Improvement of coping abilities in patients with systemic lupus erythematosus: a prospective study. Ann Rheum Dis. 2005;64(11):1618–23.
Wallace DJ. The history of antimalarials. Lupus. 1996;5(Suppl 1):S2–3.
Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis. 2010;69(1):20–8.
AlKadi HO. Antimalarial drug toxicity: a review. Chemotherapy. 2007;53(6):385–91.
Sahoo S, Kumar M, Sinha VK. Chloroquine-induced recurrent psychosis. Am J Ther. 2007;14(4):406–7.
Luijckx GJ, De Krom MC, Takx-Kohlen BC. Does chloroquine cause seizures? Presentation of three new cases and a review of the literature. Seizure. 1992;1(3):183–5.
Bertsias G, Ioannidis JP, Boletis J, Bombardieri S, Cervera R, Dostal C, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis. 2008;67(2):195–205.
Fessler BJ, Alarcon GS, McGwin G Jr, Roseman J, Bastian HM, Friedman AW, et al. Systemic lupus erythematosus in three ethnic groups: XVI. Association of hydroxychloroquine use with reduced risk of damage accrual. Arthritis Rheum. 2005;52(5):1473–80.
Wallace DJ, Metzger AL, Stecher VJ, Turnbull BA, Kern PA. Cholesterol-lowering effect of hydroxychloroquine in patients with rheumatic disease: reversal of deleterious effects of steroids on lipids. Am J Med. 1990;89(3):322–6.
Petri M. Thrombosis and systemic lupus erythematosus: the Hopkins Lupus Cohort perspective. Scand J Rheumatol. 1996;25(4):191–3.
Espinola RG, Pierangeli SS, Gharavi AE, Harris EN. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb Haemost. 2002;87(3):518–22.
Chen YM, Lin CH, Lan TH, Chen HH, Chang SN, Chen YH, et al. Hydroxychloroquine reduces risk of incident diabetes mellitus in lupus patients in a dose-dependent manner: a population-based cohort study. Rheumatology (Oxford). 2015;54(7):1244–9.
Parker B, Urowitz MB, Gladman DD, Lunt M, Donn R, Bae SC, et al. Impact of early disease factors on metabolic syndrome in systemic lupus erythematosus: data from an international inception cohort. Ann Rheum Dis. 2015;74(8):1530–6.
Jung H, Bobba R, Su J, Shariati-Sarabi Z, Gladman DD, Urowitz M, et al. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum. 2010;62(3):863–8.
Ruiz-Irastorza G, Egurbide MV, Pijoan JI, Garmendia M, Villar I, Martinez-Berriotxoa A, et al. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus. 2006;15(9):577–83.
Andrade RM, Alarcon GS, Gonzalez LA, Fernandez M, Apte M, Vila LM, et al. Seizures in patients with systemic lupus erythematosus: data from LUMINA, a multiethnic cohort (LUMINA LIV). Ann Rheum Dis. 2008;67(6):829–34.
Hanly JG, Urowitz MB, Su L, Gordon C, Bae SC, Sanchez-Guerrero J, et al. Seizure disorders in systemic lupus erythematosus results from an international, prospective, inception cohort study. Ann Rheum Dis. 2012;71(9):1502–9.
Reiss AB, Wirkowski E. Role of HMG-CoA reductase inhibitors in neurological disorders : progress to date. Drugs. 2007;67(15):2111–20.
Taylor F, Huffman MD, Macedo AF, Moore TH, Burke M, Davey Smith G, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;(1):CD004816.
Wheeler DC. Are there potential non-lipid-lowering uses of statins? Drugs. 1998;56(4):517–22.
Meroni PL, Luzzana C, Ventura D. Anti-inflammatory and immunomodulating properties of statins. An additional tool for the therapeutic approach of systemic autoimmune diseases? Clin Rev Allergy Immunol. 2002;23(3):263–77.
Pasterkamp G, van Lammeren GW. Pleiotropic effects of statins in atherosclerotic disease. Expert Rev Cardiovasc Ther. 2010;8(9):1235–7.
van Leuven SI, Mendez-Fernandez YV, Stroes ES, Tak PP, Major AS. Statin therapy in lupus-mediated atherogenesis: two birds with one stone? Ann Rheum Dis. 2011;70(2):245–8.
Elliott JR, Manzi S. Cardiovascular risk assessment and treatment in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2009;23(4):481–94.
Petri MA, Kiani AN, Post W, Christopher-Stine L, Magder LS. Lupus Atherosclerosis Prevention Study (LAPS). Ann Rheum Dis. 2011;70(5):760–5.
Schanberg LE, Sandborg C, Barnhart HX, Ardoin SP, Yow E, Evans GW, et al. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum. 2012;64(1):285–96.
Knight JS, Kaplan MJ. Cardiovascular disease in lupus: insights and updates. Curr Opin Rheumatol. 2013;25(5):597–605.
Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(7):2160–236.
Buttgereit F, da Silva JA, Boers M, Burmester GR, Cutolo M, Jacobs J, et al. Standardised nomenclature for glucocorticoid dosages and glucocorticoid treatment regimens: current questions and tentative answers in rheumatology. Ann Rheum Dis. 2002;61(8):718–22.
Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43.
Chau SY, Mok CC. Factors predictive of corticosteroid psychosis in patients with systemic lupus erythematosus. Neurology. 2003;61(1):104–7.
Zonana-Nacach A, Barr SG, Magder LS, Petri M. Damage in systemic lupus erythematosus and its association with corticosteroids. Arthritis Rheum. 2000;43(8):1801–8.
Ruiz-Arruza I, Ugarte A, Cabezas-Rodriguez I, Medina JA, Moran MA, Ruiz-Irastorza G. Glucocorticoids and irreversible damage in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2014;53(8):1470–6.
Brunner HI, Silverman ED, To T, Bombardier C, Feldman BM. Risk factors for damage in childhood-onset systemic lupus erythematosus: cumulative disease activity and medication use predict disease damage. Arthritis Rheum. 2002;46(2):436–44.
Hejaili FF, Moist LM, Clark WF. Treatment of lupus nephritis. Drugs. 2003;63(3):257–74.
Boumpas DT, Austin HA 3rd, Vaughn EM, Klippel JH, Steinberg AD, Yarboro CH, et al. Controlled trial of pulse methylprednisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet. 1992;340(8822):741–5.
Austin HA 3rd, Klippel JH, Balow JE, le Riche NG, Steinberg AD, Plotz PH, et al. Therapy of lupus nephritis. Controlled trial of prednisone and cytotoxic drugs. N Engl J Med. 1986;314(10):614–9.
Houssiau FA, Vasconcelos C, D’Cruz D, Sebastiani GD, de Ramon Garrido E, Danieli MG, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide. Ann Rheum Dis. 2010;69(1):61–4.
Katsifis GE, Tzioufas AG, Vlachoyiannopoulos PG, Voulgarelis M, Moutsopoulos HM, Ioannidis JP. Risk of myelotoxicity with intravenous cyclophosphamide in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2002;41(7):780–6.
Fraiser LH, Kanekal S, Kehrer JP. Cyclophosphamide toxicity. Characterising and avoiding the problem. Drugs. 1991;42(5):781–95.
Ognenovski VM, Marder W, Somers EC, Johnston CM, Farrehi JG, Selvaggi SM, et al. Increased incidence of cervical intraepithelial neoplasia in women with systemic lupus erythematosus treated with intravenous cyclophosphamide. J Rheumatol. 2004;31(9):1763–7.
Gonzalez LA, McGwin G Jr, Duran S, Pons-Estel GJ, Apte M, Vila LM, et al. Predictors of premature gonadal failure in patients with systemic lupus erythematosus. Results from LUMINA, a multiethnic US cohort (LUMINA LVIII). Ann Rheum Dis. 2008;67(8):1170–3.
Boumpas DT, Austin HA 3rd, Vaughan EM, Yarboro CH, Klippel JH, Balow JE. Risk for sustained amenorrhea in patients with systemic lupus erythematosus receiving intermittent pulse cyclophosphamide therapy. Ann Intern Med. 1993;119(5):366–9.
Boumpas DT, Yamada H, Patronas NJ, Scott D, Klippel JH, Balow JE. Pulse cyclophosphamide for severe neuropsychiatric lupus. Q J Med. 1991;81(296):975–84.
Baca V, Lavalle C, Garcia R, Catalan T, Sauceda JM, Sanchez G, et al. Favorable response to intravenous methylprednisolone and cyclophosphamide in children with severe neuropsychiatric lupus. J Rheumatol. 1999;26(2):432–9.
Mok CC, Lau CS, Wong RW. Treatment of lupus psychosis with oral cyclophosphamide followed by azathioprine maintenance: an open-label study. Am J Med. 2003;115(1):59–62.
Neuwelt CM, Lacks S, Kaye BR, Ellman JB, Borenstein DG. Role of intravenous cyclophosphamide in the treatment of severe neuropsychiatric systemic lupus erythematosus. Am J Med. 1995;98(1):32–41.
Ramos PC, Mendez MJ, Ames PR, Khamashta MA, Hughes GR. Pulse cyclophosphamide in the treatment of neuropsychiatric systemic lupus erythematosus. Clin Exp Rheumatol. 1996;14(3):295–9.
Stojanovich L, Stojanovich R, Kostich V, Dzjolich E. Neuropsychiatric lupus favourable response to low dose i.v. cyclophosphamide and prednisolone (pilot study). Lupus. 2003;12(1):3–7.
Barile-Fabris L, Ariza-Andraca R, Olguin-Ortega L, Jara LJ, Fraga-Mouret A, Miranda-Limon JM, et al. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann Rheum Dis. 2005;64(4):620–5.
Fernandes Moca Trevisani V, Castro AA, Ferreira Neves Neto J, Atallah AN. Cyclophosphamide versus methylprednisolone for treating neuropsychiatric involvement in systemic lupus erythematosus. Cochrane Database Syst Rev. 2013;(2):CD002265.
DiPiero J, Teng K, Hicks JK. Should thiopurine methyltransferase (TPMT) activity be determined before prescribing azathioprine, mercaptopurine, or thioguanine? Cleve Clin J Med. 2015;82(7):409–13.
Sharon E, Kaplan D, Diamond HS. Exacerbation of systemic lupus erythematosus after withdrawal of azathioprine therapy. N Engl J Med. 1973;288(3):122–4.
Oelzner P, Abendroth K, Hein G, Stein G. Predictors of flares and long-term outcome of systemic lupus erythematosus during combined treatment with azathioprine and low-dose prednisolone. Rheumatol Int. 1996;16(4):133–9.
Ginzler E, Sharon E, Diamond H, Kaplan D. Long-term maintenance therapy with azathioprine in systemic lupus erythematosus. Arthritis Rheum. 1975;18(1):27–34.
Hahn BH, Kantor OS, Osterland CK. Azathioprine plus prednisone compared with prednisone alone in the treatment of systemic lupus erythematosus. Report of a prospective controlled trial in 24 patients. Ann Intern Med. 1975;83(5):597–605.
Lim LS, Lefebvre A, Benseler S, Silverman ED. Longterm outcomes and damage accrual in patients with childhood systemic lupus erythematosus with psychosis and severe cognitive dysfunction. J Rheumatol. 2013;40(4):513–9.
Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14(Suppl 1):s2–8.
Pisoni CN, Sanchez FJ, Karim Y, Cuadrado MJ, D’Cruz DP, Abbs IC, et al. Mycophenolate mofetil in systemic lupus erythematosus: efficacy and tolerability in 86 patients. J Rheumatol. 2005;32(6):1047–52.
Appel GB, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. 2009;20(5):1103–12.
Ginzler EM, Dooley MA, Aranow C, Kim MY, Buyon J, Merrill JT, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med. 2005;353(21):2219–28.
Mok CC. Mycophenolate mofetil for non-renal manifestations of systemic lupus erythematosus: a systematic review. Scand J Rheumatol. 2007;36(5):329–37.
Ginzler EM, Wofsy D, Isenberg D, Gordon C, Lisk L, Dooley MA. Nonrenal disease activity following mycophenolate mofetil or intravenous cyclophosphamide as induction treatment for lupus nephritis: findings in a multicenter, prospective, randomized, open-label, parallel-group clinical trial. Arthritis Rheum. 2010;62(1):211–21.
Posalski JD, Ishimori M, Wallace DJ, Weisman MH. Does mycophenolate mofetil prevent extra-renal flares in systemic lupus erythematosus? Results from an observational study of patients in a single practice treated for up to 5 years. Lupus. 2009;18(6):516–21.
Mok CC, Mak A, To CH. Mycophenolate mofetil for lupus related myelopathy. Ann Rheum Dis. 2006;65(7):971–3.
Jose J, Paulose BK, Vasuki Z, Danda D. Mycophenolate mofetil in neuropsychiatric systemic lupus erythematosus. Indian J Med Sci. 2005;59(8):353–6.
Lhotta K, Wurzner R, Rosenkranz AR, Beer R, Rudisch A, Neumair F, et al. Cerebral vasculitis in a patient with hereditary complete C4 deficiency and systemic lupus erythematosus. Lupus. 2004;13(2):139–41.
Tomietto P, D’Agostini S, Annese V, De Vita S, Ferraccioli G. Mycophenolate mofetil and intravenous dexamethasone in the treatment of persistent lupus myelitis. J Rheumatol. 2007;34(3):588–91.
Saison J, Costedoat-Chalumeau N, Maucort-Boulch D, Iwaz J, Marignier R, Cacoub P, et al. Systemic lupus erythematosus-associated acute transverse myelitis: manifestations, treatments, outcomes, and prognostic factors in 20 patients. Lupus. 2015;24(1):74–81.
Bannwarth B, Labat L, Moride Y, Schaeverbeke T. Methotrexate in rheumatoid arthritis. An update. Drugs. 1994;47(1):25–50.
Valesini G, Priori R, Francia A, Balestrieri G, Tincani A, Airo P, et al. Central nervous system involvement in systemic lupus erythematosus: a new therapeutic approach with intrathecal dexamethasone and methotrexate. Springer Semin Immunopathol. 1994;16(2–3):313–21.
Zhou HQ, Zhang FC, Tian XP, Leng XM, Lu JJ, Zhao Y, et al. Clinical features and outcome of neuropsychiatric lupus in Chinese: analysis of 240 hospitalized patients. Lupus. 2008;17(2):93–9.
Wang J, Zhao Y, Zhang J, Lei H, Zhu G, Fu B. Impact analysis of autoantibody level and NR2 antibody level in neuropsychiatric SLE treated by methylprednisolone combined with MTX and DXM intrathecal injection. Cell Biochem Biophys. 2014;70(2):1005–9.
Faulds D, Goa KL, Benfield P. Cyclosporin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs. 1993;45(6):953–1040.
Yang M, Li M, He W, Wang B, Gu Y. Calcineurin inhibitors may be a reasonable alternative to cyclophosphamide in the induction treatment of active lupus nephritis: a systematic review and meta-analysis. Exp Ther Med. 2014;7(6):1663–70.
Bambauer R, Schwarze U, Schiel R. Cyclosporin A and therapeutic plasma exchange in the treatment of severe systemic lupus erythematosus. Artif Organs. 2000;24(11):852–6.
Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44(16):3823–37.
Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62(1):222–33.
Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab Study. Arthritis Rheum. 2012;64(4):1215–26.
Cobo-Ibanez T, Loza-Santamaria E, Pego-Reigosa JM, Marques AO, Rua-Figueroa I, Fernandez-Nebro A, et al. Efficacy and safety of rituximab in the treatment of non-renal systemic lupus erythematosus: a systematic review. Semin Arthritis Rheum. 2014;44(2):175–85.
Tokunaga M, Saito K, Kawabata D, Imura Y, Fujii T, Nakayamada S, et al. Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann Rheum Dis. 2007;66(4):470–5.
Narvaez J, Rios-Rodriguez V, de la Fuente D, Estrada P, Lopez-Vives L, Gomez-Vaquero C, et al. Rituximab therapy in refractory neuropsychiatric lupus: current clinical evidence. Semin Arthritis Rheum. 2011;41(3):364–72.
Dale RC, Brilot F, Duffy LV, Twilt M, Waldman AT, Narula S, et al. Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology. 2014;83(2):142–50.
Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345(10):747–55.
Bayry J, Negi VS, Kaveri SV. Intravenous immunoglobulin therapy in rheumatic diseases. Nat Rev Rheumatol. 2011;7(6):349–59.
Orange JS, Hossny EM, Weiler CR, Ballow M, Berger M, Bonilla FA, et al. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2006;117(4 Suppl):S525–53.
Toubi E, Kessel A, Shoenfeld Y. High-dose intravenous immunoglobulins: an option in the treatment of systemic lupus erythematosus. Hum Immunol. 2005;66(4):395–402.
Katz U, Achiron A, Sherer Y, Shoenfeld Y. Safety of intravenous immunoglobulin (IVIG) therapy. Autoimmun Rev. 2007;6(4):257–9.
Camara I, Sciascia S, Simoes J, Pazzola G, Salas V, Karim Y, et al. Treatment with intravenous immunoglobulins in systemic lupus erythematosus: a series of 52 patients from a single centre. Clin Exp Rheumatol. 2014;32(1):41–7.
Milstone AM, Meyers K, Elia J. Treatment of acute neuropsychiatric lupus with intravenous immunoglobulin (IVIG): a case report and review of the literature. Clin Rheumatol. 2005;24(4):394–7.
McCarthy LJ. The challenge of evidence-based medicine for apheresis. Ther Apher Dial. 2003;7(2):153–4.
McLeod BC. An approach to evidence-based therapeutic apheresis. J Clin Apher. 2002;17(3):124–32.
Basic-Jukic N, Kes P, Glavas-Boras S, Brunetta B, Bubic-Filipi L, Puretic Z. Complications of therapeutic plasma exchange: experience with 4857 treatments. Ther Apher Dial. 2005;9(5):391–5.
Schwartz J, Winters JL, Padmanabhan A, Balogun RA, Delaney M, Linenberger ML, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145–284.
Neuwelt CM. The role of plasmapheresis in the treatment of severe central nervous system neuropsychiatric systemic lupus erythematosus. Ther Apher Dial. 2003;7(2):173–82.
Bartolucci P, Brechignac S, Cohen P, Le Guern V, Guillevin L. Adjunctive plasma exchanges to treat neuropsychiatric lupus: a retrospective study on 10 patients. Lupus. 2007;16(10):817–22.
Pons-Estel GJ, Salerni GE, Serrano RM, Gomez-Puerta JA, Plasin MA, Aldasoro E, et al. Therapeutic plasma exchange for the management of refractory systemic autoimmune diseases: report of 31 cases and review of the literature. Autoimmun Rev. 2011;10(11):679–84.
Deodhar A, Norden J, So Y, Bennett R. The association of systemic lupus erythematosus and Lambert–Eaton myasthenic syndrome. J Rheumatol. 1996;23(7):1292–4.
Hsu TY, Wang SH, Kuo CF, Chiu TF, Chang YC. Acute inflammatory demyelinating polyneuropathy as the initial presentation of lupus. Am J Emerg Med. 2009;27(7):900.e3–5.
Jallouli M, Saadoun D, Eymard B, Leroux G, Haroche J, Le Thi Huong D, et al. The association of systemic lupus erythematosus and myasthenia gravis: a series of 17 cases, with a special focus on hydroxychloroquine use and a review of the literature. J Neurol. 2012;259(7):1290–7.
Illei GG, Cervera R, Burt RK, Doria A, Hiepe F, Jayne D, et al. Current state and future directions of autologous hematopoietic stem cell transplantation in systemic lupus erythematosus. Ann Rheum Dis. 2011;70(12):2071–4.
Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S, et al. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood. 2009;113(1):214–23.
Statkute L, Verda L, Oyama Y, Traynor A, Villa M, Shook T, et al. Mobilization, harvesting and selection of peripheral blood stem cells in patients with autoimmune diseases undergoing autologous hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;39(6):317–29.
Farge D, Labopin M, Tyndall A, Fassas A, Mancardi GL, Van Laar J, et al. Autologous hematopoietic stem cell transplantation for autoimmune diseases: an observational study on 12 years’ experience from the European Group for Blood and Marrow Transplantation Working Party on Autoimmune Diseases. Haematologica. 2010;95(2):284–92.
Burt RK, Traynor A, Statkute L, Barr WG, Rosa R, Schroeder J, et al. Nonmyeloablative hematopoietic stem cell transplantation for systemic lupus erythematosus. JAMA. 2006;295(5):527–35.
Alchi B, Jayne D, Labopin M, Demin A, Sergeevicheva V, Alexander T, et al. Autologous haematopoietic stem cell transplantation for systemic lupus erythematosus: data from the European Group for Blood and Marrow Transplantation registry. Lupus. 2013;22(3):245–53.
Lehnhardt FG, Scheid C, Holtik U, Burghaus L, Neveling M, Impekoven P, et al. Autologous blood stem cell transplantation in refractory systemic lupus erythematodes with recurrent longitudinal myelitis and cerebral infarction. Lupus. 2006;15(4):240–3.
Statkute L, Traynor A, Oyama Y, Yaung K, Verda L, Krosnjar N, et al. Antiphospholipid syndrome in patients with systemic lupus erythematosus treated by autologous hematopoietic stem cell transplantation. Blood. 2005;106(8):2700–9.
Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373(9678):1849–60.
Lood C, Amisten S, Gullstrand B, Jonsen A, Allhorn M, Truedsson L, et al. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: up-regulation of the type I interferon system is strongly associated with vascular disease. Blood. 2010;116(11):1951–7.
Tektonidou MG, Laskari K, Panagiotakos DB, Moutsopoulos HM. Risk factors for thrombosis and primary thrombosis prevention in patients with systemic lupus erythematosus with or without antiphospholipid antibodies. Arthritis Rheum. 2009;61(1):29–36.
Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, Brey R, Crowther M, Derksen R, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus. 2011;20(2):206–18.
Adams RJ, Albers G, Alberts MJ, Benavente O, Furie K, Goldstein LB, et al. Update to the AHA/ASA recommendations for the prevention of stroke in patients with stroke and transient ischemic attack. Stroke. 2008;39(5):1647–52.
Tuhrim S, Rand JH, Wu XX, Weinberger J, Horowitz DR, Goldman ME, et al. Elevated anticardiolipin antibody titer is a stroke risk factor in a multiethnic population independent of isotype or degree of positivity. Stroke. 1999;30(8):1561–5.
Cuadrado MJ, Bertolaccini ML, Seed PT, Tektonidou MG, Aguirre A, Mico L, et al. Low-dose aspirin vs low-dose aspirin plus low-intensity warfarin in thromboprophylaxis: a prospective, multicentre, randomized, open, controlled trial in patients positive for antiphospholipid antibodies (ALIWAPAS). Rheumatology (Oxford). 2014;53(2):275–84.
Levine SR, Brey RL, Tilley BC, Thompson JL, Sacco RL, Sciacca RR, et al. Antiphospholipid antibodies and subsequent thrombo-occlusive events in patients with ischemic stroke. JAMA. 2004;291(5):576–84.
Crowther MA, Ginsberg JS, Julian J, Denburg J, Hirsh J, Douketis J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med. 2003;349(12):1133–8.
Finazzi G, Marchioli R, Brancaccio V, Schinco P, Wisloff F, Musial J, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost. 2005;3(5):848–53.
Ruiz-Irastorza G, Hunt BJ, Khamashta MA. A systematic review of secondary thromboprophylaxis in patients with antiphospholipid antibodies. Arthritis Rheum. 2007;57(8):1487–95.
Einhaupl K, Stam J, Bousser MG, De Bruijn SF, Ferro JM, Martinelli I, et al. EFNS guideline on the treatment of cerebral venous and sinus thrombosis in adult patients. Eur J Neurol. 2010;17(10):1229–35.
D’Cruz DP, Mellor-Pita S, Joven B, Sanna G, Allanson J, Taylor J, et al. Transverse myelitis as the first manifestation of systemic lupus erythematosus or lupus-like disease: good functional outcome and relevance of antiphospholipid antibodies. J Rheumatol. 2004;31(2):280–5.
Katsiari CG, Giavri I, Mitsikostas DD, Yiannopoulou KG, Sfikakis PP. Acute transverse myelitis and antiphospholipid antibodies in lupus. No evidence for anticoagulation. Eur J Neurol. 2011;18(4):556–63.
Hughes GR, Cuadrado MJ, Khamashta MA, Sanna G. Headache and memory loss: rapid response to heparin in the antiphospholipid syndrome. Lupus. 2001;10(11):778.
Manzi S, Sanchez-Guerrero J, Merrill JT, Furie R, Gladman D, Navarra SV, et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann Rheum Dis. 2012;71(11):1833–8.
George-Chandy A, Trysberg E, Eriksson K. Raised intrathecal levels of APRIL and BAFF in patients with systemic lupus erythematosus: relationship to neuropsychiatric symptoms. Arthritis Res Ther. 2008;10(4):R97.
Hopia L, Thangarajh M, Khademi M, Laveskog A, Wallstrom E, Svenungsson E, et al. Cerebrospinal fluid levels of a proliferation-inducing ligand (APRIL) are increased in patients with neuropsychiatric systemic lupus erythematosus. Scand J Rheumatol. 2011;40(5):363–72.
Ding HJ, Gordon C. New biologic therapy for systemic lupus erythematosus. Curr Opin Pharmacol. 2013;13(3):405–12.
Harvey PR, Gordon C. B-cell targeted therapies in systemic lupus erythematosus: successes and challenges. BioDrugs. 2013;27(2):85–95.
Study of epratuzumab versus placebo in subjects with moderate to severe general systemic lupus erythematosus (SLE) (EMBODY 2). Available at: https://clinicaltrials.gov/ct2/show/NCT01261793. Accessed 22 Nov 2015.
Mathian A, Hie M, Cohen-Aubart F, Amoura Z. Targeting interferons in systemic lupus erythematosus: current and future prospects. Drugs. 2015;75(8):835–46.
Dafny N, Yang PB. Interferon and the central nervous system. Eur J Pharmacol. 2005;523(1–3):1–15.
Fragoso-Loyo H, Atisha-Fregoso Y, Nunez-Alvarez CA, Llorente L, Sanchez-Guerrero J. Utility of interferon-alpha as a biomarker in central neuropsychiatric involvement in systemic lupus erythematosus. J Rheumatol. 2012;39(3):504–9.
Santer DM, Yoshio T, Minota S, Moller T, Elkon KB. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J Immunol. 2009;182(2):1192–201.
Fragoso-Loyo H, Richaud-Patin Y, Orozco-Narvaez A, Davila-Maldonado L, Atisha-Fregoso Y, Llorente L, et al. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 2007;56(4):1242–50.
Svenungsson E, Andersson M, Brundin L, van Vollenhoven R, Khademi M, Tarkowski A, et al. Increased levels of proinflammatory cytokines and nitric oxide metabolites in neuropsychiatric lupus erythematosus. Ann Rheum Dis. 2001;60(4):372–9.
Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus. 2000;9(7):498–503.
Chen P, Vu T, Narayanan A, Sohn W, Wang J, Boedigheimer M, et al. Pharmacokinetic and pharmacodynamic relationship of AMG 811, an anti-IFN-gamma IgG1 monoclonal antibody, in patients with systemic lupus erythematosus. Pharm Res. 2015;32(2):640–53.
Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010;62(2):542–52.
Pardridge WM. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin Pharmacol Ther. 2015;97(4):347–61.
Lopez-Pedrera C, Aguirre MA, Ruiz-Limon P, Perez-Sanchez C, Jimenez-Gomez Y, Barbarroja N, et al. Immunotherapy in antiphospholipid syndrome. Int Immunopharmacol. 2015;27(2):200–8.
Signorelli F, Nogueira F, Domingues V, Mariz HA, Levy RA. Thrombotic events in patients with antiphospholipid syndrome treated with rivaroxaban: a series of eight cases. Clin Rheumatol. (Epub 30 Jul 2015).
Son M, Wypasek E, Celinska-Lowenhoff M, Undas A. The use of rivaroxaban in patients with antiphospholipid syndrome: a series of 12 cases. Thromb Res. 2015;135(5):1035–6.
Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT. Losing your nerves? Maybe it’s the antibodies. Nat Rev Immunol. 2009;9(6):449–56.
Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest. 2005;115(9):2330–40.
Wen J, Xia Y, Stock A, Michaelson JS, Burkly LC, Gulinello M, et al. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J Autoimmun. 2013;43:44–54.
Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ, Alexander JJ. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J. 2010;24(6):1682–8.
Jacob A, Hack B, Bai T, Brorson JR, Quigg RJ, Alexander JJ. Inhibition of C5a receptor alleviates experimental CNS lupus. J Neuroimmunol. 2010;221(1–2):46–52.
Alexander JJ, Bao L, Jacob A, Kraus DM, Holers VM, Quigg RJ. Administration of the soluble complement inhibitor, Crry-Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim Biophys Acta. 2003;1639(3):169–76.
Cavazzana I, Manuela N, Irene C, Barbara A, Sara S, Orietta BM, et al. Complement activation in anti-phospholipid syndrome: a clue for an inflammatory process? J Autoimmun. 2007;28(2–3):160–4.
Thurman JM, Kraus DM, Girardi G, Hourcade D, Kang HJ, Royer PA, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol Immunol. 2005;42(1):87–97.
Barilla-Labarca ML, Toder K, Furie R. Targeting the complement system in systemic lupus erythematosus and other diseases. Clin Immunol. 2013;148(3):313–21.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
César Magro-Checa, Elisabeth J. Zirkzee, Tom W. Huizinga and Gerda M. Steup-Beekman declare that they have no conflicts of interest directly relevant to the content of this review.
Funding
No sources of funding were used to support the writing of this manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Magro-Checa, C., Zirkzee, E.J., Huizinga, T.W. et al. Management of Neuropsychiatric Systemic Lupus Erythematosus: Current Approaches and Future Perspectives. Drugs 76, 459–483 (2016). https://doi.org/10.1007/s40265-015-0534-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-015-0534-3