
Abstract
Introduction and Objective
Signals of adverse drug reactions (ADRs) can be supported by reports of ADRs and by interventional and non-interventional studies. The evidence base and features of ADR reports that are used to support signals remain to be comprehensively described. To this end, we have undertaken a scoping review.
Methods
We searched the following databases: PubMed, EMBASE, PsycINFO, Web of Science, and Google Scholar, without language or time restrictions. We also hand searched the bibliographies of relevant studies. We included studies of any design if the results were described as signals. We assessed the levels of evidence using the Oxford Centre for Evidence-Based Medicine (OCEBM) criteria and coded features of reports of ADRs using the Bradford Hill guidelines.
Results
Overall, 1974 publications reported 2421 studies of signals; 1683/2421 were clinical assessments of anecdotal reports of ADRs, but only 225 (13%) of these included explicit judgments on which features of the ADR reports were supportive of a signal. These 225 studies yielded 228 signals; these were supported by features, which were: ‘experimental evidence’ (i.e., positive dechallenge or rechallenge, 154 instances [68%]), ‘temporality’ (i.e., time to onset, 130 [57%]), ‘exclusion of competing causes’ (49 [21%]), and others (40 [17%]). Positive dechallenge/rechallenge often co-occurred with temporality (77/228). OCEBM 4 (i.e., case series and case-control studies) was the most frequent level of evidence (2078 studies). Between 2013 and 2019, there was a three-fold increase in clinical assessments of reports of ADRs compared with a less than two-fold increase in studies supported by higher levels of evidence (i.e., OCEBM 1–3). We identified an increased rate between 2013 and 2019 in disproportionality analyses (about 15 studies per year), mostly from academia.
Conclusions
Most signals were supported by temporality and dechallenge/rechallenge, but clear reporting of judgments on causality remains infrequent. The number of studies supported only by anecdotal reports of ADRs increased from year to year. The impact of a growing number of signals of disproportionate reporting communicated without an accompanying clinical assessment should be evaluated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In a scoping review, we have investigated the types of evidence that have been used to support 2421 pharmacovigilance signals communicated internationally. |
Signals based only on anecdotal reports have become increasingly common and dominate the literature (70%). |
Most signals based only on anecdotal reports are supported by evidence involving temporality and dechallenge/rechallenge, but clear reporting of how judgments of association or causality are made is uncommon (13%). |
1 Introduction
Interventional and observational studies, along with anecdotal reports, can offer evidence that an event or a group of events is causally linked to one or more medicinal products. Such indications are typically referred to as signals of adverse drug reactions (ADRs, [1]). When they are solely supported by ratios of observed over expected counts of reports of ADRs, they are called signals of disproportionate reporting (SDRs) [1]. A signal can be described as a hypothesis, supported by data and arguments, that there is a causal association between a medicinal product and an adverse event [2] that may justify further investigations aimed at quantifying the risk of an ADR. One of the objectives of pharmacovigilance is the detection of such signals, and it is carried out by stakeholders such as regulatory agencies, research organizations, academia, pharmaceutical companies, and others. Overviews of the evidence underpinning signals, limited to the European Union (EU) [3], have shown that detected signals were chiefly based on case reports and were more likely to lead to regulatory action when supported by additional study designs [4]. Combined qualitative/quantitative algorithms for signal detection have suggested that the recency of reports of ADRs, their geographical spread, and the availability of narratives [5] were factors predictive of signals that led to regulatory action, such as inclusion in Summaries of Products Characteristics. However, further understanding of features (such as positive dechallenge or temporality) that are considered during clinical assessments of reports may complement such findings. Additionally, strategies to detect signals have progressively improved, reducing the time to signal detection, by adopting machine-readable Summaries of Products Characteristics, or, to a lesser extent, using clinically related concepts to specify outcomes of interest, as summarized in the PROTECT guidelines [6]. Nevertheless, the median time it takes for a signal to be communicated (time to communication, TTC) remains to be quantified. We sought to review the global published evidence that has been used to support signals, and chose a scoping review design.
The aims of the present review were:
-
(a)
To provide regulatory decision makers with a synthesis of the evidence that is used to support signals of ADRs.
-
(b)
To describe the frequencies of the features of reports of ADRs explicitly referred to as supportive of signals in clinical assessments.
-
(c)
To determine and compare the levels of evidence of signals/SDRs across stakeholders in pharmacovigilance.
-
(d)
To obtain insights into the TTC and to discover whether it has changed over time.
2 Methods
This study follows the 2018 Preferred Reporting Items for Systematic Reviews and Meta-analyses extension for Scoping Reviews [7]. We have previously published [8] and registered the protocol of this work on the Open Science Framework (registration number: osf.io/a4xns). We report all protocol deviations and additional information in the Electronic Supplementary Material (ESM).
In brief, in the first week of September 2020, we searched PubMed (https://pubmed.ncbi.nlm.nih.gov/), EMBASE (OvidSP), PsycINFO (OvidSP), Science Citation Index (Web of Science Core Collection), and Google Scholar (https://scholar.google.co.uk/), and in April 2021, we searched OpenGrey (https://opengrey.eu/) and GreyNet International (https://www.greynet.org/), the English websites of 35 regulatory agencies/authorities, drug bulletins, and the SIGNAL Document of the Uppsala Monitoring Centre (from the start of each source until 31 August, 2020). We complemented searches with backward citation screening. We sent Freedom of Information requests to the US Food and Drug Administration (FDA) and the European Medicines Agency, and contacted other regulators, to obtain full lists of signals. In this way, we retrieved large numbers of signals from Malaysia, Japan, Canada, and New Zealand, including items that, while not explicitly described as signals, were deemed as such by the regulators. When we did not retrieve records that were described using expressions other than “signal”, we ensured that our retrieval strategy was consistent, by confirming with regulators whether their lists of communications included both signals and other forms of communications of risks. The complete search strategy is detailed in table 1 of the ESM.
We included associations detected in clinical assessments of reports of ADRs and observational/interventional studies whose original authors used the term “signal” in the abstract/full text, if the ADRs had not been documented previously. We considered an ADR as previously undocumented if it was:
-
(1)
Unlisted on Summaries of Product Characteristics or equivalent documents in the warnings, contraindications, or untoward effects sections, as reported by the original authors at the time of communication; or
-
(2)
Not previously communicated via direct healthcare professional communications or other country-specific channels, as reported by the original authors at the time of communication.
If information on (1) or (2) above was unavailable in peer-reviewed publications, we verified that an ADR had not been described in the cited references of each included record in interventional or observational studies, i.e., that its risk had not been previously quantified. When none of this information was available, we adopted an agnostic approach, to avoid omitting possibly relevant publications, and analyzed their findings separately, but did not calculate the TTC.
We also included papers that described SDRs. We required SDRs to present complete thresholds for detection, requesting them from original authors if missing. We retained the earliest version of repeated communications of the same signal by the same stakeholder (the first author of a communication) and combined new information appearing in repeated communications with earlier information.
We excluded records that lacked full texts or where the authors explicitly stated that the evidence did not support a signal or that no signals were detected, or that we could not translate. We omitted signals/SDRs concerning the following: medical devices without active ingredients, supplements, lack of efficacy, medication errors, or beneficial effects.
We coded features of reports of ADRs to a codebook developed iteratively (see the ESM). Related codes were coalesced, or grouped according to the Bradford Hill guidelines, where feasible.
We attributed levels of evidence, applying the Oxford Centre for Evidence-Based Medicine (OCEBM) tool [9], to studies supporting signals. The OCEBM levels of evidence tool ranks study designs from 1 to 5, based on whether they are likely to provide the best available evidence for decision making, 1 being the highest level and 5 the lowest. We followed the row “What are the rare harms?” and omitted level 5 (mechanism-based reasoning) [10]. Thus, systematic reviews of randomized trials or n-of-1 trials were OCEBM level 1; examples of level 2 study designs included individual randomized trials, of level 3 prospective cohort studies with appropriate follow-up, and of level 4 series of case reports or case-control studies. For studies that fell outside the OCEBM levels, we postulated subtypes. Where we could not, we assigned no level of evidence. If multiple studies supported a signal, the highest quality level of evidence applied. Please see ESM.
We used two units of analysis: (1) studies and (2) signals.
-
(1)
Multiple studies of any design or any level of evidence supporting a signal were counted as one, as was a single study supporting multiple signals. This unit was distinct from publications, which could report more than one study. Studies were used in the characterization of the level of evidence.
-
(2)
Signals, as drug-event or drug-drug event combinations, were used to count the features of ADR reports.
All data were analyzed descriptively in primary and secondary analyses. The primary analysis focused on previously undocumented signals/SDRs (see the ESM) and characterized the level of evidence and stakeholder over time (counts by studies), as well as the features of reports of ADRs (counts by signals).
For each drug-event or drug–drug event combination in the primary analysis, we selected the ‘first report’ of an ADR based on the earliest of two dates, either the first date on which a report of the ADR was entered into VigiBase (data lock point: 30/08/2020), the World Health Organization’s global database of reports of suspected ADRs (minimum value of the E2b field FirstDateDatabase), or the first date on which a report of ADR was received by a regional/national pharmacovigilance center (minimum value of the E2b field: ReceiveDate). By subtracting from the year of communication of a signal/SDR that of the first report in VigiBase, we calculated the TTC. This approach was similar to that used in a previously published set of systematic reviews on withdrawals of marketing authorizations [11, 12], but which relied on unpublished, instead of peer-reviewed reports of ADRs.
The secondary analysis encompassed: (a) SDRs with ambiguous thresholds; (b) signals/SDRs whose prior ADR documentation was unclear; and (c) “laboratory signals” or signals from hospital monitoring. In it, we provided counts by studies for the level of evidence and stakeholders.
Records were managed in EndNote (version 8.2). One author (DS) retrieved, screened, and charted the data, a second cross-validated the findings (IJO), and a third settled disagreements after discussion (JKA). A member of the Uppsala Monitoring Centre retrieved the dates of the reports of ADRs to calculate the TTC. All data were charted and analyzed in Microsoft Excel.
3 Results
3.1 Overview of Publications and Their Distribution Over Primary/Secondary Analyses
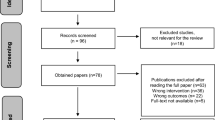
We identified 9525 non-duplicate citations, of which 1509 were considered eligible, based on title/abstract screening (see Fig. 1). Through hand searches, we identified 2260 eligible publications and excluded 522 after a full-text review. In all, we included 2132 publications for analysis, corresponding to 2591 studies, 9167 signals, and 4881 SDRs. Over 5000 signals came from a single study on drug–drug interactions [13]. Table 1 shows the distribution of studies and publications between the primary and secondary analyses. See the ESM for responses to our requests to regulators, for the full list of included and excluded records, and for the data charting form.

Preferred reporting items for systematic reviews and meta-analyses flow chart of the scoping review. 1Includes: 59 abstracts subsequently published as papers, 42 previously communicated signals, 3 duplicate publications. 2Includes: 41 non-systematic (narrative) reviews, 17 descriptive (quantitative) analyses, 9 measurements of usefulness of sources of information or biases affecting disproportionality analyses, 4 commentaries, 1 creation of an interventional program, 1 evidence mapping. 3Includes: 16 records of beneficial effects or drug repurposing, 18 of medication errors, false-positive laboratory abnormalities, or increases in plasma concentrations of a medicinal product after suspected drug–drug interactions. 4Includes: 9 not concerning medicinal products, 4 without data in humans (i.e., simulation studies), 1 withdrawn publication. 51728 from: 16 cited references in electronic records, 4 from Google Scholar, 2 from original authors, 585 from cited reference in the gray literature, 999 from websites, 122 from organizations. ADR adverse drug reaction
3.2 Primary Analysis
The primary analysis included 1974 publications, or 2421 studies, that communicated 9000 signals and 1861 SDRs; 2242 signals originated exclusively from clinical assessments of ADR reports (1683 studies or 941 publications). For 225 (13%) clinical assessments of ADR reports, presenting 228 signals, there were explicit judgments on the features of reports of ADRs (e.g., plausible time to onset) that supported signals. Only three signals in this subset were supported by multiple types of evidence, while the rest were exclusively supported by one.
3.2.1 Features of the Reports of ADRs Supporting Signals
Across the 228 signals, we recorded 12 distinct supporting features, which were positive dechallenge, temporality, positive rechallenge, exclusion of competing causes, single suspected drug, case ascertainment, consistency, biological gradient, specificity, coherence, and reporter type and reported causality assessment. Positive dechallenge/rechallenge were included in ‘experimental evidence’, and ‘single suspected drug’ in ‘exclusion of competing causes’ (see Table 2).
We further categorized the signals as supported by individual or multiple features: 89 were supported by one feature, 79 by two, 48 by three, and 12 by more than three. When supported by only one feature, the most frequent were temporality (n = 37/89), positive dechallenge (n = 23/89), and positive rechallenge (n = 13/89). Overall, temporality combined with positive dechallenge was the most frequent co-occurrence (72/228), followed by positive dechallenge with rechallenge (63/228); positive dechallenge and/or rechallenge co-occurred with temporality in 77/228 signals. See Fig. 2 for signals supported by two or three features (full data available on request).

Schematic representation of 79 and 48 signals supported by at least two or three features. Each node is labeled after the features invoked in clinical assessments of adverse drug reaction reports; linked nodes indicate features that co-occurred. In brackets, count of signals per feature. Excl. excluding
3.2.2 Levels of Evidence
There were 1974 publications in the primary analysis, corresponding to 2421 studies: regulators communicated 1563, private foundations 672, academia 168, healthcare workers 10, and pharmaceutical companies 8. OCEBM level 4 was the most frequent. In 71 cases, the study design was unclear.
Analysis by OCEBM subtypes showed that within the main categories, the largest contributors to level 4 were studies matching this level and mostly (1683/1778) comprised qualitative analyses of reports of ADRs alone, followed by disproportionality analyses (DAs) and analyses including studies with unclear designs. Disproportionality analyses mostly (n = 111/181, 61%) came from academia, fewer (n = 43/181, 24%) from regulators. OCEBM 1 and 2 mostly comprised randomized controlled trials (RCTs), either pooled or individual, that did not prespecify the outcomes of interest. Most of the OCEBM 3 studies matched this level (see Table 3).
Over time, for any stakeholders, the number of studies per year remained at around 50 from 1986 (first recorded date) to 2010, with an initial increase beginning in 2011 and a larger increment from 2013 onwards (see Fig. 3). Considering clinical assessments of reports of ADRs alone, we recorded the yearly average of 36 studies in 2000–2012 and 116 in 2013–2019, corresponding to about a three-fold increase (3.2). This was a 3.3-fold increase for studies matching OCEBM 4 as a whole. For OCEBM 1–3, the average was 10 for the first period and 20 for the second, i.e., a near two-fold increase (1.8). Out of 181 DAs, 107 (59%) were communicated in 2013–19 (25 in 2013–2015 and 82 in 2016–2019; data available on request). There were 2.8 studies per year on average in 2000–2012, and 15 per year in 2013–2019 (ratio 5.4). During this second period, academia communicated on average 11 DAs per year.

Stacked bar graph, showing the numbers of unique studies grouped by year and by Oxford Centre for Evidence-Based Medicine (OCEBM) levels, irrespective of stakeholder, concerning 2350 studies (71 classified as N/A were omitted). In red OCEBM 1, yellow OCEBM 2, blue OCEBM 3, and dark green OCEBM 4 (excluding subtype 4, i.e., disproportionality analyses [DAs]); in light green, OCEBM subtype 4
3.2.3 TTC of Signals
We computed, where possible, the TTC for signals/SDRs in the primary analysis. 7992/10,861 had TTC ≥ 0 and 762 < 0. Across all OCEBM levels and subtypes, the TTC was 0–51 years. There were 6200 signals with a TTC between 0 and 15 years, 1396 with a TTC between 16 and 30 years, and 396 with a TTC ≥30 years, with a median of 9 years. The median value was 8 years in 2000–2012 and 9 years in 2013–2019. See the ESM for a list of all the years of first reports in VigiBase.
3.3 Secondary Analysis: Levels of Evidence in Signals/SDRs Whose Prior Documentation was Unclear
We analyzed the level of evidence of 170 studies separately (158 publications, 3187 signals/SDRs): for 121 (71%), we could not ascertain whether the ADRs of interest had been previously documented, 26 (16%) did not report a threshold, and we received no replies to requests for clarifications (among which 14 had unclear prior documentation of the ADR), 9 (5.3%) reported incomplete values of disproportionality, 8 (4.7%) detected signals from laboratory values (1 in hospital monitoring), in 3 (1.8%) we recorded a mismatch between the reported threshold and entries regarded as SDRs, and for 3 no values of disproportionality were reported.
The most frequent OCEBM level was 4 (n = 161), followed by studies for which we could not assign a level of evidence (n = 7) and that consisted of laboratory signals/hospital monitoring. Analysis of OCEBM sub-levels showed that the largest contributors were DAs (n = 140, 82%).
4 Discussion
4.1 Summary of Main Findings
Reporting of judgments in clinical assessments is infrequent (225/1683 studies). Our results show that the most frequent features of reports of ADRs discussed in 228 signals were ‘experimental evidence,’ ‘temporality,’ and ‘exclusion of competing causes’. Temporality and positive dechallenge often co-occurred, as did positive dechallenge and rechallenge. We found 2591 studies across websites, bulletins, and electronic databases; OCEBM level 4 was the most frequent overall, with clinical assessments of reports of ADRs making up most of this category. In 2013–2019, these increased three-fold, while other studies (OCEBM 1–3) doubled compared with previous years (2000–2012). Disproportionality analyses without clinical assessments were communicated about five times more often in later years and mostly by academia.
4.2 Features of Reports of ADRs
The features we identified are generally in keeping with previous research; they may reflect the ‘strength of evidence’ of reports of ADRs [14]. We have shown how ‘experimental evidence’ was among one of the most frequently considered features. The presence of positive dechallenge/rechallenge has been linked to a higher likelihood of amendments to section 4.8 of product information in the EU [4]. Similarly, a predictive model for the utility of case reports, developed by the FDA [15], suggested that positive dechallenge, positive rechallenge, and ‘designated medical events’ were among the strongest predictors of inclusion of reports of ADRs in assessments of case series. The ‘exclusion of competing causes’ may be understood by considering that reports containing more than one suspected medicinal product tended to be excluded from assessments [15].
In contrast, other features were infrequent, such as ‘biological gradient,’ ‘specificity,’ and ‘coherence’. This may be partly explained by the degree of completeness of reports of ADRs [16]. A review of the features of reports of ADRs of the FDA’s Potential Signals of Serious Risks suggested that the number of reports and prior documentation of an ADR in the year before regulatory action were predictive of regulatory actions [17]; neither appeared in our list of features. This study drew features of reports by analyzing the FDA Adverse Event Reporting System, while we extracted them directly from the clinical assessments, which may explain the discrepancy.
Few studies clearly presented judgments in support of signals. For context, these were available in 225/1683 clinical assessments of ADR reports (see Table 1). Current guidelines encourage consideration of features of reports in clinical assessments [18], thus, in the included studies, it is possible that while judgments were not clearly reported, features were accounted for. For instance, ‘reported causality’ (i.e., the availability of information on single-case causality assessments in structured fields) was one of the least frequent features. However, this feature was described as helpful in a decision-support system for signal validation when SDRs were confounded by indication [19]. To facilitate a critical interpretation of signals, judgments on features of ADR reports may be made clearer in clinical assessments (e.g., [20, 21]).
4.3 Levels of Evidence Underpinning Signals
Consistent with previous reviews [22,23,24], OCEBM level 4 was the most frequent. Between 2010 and 2018, RCTs formed the basis of 36% of FDA Drug Safety Communications [24], giving some support to the lower frequencies of OCEBM 1–2. The recent (2013–2019) increasing trend in OCEBM 4 studies, partly mirrored by OCEBM 1–3, continues to underscore the importance of reports of ADRs in pharmacovigilance and a growing relevance of interventional studies or systematic reviews of RCTs for signal detection. Well-reported RCTs often provide the first indications of the most frequent adverse effects of an intervention [25]; an increase in the use of RCTs as supporting evidence for signals may call for clearer reporting of detected signals in these study designs.
We found that we might have risked providing a biased overview of higher quality types of evidence had we assigned the same levels of evidence as prescribed by the OCEBM tool. For instance, the highest level of evidence, level 1, comprises four subtypes. From a critical perspective, a meta-analysis that includes trials that prespecify the outcome of interest provides a higher quality of evidence than one that does not. The use of subtypes allowed us to disentangle records that concerned disproportionality analyses alone from clinical assessments of reports of ADRs, as they would both be formally categorized as OCEBM level 4. In turn, we were able to show how DAs alone are rarely communicated by regulatory agencies and are primarily published by academia. While there were examples of published DAs [26,27,28] triggering regulatory procedures [29,30,31], further analyses may be needed to understand to what extent published DAs have a bearing on regulation in pharmacovigilance and whether a growing use of these methods is warranted to answer questions on drug-induced harms. We have shown that, despite a five-fold increase in DAs in recent years, regulators seldom rely on DAs alone and that some DAs lack information on prior documentation of ADRs. Earlier research has shown that DAs alone rarely support and may follow/confirm regulatory decisions [32], in addition to suffering from spin or overinterpretation [33], and that determining if SDRs merit further action further demands substantial resources [34].
4.4 TTC of Signals
The calculation of TTC could give rise to four scenarios: (1) TTC > 0, (2) TTC = 0, (3) TTC < 0, or (4) no TTC value. The first two would indicate that reports in VigiBase for a given signal/SDR were available at the time of communication, the third that while there were reports in VigiBase, they may have been entered in the database only after the year of communication. The last would indicate that there were no reports in VigiBase for a given medicinal product and event as of the data lock point.
A median TTC of 9 years is consistent with similar analyses of time to signal detection, showing a median time of 10 years [35]. There was a difference of 1 year in median TTC for the periods 2000–2012 and 2013–2019. However, prior documentation of ADRs may affect the TTC [36], as may the time on market of medicinal products [37] or the public health impact/intensity of ADRs in signal prioritization [4, 14]. The number of reports of suspected ADRs has grown with time, and VigiBase holds over 30 million reports as of 2022. Reports, however, still lack quality, with a large proportion missing narratives [38]. Lack of guidance on improving the quality of reports has previously been highlighted [39]. As such, our findings may suggest that there are delays before a minimum number of sufficiently complete reports are received by the competent regulators; reducing the TTC may require strategies to emphasize the usefulness of complete reports of ADRs in signal detection, targeting the reporters. A follow-up study to investigate whether completeness of reports of ADRs and other variables may affect the TTC is in progress.
4.5 Strengths and Limitations
The key strengths of this study lie in its exhaustiveness, absence of time restrictions, and use of inclusion criteria allowing retrieval of signals/SDRs, rather than established or identified risks (as defined in [40]). To the best of our knowledge, this is the first attempt at systematically collating and describing signals of ADRs globally.
Our primary aim was to characterise the evidence on which signals are based. To succinctly present the results of the study, we did not investigate the ensuing regulatory actions following signals. However, we plan to address this area in a future review.
Our results should be viewed in the light of the decision not to consider ‘biological plausibility’ or interpret disproportionality measures as evidence for ‘strength of association,’ whereas some signals may solely be supported by these considerations. It is also reasonable to assume that unreported judgments about features of reports of ADRs do not necessarily entail absence of judgments altogether.
We merged updates to communications of signals (see “Methods”), so the evidence-base underpinning signals may have accrued for communications updated over time. In other words, the follow-up time for communications may be unevenly distributed, particularly for later years (e.g., 2015–2020); thus, studies dated earlier (e.g., 2010–2014) may yield higher quality evidence. Other factors that might affect trends of OCEBM levels could be the completeness of the eligible studies. For example, signals could be included in full assessment reports or their summaries, depending on the regulator (see the ESM). Such differences may partly explain the presence of OCEBM subtype 4. Low frequencies of signals/SDRs primarily communicated by pharmaceutical companies may have arisen because we defined the first author as the communicator (see the ESM); an FDA review in fact suggested that most changes to product information in the USA were initiated by marketing authorization holders [22].
Misclassifications of signals, already noted in the early 2000s [41, 42], persist. Not only were signals reported across different webpages on websites of regulators, but they were also described using expressions defined otherwise as per the Council for International Organizations of Medical Science [43] (see the ESM). While misclassifications may have led to the omission of relevant records, as we required findings to be explicitly described as “signal(s)”, we mitigated their influence by reaching out to regulators and, in keeping with our inclusion criteria, asked which communications constituted signals, irrespective of the expressions used to qualify them (e.g., ‘safety issue’). Further harmonization may be needed to collect all signals in dedicated sections of drug regulatory websites.
A possible concern could also be that some methods papers that detected undocumented SDRs were included. To this end, we categorized papers, based on their aims, as to whether they developed or evaluated methods, but found only a small proportion (data not shown).
5 Conclusions
Judgments are clearly presented in only a small proportion of clinical assessments of ADR reports. When they are, experimental evidence, temporality, and exclusion of competing causes are key features supporting signals. Clinical assessments of reports of ADRs alone have increasingly supported signals in recent years. For the same period, the number of signals based on evidence of better quality has also grown but not at the same rate. Disproportionality analyses have been published particularly by academia over 2013–2019, while regulators rely more often on OCEBM levels 1–4 than DAs. Future research should evaluate the usefulness of DAs for decision makers.
Change history
29 March 2023
The original article has been corrected. Supplementary materials were missing from this article and have now been uploaded.
References
Hauben M, Aronson JK. Defining “signal” and its subtypes in pharmacovigilance based on a systematic review of previous definitions. Drug Saf. 2009;32(2):99–110. https://doi.org/10.2165/00002018-200932020-00003.
Meyboom RH, Egberts AC, Edwards IR, Hekster YA, de Koning FH, Gribnau FW. Principles of signal detection in pharmacovigilance. Drug Saf. 1997;16(6):355–65. https://doi.org/10.2165/00002018-199716060-00002.
Farcaş A, Măhălean A, Bulik NB, Leucuta D, Mogoșan C. New safety signals assessed by the pharmacovigilance risk assessment committee at EU level in 2014–2017. Expert Rev Clin Pharmacol. 2018;11(10):1045–51. https://doi.org/10.1080/17512433.2018.1526676.
Insani WN, Pacurariu AC, Mantel-Teeuwisse AK, Gross-Martirosyan L. Characteristics of drugs safety signals that predict safety related product information update. Pharmacoepidemiol Drug Saf. 2018;27(7):789–96. https://doi.org/10.1002/pds.4446.
Caster O, Juhlin K, Watson S, Norén GN. Improved statistical signal detection in pharmacovigilance by combining multiple strength-of-evidence aspects in vigiRank. Drug Saf. 2014;37(8):617–28. https://doi.org/10.1007/s40264-014-0204-5.
Wisniewski AFZ, Bate A, Bousquet C, Brueckner A, Candore G, Juhlin K, et al. Good signal detection practices: evidence from IMI PROTECT. Drug Saf. 2016;39(6):469–90. https://doi.org/10.1007/s40264-016-0405-1.
Tricco AC, Lillie E, Zarin W, O’Brien KK, Colquhoun H, Levac D, et al. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169(7):467–73. https://doi.org/10.7326/M18-0850.
Sartori D, Aronson JK, Onakpoya IJ. Signals of adverse drug reactions communicated by pharmacovigilance stakeholders: protocol for a scoping review of the global literature. Syst Rev. 2020;9(1):180. https://doi.org/10.1186/s13643-020-01429-z.
OCEBM Levels of Evidence Working Group, Howick J, Chalmers I, Glasziou P, Greenhalgh T, Heneghan C, et al. The Oxford Levels of Evidence 2. 2011. Available from: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/ocebm-levels-of-evidence. [Accessed 2 Apr 2022].
Aronson JK. Defining aspects of mechanisms: evidence-based mechanism (evidence for a mechanism), mechanism-based evidence (evidence from a mechanism), and mechanistic reasoning. In: LaCaze A, Osimani B, editors. Uncertainty in pharmacology: epistemology, methods, and decisions. Cham: Springer International Publishing; 2020. p. 3–38.
Onakpoya IJ, Heneghan CJ, Aronson JK. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med. 2016;14:10. https://doi.org/10.1186/s12916-016-0553-2.
Onakpoya IJ, Heneghan CJ, Aronson JK. Post-marketing withdrawal of analgesic medications because of adverse drug reactions: a systematic review. Expert Opin Drug Saf. 2018;17(1):63–72. https://doi.org/10.1080/14740338.2018.1398232.
Iyer SV, Harpaz R, LePendu P, Bauer-Mehren A, Shah NH. Mining clinical text for signals of adverse drug-drug interactions. J Am Med Inform Assoc. 2013;21(2):353–62. https://doi.org/10.1136/amiajnl-2013-001612.
Pacurariu AC, Coloma PM, Gross-Martirosyan L, Sturkenboom MC, Straus SM. Decision making in drug safety: a literature review of criteria used to prioritize newly detected safety issues. Pharmacoepidemiol Drug Saf. 2017;26(3):327–34. https://doi.org/10.1002/pds.4128.
Muñoz MA, Dal Pan GJ, Wei YJ, Delcher C, Xiao H, Kortepeter CM, et al. Towards automating adverse event review: a prediction model for case report utility. Drug Saf. 2020;43(4):329–38. https://doi.org/10.1007/s40264-019-00897-0.
Bergvall T, Norén GN, Lindquist M. vigiGrade: a tool to identify well-documented individual case reports and highlight systematic data quality issues. Drug Saf. 2014;37(1):65–77. https://doi.org/10.1007/s40264-013-0131-x.
Fukazawa C, Hinomura Y, Kaneko M, Narukawa M. Factors influencing regulatory decision-making in signal management: analysis based on the signals identified from the FAERS. Ther Innov Regul Sci. 2021;55(4):685–95. https://doi.org/10.1007/s43441-021-00265-0.
Pacurariu A, van Haren A, Berggren AL, Grundmark B, Zondag D, Harder H, et al. SCOPE Work Package 5. Signal management: best practice guide. 2016. Available from: https://www.walkgroveonline.com/scope_training/WP5/Signal%20Management%20Best%20Practice%20Guide.pdf. [Accessed 2 Apr 2022].
Imran M, Bhatti A, King DM, Lerch M, Dietrich J, Doron G, et al. Supervised machine learning-based decision support for signal validation classification. Drug Saf. 2022;45(5):583–96. https://doi.org/10.1007/s40264-022-01159-2.
Perrio M, Voss S, Shakir SA. Application of the Bradford Hill criteria to assess the causality of cisapride-induced arrhythmia: a model for assessing causal association in pharmacovigilance. Drug Saf. 2007;30(4):333–46. https://doi.org/10.2165/00002018-200730040-00006.
Russom M, Fitsum Y, Debesai M, Russom N, Bahta M. Tamsulosin and risk of priapism: a causality assessment using Austin Bradford Hill criteria. Pharmacol Res Perspect. 2022;10(2): e00934. https://doi.org/10.1002/prp2.934.
Croteau D, Pinnow E, Wu E, Muñoz M, Bulatao I, Dal Pan G. Sources of evidence triggering and supporting safety-related labeling changes: a 10-year longitudinal assessment of 22 new molecular entities approved in 2008 by the US Food and Drug Administration. Drug Saf. 2022;45(2):169–80. https://doi.org/10.1007/s40264-021-01142-3.
Lester J, Neyarapally GA, Lipowski E, Graham CF, Hall M, Dal Pan G. Evaluation of FDA safety-related drug label changes in 2010. Pharmacoepidemiol Drug Saf. 2013;22(3):302–5. https://doi.org/10.1002/pds.3395.
Tau N, Shochat T, Gafter-Gvili A, Tibau A, Amir E, Shepshelovich D. Association between data sources and US food and drug administration drug safety communications. JAMA Intern Med. 2019;179(11):1590–2. https://doi.org/10.1001/jamainternmed.2019.3066.
Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340: c869. https://doi.org/10.1136/bmj.c869.
Aggarwal N. Drug-induced subacute cutaneous lupus erythematosus associated with proton pump inhibitors. Drugs Real World Outcomes. 2016;3(2):145–54. https://doi.org/10.1007/s40801-016-0067-4.
Béné J, Moulis G, Bennani I, Auffret M, Coupe P, Babai S, et al. Bullous pemphigoid and dipeptidyl peptidase IV inhibitors: a case–noncase study in the French Pharmacovigilance Database. Br J Dermatol. 2016;175(2):296–301. https://doi.org/10.1111/bjd.14601.
Poluzzi E, Raschi E, Godman B, Koci A, Moretti U, Kalaba M, et al. Pro-arrhythmic potential of oral antihistamines (H1): combining adverse event reports with drug utilization data across Europe. PLoS ONE. 2015;10(3): e0119551. https://doi.org/10.1371/journal.pone.0119551.
European Medicines Agency—Pharmacovigilance Risk Assessment Committee. Minutes of the PRAC meeting on 11-14 January 2016. 2016. Available from: https://www.ema.europa.eu/en/documents/minutes/minutes-prac-meeting-11-14-january-2016_en.pdf. [Accessed 25 Jan 2022].
European Medicines Agency—Pharmacovigilance Risk Assessment Committee. Minutes of the PRAC meeting on 04-08 July 2016. 2016. Available from: https://www.ema.europa.eu/en/documents/minutes/minutes-prac-meeting-04-08-july-2016_en.pdf. [Accessed 25 Jan 2022].
Health Canada. Summary safety review: proton pump inhibitors (PPIs): assessing the risk of a type of skin reaction [subacute cutaneous lupus erythematosus (SCLE)]. 2017. Available from: https://hpr-rps.hres.ca/reg-content/summary-safety-review-detail.php?lang=en&linkID=SSR00182. [Accessed 7 Feb 2022].
Dias P, Penedones A, Alves C, Ribeiro CF, Marques FB. The role of disproportionality analysis of pharmacovigilance databases in safety regulatory actions: a systematic review. Curr Drug Saf. 2015;10(3):234–50. https://doi.org/10.2174/1574886310666150729112903.
Mouffak A, Lepelley M, Revol B, Bernardeau C, Salvo F, Pariente A, et al. High prevalence of spin was found in pharmacovigilance studies using disproportionality analyses to detect safety signals: a meta-epidemiological study. J Clin Epidemiol. 2021;138:73–9. https://doi.org/10.1016/j.jclinepi.2021.06.022.
Alvarez Y, Hidalgo A, Maignen F, Slattery J. Validation of statistical signal detection procedures in EudraVigilance post-authorization data. Drug Saf. 2010;33(6):475–87. https://doi.org/10.2165/11534410-000000000-00000.
Rolfes L, van Hunsel F, Caster O, Taavola H, Taxis K, van Puijenbroek E. Does patient reporting lead to earlier detection of drug safety signals? A retrospective comparison of time to reporting between patients and healthcare professionals in a global database. Br J Clin Pharmacol. 2018;84(7):1514–24. https://doi.org/10.1111/bcp.13576.
Hashiguchi M, Imai S, Uehara K, Maruyama J, Shimizu M, Mochizuki M. Factors affecting the timing of signal detection of adverse drug reactions. PLoS ONE. 2015;10(12): e0144263. https://doi.org/10.1371/journal.pone.0144263.
Fukazawa C, Hinomura Y, Kaneko M, Narukawa M. Significance of data mining in routine signal detection: analysis based on the safety signals identified by the FDA. Pharmacoepidemiol Drug Saf. 2018;27(12):1402–8. https://doi.org/10.1002/pds.4672.
Karimi G, Star K, Lindquist M, Edwards IR. Clinical stories are necessary for drug safety. Clin Med. 2014;14(3):326–7. https://doi.org/10.7861/clinmedicine.14-3-326.
Edwards IR. Good pharmacovigilance practice and the curate’s egg. Drug Saf. 2012;35(6):429–35. https://doi.org/10.2165/11634410-000000000-00000.
Council for International Organizations of Medical Science (CIOMS) - Working Group VIII. In: Raine J, Close P, Dal Pan G, Edwards IR, Gregory B, Hauben M, et al., editors. Practical aspects of signal detection in pharmacovigilance. Geneva: ; 2010: p. 146.
Hauben M, Reich L. Communication of findings in pharmacovigilance: use of the term “signal” and the need for precision in its use. Eur J Clin Pharmacol. 2005;61(5):479–80. https://doi.org/10.1007/s00228-005-0951-4.
Lindquist AM. Seeing and observing in international pharmacovigilance: achievements and prospects in worldwide drug safety. Uppsala: Uppsala Monitoring Centre; 2003.
Council for International Organizations of Medical Sciences (CIOMS). CIOMS cumulative pharmacovigilance glossary. Geneva, Switzerland. 2021. Available from: https://cioms.ch/wp-content/uploads/2021/03/CIOMS-Cumulative-PV-Glossary-v1.0.pdf. [Accessed 18 Oct 2022].
Acknowledgments
The authors are indebted to the national centers that make up the World Health Organization Programme for International Drug Monitoring and provide reports to VigiBase. However, the opinions and conclusions of this study are not necessarily those of the various centers nor of the World Health Organization. MedDRA trademark® is registered by ICH. We thank Nia Roberts, outreach librarian at the Bodleian Health Care Libraries of Oxford, for substantial contributions to the design of the search strategy and its adaptation to the electronic databases, and Sara Vidlin at the UMC, for retrieving the dates of first reports in VigiBase. We further thank Lucie Gattepaille (ex-UMC) for piloting the retrieval of the dates of reports of ADRs. Finally, we thank Henric Taavola (UMC) for aiding DS in interpreting some of the retrieved data mining studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work is part of Daniele Sartori’s DPhil in Evidence-Based Medicine at the University of Oxford (Nuffield Department of Primary Care Health Sciences and the Department of Continuing Education), UK. The DPhil is fully funded by the UMC.
Conflicts of Interest
JKA has written papers on adverse drug reactions in peer-reviewed journals and has received royalties from textbooks that he has edited or co-edited; he has often acted as an expert witness in cases involving adverse drug reactions, including opioids, most often in Coroners’ courts. DS and GNN are employed by the UMC, which manages VigiBase and holds the archives of the Signal Document used to retrieve part of the included studies. Part of the reviewed materials was published in peer-reviewed journals, drug bulletins, and abstracts or posters by researchers presently or formerly belonging to this foundation. Specifically, DS has authored signals published on drug bulletins, as abstracts or posters, some of which were included in this review, and received funding for doctoral studies from the UMC. IJO has no competing interests.
Ethics Approval
This research relied on publicly available, previously collected, anonymized, non-National Health Service data. As such, ethics approval was not required.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Original data, including coding of medicinal products and adverse events to standardized dictionaries, will be made available upon request.
Code Availability
The code used to retrieve the dates of the reports from VigiBase will be made available on reasonable request.
Authors’ Contributions
DS designed the study, with support from IJO, JKA, and specific inputs from GNN. GNN joined the research team later, during data analysis and production of the manuscript, providing support for analysis and substantial contribution to the overall structure of this text. This manuscript was written by DS with support from IJO, GNN, and JKA. All authors read and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Sartori, D., Aronson, J.K., Norén, G.N. et al. Signals of Adverse Drug Reactions Communicated by Pharmacovigilance Stakeholders: A Scoping Review of the Global Literature. Drug Saf 46, 109–120 (2023). https://doi.org/10.1007/s40264-022-01258-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-022-01258-0