
Abstract
Introduction
Eslicarbazepine acetate was first approved in the European Union in 2009 as adjunctive therapy in adults with partial-onset seizures with or without secondary generalization.
Objective
The objective of this study was to review the safety profile of eslicarbazepine acetate analyzing the data from several clinical studies to 6 years of post-marketing surveillance.
Methods
We used a post-hoc pooled safety analysis of four phase III, double-blind, randomized, placebo-controlled studies (BIA-2093-301, -302, -303, -304) of eslicarbazepine acetate as add-on therapy in adults. Safety data of eslicarbazepine acetate in special populations of patients aged ≥65 years with partial-onset seizures (BIA-2093-401) and subjects with moderate hepatic impairment (BIA-2093-111) and renal impairment (BIA-2093-112) are also considered. The incidences of treatment-emergent adverse events, treatment-emergent adverse events leading to discontinuation, and serious adverse events were analyzed. The global safety database of eslicarbazepine acetate was analyzed for all cases from post-marketing surveillance from 1 October, 2009 to 21 October, 2015.
Results
From a pooled analysis of four phase III studies, it was concluded that the incidence of treatment-emergent adverse events, treatment-emergent adverse events leading to discontinuation, and adverse drug reactions were dose dependent. Dizziness, somnolence, headache, and nausea were the most common treatment-emergent adverse events (≥10% of patients) and the majority were of mild-to-moderate intensity. No dose-dependent trend was observed for serious adverse events and individual serious adverse events were reported in less than 1% of patients. Hyponatremia was classified as a possibly related treatment-emergent adverse event in phase III studies (1.2%); however, after 6 years of post-marketing surveillance it represents the most frequently (10.2%) reported adverse drug reaction, with more than half of these cases occurring with eslicarbazepine acetate at daily doses of 1200 mg. Other adverse drug reactions reported in post-marketing surveillance are seizure (5.8%), dizziness (4.1%), rash (2.6%), and fatigue (2.1%). The safety profile of eslicarbazepine acetate in renal and hepatic impairment subjects (phase I studies) and in elderly patients (phase III study) did not raise any specific concern.
Conclusion
After 6 years of post-marketing surveillance, eslicarbazepine acetate maintains a similar safety profile to that observed in pivotal clinical studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The most common treatment-emergent adverse events (dizziness, somnolence, headache, and nausea) were dose dependent and the majority were of mild-to-moderate intensity. |
Most of the hyponatremia cases in post-marketing surveillance have been related to high doses of eslicarbazepine acetate (>1200 mg), drug administration errors, concomitant medications, and/or inter-current illness. |
After 6 years of real-life clinical experience, eslicarbazepine acetate maintains the safety profile obtained in clinical studies, including four phase III studies. |
1 Introduction
Eslicarbazepine acetate (ESL) is a once-daily (QD) oral antiepileptic drug (AED) approved by the European Medicines Agency (EMA) on 21 April, 2009 (Zebinix®; BIAL-Portela & Ca, S.A., S. Mamede do Coronado, Portugal), as adjunctive therapy in adults with partial-onset seizures (POS), focal seizures according to the new classification of the International League Against Epilepsy, with or without secondary generalization [1]. Currently, ESL is approved in 43 countries (31 of the European Economic Area and 12 non-European Economic Area countries, including USA and Canada), and is currently marketed in 22. Eslicarbazepine acetate (Aptiom®; Sunovion Pharmaceuticals Inc., Marlborough, MA, USA) has also been approved by the US Food and Drug Administration for adjunctive therapy (November 2013) and monotherapy (August 2015) in adults with POS [2]. Recently, the EMA approved ESL (Zebinix®) for monotherapy in the treatment of POS in adults and as adjunctive therapy in children aged above 6 years with POS.
During the clinical development program, four pivotal phase III, randomized, controlled studies (BIA-2093-301, -302, -303, -304) assessed the efficacy and safety of adjunctive ESL for adult patients with refractory POS [3,4,5,6]. An analysis of pooled (pool reviewed and approved by the EMA) safety data (-301, -302, and -303) has been previously reported [7] and, more recently, Biton et al. [8] published a pooled analysis of three phase III studies (-301, -302, and -304) [pool reviewed and approved by the Food and Drug Administration].
The main purpose of this work is to provide an overview on the safety profile of ESL by expanding the information obtained from several clinical studies including two phase I studies (renal or hepatic impairment subjects), one phase III study (elderly patients), and a post-hoc pooled analysis of safety data of four phase III studies (-301, -302, -303, -304) to that obtained by the pharmacovigilance activities gathered during 6 years of real-life clinical experience. To the best of our knowledge, this is the first time that an overview of the safety of ESL from clinical studies to post-marketing experience is reported. Moreover, we discuss the safety profile of ESL in special populations such as renal and hepatic impairment subjects, elderly patients, and pregnant women.
1.1 Pharmacokinetics and Pharmacodynamics of Eslicarbazepine Acetate
After oral administration, ESL undergoes extensive first-pass hydrolysis to its major active metabolite eslicarbazepine, representing approximately 95% of circulating active moieties [9,10,11]. Maximum plasma concentrations of eslicarbazepine are attained 2–3 h post-dose and steady-state plasma concentrations are achieved after 4–5 days of QD dosing, consistent with an effective half-life in the order of 20–24 h. In studies in healthy subjects and adult patients with epilepsy, the eslicarbazepine apparent half-life of elimination was 10–20 and 13–20 h, respectively. A study in healthy volunteers showed that in the cerebrospinal fluid (CSF) there was a smaller apparent peak-to-trough fluctuation of eslicarbazepine (1.5) compared with plasma (2.9), with a longer apparent half-life (24.8 ± 8.1 h), and with just minor exposure to both oxcarbazepine or R-licarbazepine in plasma or CSF [9]. The smaller peak-trough fluctuation in the CSF compared with plasma is explained by a slower brain disposition and elimination rates and a smaller difference between the rates of these processes in relation to the plasma compartment. Indeed, the maximum plasma concentration in CSF is achieved at 12 h, that is half of the time range (24 h), meaning that the amount of ESL eliminated from the CSF balances the amount 12 h after (time to maximum plasma concentration = 12 h, half-life = 24.8 h). However, in plasma, the maximum plasma concentration is attained at 2.4 h and a fast disposition was observed compared with the elimination phase (time to maximum plasma concentration = 2.4 h, half-life = 15.9 h) [9].
Food did not significantly affect the pharmacokinetics of eslicarbazepine, meaning that ESL tablets may be taken with or without food [1, 12]. Eslicarbazepine acetate tablets can be taken either as whole or as crushed, as was shown in a bioequivalence study with ESL 800 mg crushed or intact tablets in healthy volunteers [2, 13].
Studies in humans have shown that drug–drug interactions between ESL and other AEDs are unlikely to be clinically relevant in most cases, although dosage adjustments may be required if ESL is administered concomitantly with carbamazepine and phenytoin [11]; ESL also may decrease the effectiveness of hormonal contraceptives and, therefore, additional non-hormonal forms of contraception are recommended [14, 15]. Regarding statins, there was a decrease in exposure to simvastatin and rosuvastatin when co-administered with ESL [1, 2, 11, 16].
Eslicarbazepine is thought to act by stabilizing the inactive state of voltage-gated sodium channels, with greater selectivity for the inactive state of the channel vs. the resting state, compared with carbamazepine or oxcarbazepine [17, 18]. Additionally, eslicarbazepine does not share with carbamazepine the ability to alter fast inactivation of voltage-gated sodium channels, but rather appears to modify the kinetics and voltage dependence of slow inactivation states [19, 20]. Eslicarbazepine also shows a 10- to 60-fold higher potency for the blockade of low- and high-affinity hCav3.2 inward currents when compared with carbamazepine [21]. Pharmacokinetic/pharmacodynamic analysis demonstrated that as the ESL dosage and eslicarbazepine plasma concentrations increase, the seizure frequency decreases [22].
2 Methods
2.1 Pooled Analysis of Four Phase III Studies
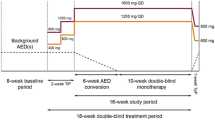
This is a post-hoc pooled analysis of safety data from four pivotal phase III epilepsy studies (BIA-2093-301 [NCT00957684], BIA-2093-302 [NCT00957047], BIA-2093-303 [NCT00957372], and BIA-2093-304 [NCT00988429]; registered at ClinicalTrials.gov). Of note, as major protocol violations raised doubts on the reliability of the study results in Study BIA-2093-303, this particular study was not included in the US Food and Drug Administration New Drug Application for ESL. Each study was a multinational, randomized, double-blind, placebo-controlled trial in patients with treatment-refractory POS. Details of the study population, inclusion and exclusion criteria, efficacy assessments, and safety and tolerability assessments have been published elsewhere [3,4,5,6]; briefly after an 8-week baseline period, patients were randomized in a 1:1:1:1 ratio to one of the following four QD treatment groups: ESL 400 mg (only studies -301 and -302), ESL 800 mg, ESL 1200 mg, or placebo. The double-blind phase comprised a 2-week titration period (no titration in ESL 400-mg and ESL 800-mg arms for study -302), a 12-week maintenance period, and a 4-week tapering-off period (no tapering-off in study -302). At randomization, patients were meant to have four or more POS (three for study 304) per 4 weeks during the baseline period, while receiving treatment with one or two concomitant AEDs (studies -301, -303, and -304) or one to three AEDs (study -302), except with oxcarbazepine and felbamate [3,4,5,6]. All studies were approved by the appropriate ethics committees or institutional review boards and were conducted according to the international and local regulations of the countries where they were performed. Patients gave their written consent prior to enrolment.
Outcomes of safety data included the incidence of treatment-emergent adverse events (TEAEs), TEAEs leading to discontinuation, serious adverse events (SAEs) and clinical laboratory tests (hematology, coagulation, biochemistry, thyroid function, and urinalysis). These data were analyzed through descriptive statistics. Treatment-emergent adverse events were recorded by the investigator and assessed for severity (mild, moderate, or severe), causality, and seriousness. These TEAEs were also re-analyzed by the sponsor, nevertheless the investigator’s opinion was binding. Treatment-emergent adverse events were classified using the Medical Dictionary for Regulatory Activities (MedDRA®) Version 10.0 (for studies -301, -302, and -303) or 13.1 (for study -304).
2.2 Special Populations
2.2.1 Renal Impairment
The pharmacokinetics of ESL in subjects with various degrees of renal function was evaluated in an open-label, single-dose (ESL 800 mg), phase I study (BIA-2093-112 [NCT02281422]) including 40 subjects (eight subjects per each of the five renal function groups): normal renal function (creatinine clearance >80 mL/min); mild (creatinine clearance 50–80 mL/min); moderate (creatinine clearance 30–50 mL/min); severe renal impairment (creatinine clearance <30 mL/min); and end-stage renal disease (requiring hemodialysis).
2.2.2 Hepatic Impairment
An open-label, multiple-dose, single-center study (BIA-2093-111 [NCT02281526]) was implemented to investigate the pharmacokinetics of ESL in subjects with moderate hepatic impairment. Healthy subjects (n = 8) and moderately liver-impaired patients (n = 8) were treated with ESL 800 mg QD for 8 consecutive days.
2.2.3 Elderly Population
A post-approval commitment with the EMA, a multicenter, open-label, non-controlled, single-arm study (BIA-2093-401 [NCT01422720]) with flexible doses of ESL between 400 and 1200 mg QD was performed to evaluate the safety, tolerability, and efficacy of ESL as adjunctive therapy in patients aged ≥65 years with POS, over a 26-week treatment period (n = 72) [23].
The global safety database of ESL was reviewed to identify pregnancy cases with exposure to ESL reported up to 21 October, 2015. The EMBASE and MEDLINE databases were also searched to identify literature reports on the use of ESL during pregnancy published between 1 October, 2009 and 21 October, 2015.
2.3 Post-Marketing Surveillance
The safety data were obtained from spontaneous reports, health authority reports, literature reports, reports from non-interventional studies, and other solicited sources, which were collected worldwide as part of the routine pharmacovigilance activities of the marketing authorization holder. All individual case safety reports collected are included in the master safety database (Argus Safety™) established and maintained by the marketing authorization holder. The Argus Safety™ database was searched for all cases from post-marketing surveillance (PMS), from the date of first launch (1 October, 2009) up to 21 October, 2015 (6 years). All adverse drug reactions (ADRs) were coded using the MedDRA® Version 18.0, assessed for seriousness and listedness. Data were pooled together from all spontaneous and literature reports, whilst solicited reports were pooled separately. For spontaneous and literature reports, an at least possible causality was defined, while for solicited reports only cases assessed as at least possibly related to ESL by the reporter were included in the analysis.
Cumulative summary tabulations of terms arranged by primary system organ class (SOC) were retrieved and sorted according to the European Union summary of product characteristics (SmPC) guideline. MedDRA® preferred terms within each SOC were sorted by decreasing frequency and the results were compared with the approved SmPC.
Patient exposure to marketed ESL was estimated on the basis of worldwide ex-factory sales for the period from 1 October, 2009 until 31 August, 2015. For estimation of patient exposure, it was assumed that the ex-factory amounts delivered were entirely dispensed and actually all administered, and were used at the dosage regimen of one tablet per day, regardless of dose strength as recommended in the ESL SmPC [1]. Because ESL is intended for long-term therapy, exposure is calculated in patient-months (units divided by 30) and patient-years, rather than in the number of treated patients. During this period, ex-factory amounts reached a total of 33,289,762 units (one unit = one tablet). This corresponds to an estimated patient cumulative exposure of 1,109,658 patient-months, corresponding to 92,471.5 patient-years [24]. As described in Table 1, patient exposure to marketed product is currently distributed mainly in Spain, Germany, Portugal, France, and USA.
3 Results
3.1 Safety Findings from Pooled Analysis of Four Phase III Studies
More than 1700 patients were enrolled in the four pivotal, phase III, double-blind, randomised, placebo-controlled studies [3,4,5,6], the methods of which were briefly described above, and 1699 were included in the integrated safety population [24]. During the 14 weeks of double-blind treatment period, there was an apparent dose-dependent increase in the overall incidence of TEAEs with the increase in ESL dosage (Table 2) [24]. The same trend was observed for the occurrence of ADRs [TEAEs assessed as at least possibly related by the investigator] and TEAEs leading to discontinuation, but no dose-dependence relationship was observed for the occurrence of serious TEAEs (Table 2). Similar conclusions were achieved by other ESL pooled analyses that considers three phase III studies [7, 8].
In the overall safety population, the most common TEAEs (affecting ≥10% of patients in any treatment group) were dizziness, somnolence, headache, and nausea (Table 3) and all were dose dependent. The most frequent TEAEs leading to discontinuation (affecting ≥3% of patients in any group: placebo; ESL 400 mg; ESL 800 mg; ESL 1200 mg) were: dizziness (0.8; 1.0; 4.8 and 8.2%, respectively), ataxia (0; 1.5; 1.8 and 3.9%, respectively), nausea (0; 0; 2.2 and 5.3%, respectively), and vomiting (0.2; 1.0; 2.0 and 4.3%, respectively) [24]. Similar safety profiles were obtained in the pooled analysis of clinical studies -301, -302, and -303 [7], as well as in the pooled population obtained from studies -301, -302, and -304 [8].
The majority of TEAEs were of mild or moderate intensity in any treatment group (mild: placebo, 39.8%; ESL, 34.7%; moderate: placebo, 48.3%; ESL, 45.5%; severe: placebo, 11.9%; ESL, 19.8%). The overall incidence of SAEs was low in any treatment group. There was a higher incidence of SAEs in the ESL group (3.8%) than in the placebo group (2.1%). No individual SAE was reported in more than 1% of patients [24]. Indeed, the only SOCs in which two or more subjects per group reported SAEs were nervous system disorders (placebo: four subjects [0.8%], ESL: 22 subjects [1.9%]), injury, poisoning and procedural complications (placebo: three subjects [0.6%], ESL: nine subjects [0.8%]), and gastrointestinal disorders (placebo: one subject [0.2%], ESL: eight subjects [0.7%]) [24].
Laboratory parameters, vital signs, physical examination, and electrocardiograms did not raise any major safety issue. Hyponatremia (<125 mEq/L), as an adverse event (AE), was reported in 17 patients (placebo n = 0; ESL 400 mg n = 1 (0.5%); ESL 800 mg n = 6 (1.2%); and ESL 1200 mg n = 10 (2.0%)) and it was classified as a possibly related TEAE in 16 patients (1.2%) treated with ESL. Hyponatremia or decreased blood sodium levels led to the discontinuation of study medication in 0.3 and 0.1% of ESL-treated patients and placebo treated patients, respectively [24].
In this full integrated analysis, three deaths occurred; two in the placebo group (acute respiratory failure and possible sudden unexpected death in epilepsy), and one in the ESL 800-mg group [while taking ESL 400 mg during titration (status epilepticus, subsequently classified as probable sudden unexpected death in epilepsy)]. One patient drowned during the baseline period of study 304 (without having taken ESL).
3.2 Safety Findings from Special Populations
In the study for the evaluation of pharmacokinetics of ESL in patients with impaired renal function, safety was evaluated as a secondary endpoint. Twelve subjects (30%) reported 15 AEs assessed as being at least possibly related to study medication: normal renal function group: headache (n = 5) and somnolence (n = 1); mild renal impairment group: headache (n = 4), somnolence (n = 3), and dizziness (n = 1); and in the severe renal impairment: somnolence (n = 1). Overall, AEs were mild to moderate in intensity. Two AEs were assessed by the investigator as severe in intensity: fatigue (unlikely related) and headache (possibly related). Mean changes from baseline for hematology, clinical chemistry, and clotting profile variables raised no concerns in general for all treatment groups.
Safety was also assessed in subjects with hepatic impairment and compared with healthy subjects. In general, ESL was well tolerated by subjects in both groups. Of the 36 AEs reported, three were considered to be drug related: nausea in a subject in the hepatic impairment group and two events of fatigue in the healthy control group. Seven AEs were assessed to be of moderate intensity and 28 to be of mild intensity. There was one SAE described as hepatic encephalopathy secondary to increased protein intake (unlikely related) in the liver-impaired group. Mean changes from baseline for hematology, clinical chemistry, and clotting profile variables raised no concerns, in general.
Overall, 78 ESL exposures during pregnancy were reported up to 21 October, 2015. Of these, 40 patients became pregnant during clinical studies and 38 pregnancies were reported from post-marketing surveillance [24]. In about half of pregnancies, ESL was used as part of an AED add-on regimen. The available data are insufficient in both quantity and detail level to allow conclusions about the safety of ESL in pregnant women. There are no case reports involving drug exposure during lactation.
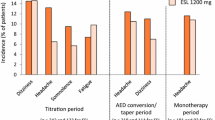
Regarding the elderly population (aged ≥65 years, n = 72), the majority of subjects were exposed to doses not higher than 800 mg QD; only ten subjects (13.9%) were exposed to ESL 1200 mg QD. The overall frequency of TEAEs was 65.3%, similar to pooled population of phase III studies (ESL 800 mg, 67%, see Table 3). The most frequent TEAEs were dizziness (12.5%), somnolence (9.7%), and fatigue, convulsion, and hyponatremia (8.3%, each), while rash was uncommon, occurring only in two patients (2.8%). Figure 1 describes the incidence of TEAEs observed in non-elderly patients and those obtained from pooled analysis of four phase III studies. Fatigue, convulsion, and hyponatremia were more frequent in the elderly population, in addition to the incidence of SAEs (13.9%), and TEAEs leading to study discontinuation (22.2%) was higher than those the observed for pooled phase III studies (4.8 and 12.2% for ESL 800 mg, respectively, Fig. 1). However, dizziness and somnolence were more frequent in the non-elderly population (Fig. 1). The majority of TEAEs were of mild or moderate intensity and the TEAEs leading to the discontinuation of more than one patient were hyponatremia (n = 3), dizziness (n = 2), and fatigue (n = 2). Three deaths were reported during this study as a result of cardiac failure, glioblastoma multiforme, and ischemic stroke, nevertheless none of the fatal cases was considered related to study medication by the investigator nor the sponsor.

Incidence of treatment-emergent adverse events (TEAEs) in pooled four phase III studies (-301, -302, -303, -304) of eslicarbazepine acetate (ESL) as add-on therapy in adults (non-elderly patients) and in an elderly population (BIA-2093-401). Serious adverse events (SAE) and TEAEs leading to discontinuation of medication obtained for both populations are also presented
3.3 Safety Findings from Post-Marketing Surveillance
Overall, 702 serious and 1273 non-serious ADRs from health authorities, literature, and spontaneous reports, and 47 serious ADRs from post-marketing non-interventional studies were reported during the 6 years of ESL marketing. Based on the current sales data, this corresponds to a reporting rate of 0.18 events per 100 patient-months, considering only non-solicited sources (Table 4) [24].
Most commonly, ADRs were listed and reported in the SOCs nervous system disorders (23.4%), general disorders and administration-site conditions (13.6%), metabolism and nutrition disorders (11.3%), skin and subcutaneous tissue disorders (8.7%), psychiatric disorders (7.4%), and gastrointestinal disorders (6.5%). The most frequent reported ADR terms were hyponatremia (10.2%), seizure (5.8%), dizziness (4.1%), rash (2.6%), fatigue (2.1%), and nausea (1.8%) (Table 4) [24]. Cases of off-label use included overdose (n = 111), unapproved indication (n = 92), use in monotherapy (n = 65, not approved within timeframe of data analysis), inappropriate starting dose (n = 46), use in pediatric population (n = 40, not approved within timeframe of data analysis), and drug administration error (n = 15) [24]. A total of 239 cases were reported as ‘hyponatremia’ or ‘blood sodium decreased’ (188 serious and 51 non-serious) [Table 4].
Serious and non-serious adverse events classified within the SOC psychiatric disorders represented 7.4% of all ADRs reported. Confusional state (n = 14), depression (n = 12), aggression (n = 10), and suicidal ideation (n = 10) were the most frequent events. Four cases of suicide attempt (two fatal cases) had been observed. Cardiac disorders were also infrequently reported (21 cases, mostly bradycardia) [24]. Considering the skin and subcutaneous disorders, rash (generalized and pruritic) represented about 3.8% of all ADRs registered.
4 Discussion
Pooled safety data from the four phase III studies and from specific studies in special populations together with those obtained from 6 years of post-marketing experience were analyzed to confirm the assessment of the safety profile of ESL. This approach allows the assessment of safety data in a broader population and in more ‘realistic clinical conditions’ because it combines the data obtained from strict controlled, double-blind, phase III studies with uncontrolled spontaneous reports from post-marketing [7, 8].
A dose-dependent increase in the overall incidence of TEAEs, TEAEs leading to discontinuation, and ADRs with the increase in ESL dosage was observed in this full integrated analysis. These results are in agreement with those obtained from individual phase III trials [3,4,5,6] and corroborate with previous pooled analysis [7, 8]. The total discontinuation rate for ESL patients (n = 1186) was 15.8% (Table 2), which is low compared with withdrawal rates reported with oxcarbazepine, which ranged from 12 to 67% [25]. Biton et al. [8], which does not include study -303, reported a similar ESL discontinuation rate (17.5%). Moreover, despite somnolence and headache being common TEAEs, their relationship to discontinuation was minor, when compared with dizziness, ataxia, nausea, and vomiting.
Beyond these four phase III studies, the safety profile of ESL in adult patients was also evaluated in special populations, with special attention to renal and hepatic impairment and elderly patients. The phase I study performed in subjects with renal impairment demonstrated that clearance of ESL is dependent on renal function and, therefore, during treatment with ESL, dose adjustment is recommended in patients with creatinine clearance <60 mL/min. Hemodialysis removes ESL metabolites from plasma [26], and in patients with creatinine clearance <30 mL/min, ESL use is not recommended. Despite the fact that ESL undergoes extensive hepatic first-pass hydrolysis, moderate hepatic impairment did not affect the pharmacokinetics of ESL. No dose adjustment is needed in patients with mild-to-moderate liver impairment. Patients with severe hepatic impairment have not been evaluated; therefore, its use is not recommended in this group of patients [27].
During its clinical development, the use of ESL in elderly patients (aged ≥65 years) was limited (n = 9, in a pooled population of four phase III studies) and, as such, a multicenter, open-label, non-controlled, single-arm study was performed (BIA-2093-401) [23]. The highest incidence of hyponatremia, SAEs, and TEAEs leading to study discontinuation in elderly patients was not unexpected given the higher co-morbidities in this age group. Nevertheless, no special concern is adopted to elderly patients when compared with other aged groups and no dose adjustment of ESL is needed in the elderly population if the renal function is not disturbed [23]. Finally, current data of ESL exposure in pregnant women are limited, and for that reason it is recommended that if women receiving ESL become pregnant or plan to become pregnant, the use of ESL should be carefully re-evaluated [1].
The incidence of rash, which is a common idiosyncratic reaction with AEDs, seems to be low in patients treated with ESL and dose dependent (Table 3). In the post-marketing, it occurred in 3.8% of the reports, which is in accordance with incidence values found for pooled population of phase III studies (1.5 and 4.9% for ESL 800 and 1200 mg, respectively) and elderly patients (2.8%).
There were two cases of potential drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome that occurred during the clinical trial (BIA-2093-301) and two other cases from PMS. The first two patients, recently described by Biton et al. [8], had combinations of AEs that may potentially met the RegiSCAR criteria for DRESS syndrome. Both cases were identified as ‘potential DRESS’ as no confirmation on diagnosis was identified by the reporter.
Regarding post-marketing DRESS cases, the first refers to a 60-year-old male patient, bedridden as a result of multi-infarction dementia, with a history of several episodes of febrile exanthema and allergy to paracetamol and levetiracetam. The patient had been taking ESL for 5 days and further developed febrile exanthema, which worsened 10 days later owing to a hemorrhagic exanthema. Causality cannot be assessed properly because of multi-drug treatment, pre-existent allergy to levetiracetam, and recurring febrile exanthemas in the past. The dermatological expert confirmed that DRESS was a probable diagnosis in this case with concomitantly used drugs or infection as plausible alternative causes. The second PMS case refers to a 16-year-old female patient who developed ‘cutaneous eruption’ primarily on the trunk and left arm associated with face edema, headache, and hyperthermia 12 days after exposure to ESL (Zebinix®). Concomitant medication included lamotrigine and clobazam. The dermatological expert confirmed that DRESS was a possible diagnosis in this case, possibly related to ESL.
In phase III studies, hyponatremia <125 mmol/L was reported in ≤2% of patients taking ESL, whilst in post-marketing hyponatremia/blood sodium decreased represented 14.1% of ADRs reported. Most of the PMS cases were commonly in the range of 120–129 mmol/L, nevertheless in 33 (13.8% of these cases), the sodium levels were below 120 mmol/L and complications including seizure, confusional state, nausea, vomiting, and hypertension were reported in about 50% of these cases. More than half of the hyponatremia cases (n = 140, 58.6%) occurred when ESL was given at daily doses higher than 1200 mg. Other cases occurred with drug administration errors, including no up-titration, unapproved use, or the result of concomitant medication (e.g., diuretics) or intercurrent illness (e.g., renal disease) [24]. Changes in mean clinical laboratory parameters did not reveal clinically relevant findings.
Data from literature demonstrate an interaction of ESL with other AEDs by an inducing effect on the metabolism of drugs (carbamazepine, phenobarbital, topiramate), which are primarily eliminated by metabolism through CYP3A4 or conjugation through the UDP–glucuronosyl (lamotrigine) or inhibiting properties against CYP2C9 (phenytoin) [11]. Plasmatic concentrations of oral contraceptives and simvastatin may also be affected by ESL most likely caused by an induction of CYP3A4 [14, 16]. The PMS safety data collected during these 6 years did not identify any new potential interaction beyond those identified in the clinical development program and described in the literature.
Psychiatric AEs such as anxiety, aggressive behavior, irritability, and depression have been associated with AED therapy. Recently, published data demonstrated that patients receiving sodium channel-blocking AEDs (as ESL) were statistically less likely to develop intolerable psychiatric problems, compared with those taking AEDs possessing other mechanisms of action [28]. From the four phase III ESL studies, the most frequently reported psychiatric TEAEs were depression and irritability, both demonstrated no dose relationship with ESL (Table 3). From post-marketing experience, confusional state, depression, aggression, and suicidal ideation represented 0.82, 0.71, 0.59, and 0.59% of reported ADRs, respectively. All cases of suicidal ideation were classified as serious because of medical significance, nevertheless for most of them alternative explanations are available, mainly depression and concomitant AEDs. Cognition-related TEAEs were reported uncommonly with ESL treatment during post-marketing experience, similar to that described in the literature [29].
The main limitations of this work is the analysis of TEAE incidence of four phase III studies because descriptive statistics was performed and the underlying limitations of post-marketing reports.
5 Conclusions
Based on four phase III clinical trials and a review of post-marketing safety data, adjunctive therapy with ESL is generally well tolerated, with most AEs being of mild-to-moderate severity. The most common TEAEs (dizziness, somnolence, headache, and nausea) were dose dependent. Dose adjustment of ESL is recommended in patients with creatinine clearance <60 mL/min, whilst no dose adjustment is needed in patients with mild-to-moderate liver impairment (phase I studies). Elderly patients had the highest incidence of hyponatremia and SAEs when compared with non-elderly patients; nevertheless, no special concern is needed for the elderly population if renal function is not disturbed. Most of the hyponatremia cases in post-marketing experience have been related to high doses of ESL (>1200 mg), drug administration errors, concomitant medications or/and inter-current illness. Limited data are available to draw conclusions about the safety of ESL in pregnant women. Despite the limitations of the global safety databases, the data available support the safety profile of ESL according to the information gathered during clinical development and continue to support the favorable benefit-risk profile of the product.
References
European Medicines Agency. Zebinix (eslicarbazepine acetate): EU summary of product characteristics. 2017. Available from: http://www.ema.europa.eu/ema/. Accessed 15 July 2017.
Sunovion Pharmaceuticals Inc. Aptiom® (eslicarbazepine acetate) tablets, for oral use: US prescribing information. 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022416s001lbl.pdf. Accessed 15 July 2017.
Elger C, Halasz P, Maia J, Almeida L, Soares-da-Silva P. Efficacy and safety of eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures: a randomized, double-blind, placebo-controlled, parallel-group phase III study. Epilepsia. 2009;50(3):454–63.
Ben-Menachem E, Gabbai AA, Hufnagel A, Maia J, Almeida L, Soares-da-Silva P. Eslicarbazepine acetate as adjunctive therapy in adult patients with partial epilepsy. Epilepsy Res. 2010;89(2–3):278–85.
Gil-Nagel A, Lopes-Lima J, Almeida L, Maia J, Soares-da-Silva P. Efficacy and safety of 800 and 1200 mg eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures. Acta Neurol Scand. 2009;120(5):281–7.
Sperling MR, Abou-Khalil B, Harvey J, Rogin JB, Biraben A, Carlo A, et al. Eslicarbazepine acetate as adjunctive therapy in patients with uncontrolled partial-onset seizures: results of a phase III, double-blind, randomized, placebo-controlled trial. Epilepsia. 2015;56(2):244–53.
Gil-Nagel A, Elger C, Ben-Menachem E, Halasz P, Lopes-Lima J, Gabbai AA, et al. Efficacy and safety of eslicarbazepine acetate as add-on treatment in patients with focal-onset seizures: integrated analysis of pooled data from double-blind phase III clinical studies. Epilepsia. 2013;54(1):98–107.
Biton V, Rogin JB, Krauss G, Abou-Khalil B, Rocha JF, Moreira J, et al. Adjunctive eslicarbazepine acetate: a pooled analysis of three phase III trials. Epilepsy Behav. 2017;72:127–34.
Nunes T, Rocha JF, Falcao A, Almeida L, Soares-da-Silva P. Steady-state plasma and cerebrospinal fluid pharmacokinetics and tolerability of eslicarbazepine acetate and oxcarbazepine in healthy volunteers. Epilepsia. 2013;54(1):108–16.
Elger C, Bialer M, Falcao A, Vaz-da-Silva M, Nunes T, Almeida L, et al. Pharmacokinetics and tolerability of eslicarbazepine acetate and oxcarbazepine at steady state in healthy volunteers. Epilepsia. 2013;54(8):1453–61.
Bialer M, Soares-da-Silva P. Pharmacokinetics and drug interactions of eslicarbazepine acetate. Epilepsia. 2012;53(6):935–46.
Maia J, Vaz-da-Silva M, Almeida L, Falcão A, Silveira P, Guimarães S, et al. Effect of food on the pharmacokinetic profile of eslicarbazepine acetate (BIA 2-093). Drugs R D. 2005;6(4):201–6.
Kharidia J, Blum D, Cheng H. A pharmacokinetic study comparing eslicarbazepine acetate administered orally as crushed or intact tablet in healthy volunteers. Epilepsy Curr. 2013;13(Suppl. 1):106.
Falcao A, Vaz-da-Silva M, Gama H, Nunes T, Almeida L, Soares-da-Silva P. Effect of eslicarbazepine acetate on the pharmacokinetics of a combined ethinylestradiol/levonorgestrel oral contraceptive in healthy women. Epilepsy Res. 2013;105(3):368–76.
Reimers A, Brodtkorb E, Sabers A. Interactions between hormonal contraception and antiepileptic drugs: clinical and mechanistic considerations. Seizure. 2015;28:66–70.
Falcao A, Pinto R, Nunes T, Soares-da-Silva P. Effect of repeated administration of eslicarbazepine acetate on the pharmacokinetics of simvastatin in healthy subjects. Epilepsy Res. 2013;106(1–2):244–9.
Almeida L, Soares-da-Silva P. Eslicarbazepine acetate (BIA 2-093). Neurotherapeutics. 2007;4(1):88–96.
Bonifacio MJ, Sheridan RD, Parada A, Cunha RA, Patmore L, Soares-da-Silva P. Interaction of the novel anticonvulsant, BIA 2-093, with voltage-gated sodium channels: comparison with carbamazepine. Epilepsia. 2001;42(5):600–8.
Hebeisen S, Pires N, Loureiro AI, Bonifacio MJ, Palma N, Whyment A, et al. Eslicarbazepine and the enhancement of slow inactivation of voltage-gated sodium channels: a comparison with carbamazepine, oxcarbazepine and lacosamide. Neuropharmacology. 2015;89:122–35.
Hebeisen S, Soares-da-Silva P. Slow and fast inactivation of voltage-gated sodium channels by eslicarbazepine, carbamazepine, oxcarbazepine and lacosamide. Epilepsy Curr. 2012;13(Suppl. 1):414–5.
Brady K, Hebeisen S, Konrad D, Soares-da-Silva P. The effects of eslicarbazepine, R-licarbazepine, oxcarbazepine and carbamazepine on ion transmission Cav3.2 channels. Epilepsia. 2011;52(Suppl. 6):260.
Falcao A, Fuseau E, Nunes T, Almeida L, Soares-da-Silva P. Pharmacokinetics, drug interactions and exposure-response relationship of eslicarbazepine acetate in adult patients with partial-onset seizures: population pharmacokinetic and pharmacokinetic/pharmacodynamic analyses. CNS Drugs. 2012;26(1):79–91.
Costa R, Oliveira C, Lopes N, Gama H, Sousa R, Nunes T, et al. Safety and efficacy of eslicarbazepine acetate treatment in elderly patients. Epilepsia. 2014;55:112.
Bial–Portela & Cª, S.A. Data on file; 2015.
Zaccara G, Giovannelli F, Maratea D, Fadda V, Verrotti A. Neurological adverse events of new generation sodium blocker antiepileptic drugs: meta-analysis of randomized, double-blinded studies with eslicarbazepine acetate, lacosamide and oxcarbazepine. Seizure. 2013;22(7):528–36.
Almeida L, Potgieter JH, Maia J, Soares-da-Silva P. Pharmacokinetics of eslicarbazepine acetate in patients with renal impairment. J Clin Pharmacol. 2007;47:1183–211.
Almeida L, Potgieter JH, Maia J, Potgieter MA, Mota F, Soares-da-Silva P. Pharmacokinetics of eslicarbazepine acetate in patients with moderate hepatic impairment. Eur J Clin Pharmacol. 2008;64(3):267–73.
Stephen LJ, Wishart A, Brodie MJ. Psychiatric side effects and antiepileptic drugs: observations from prospective audits. Epilepsy Behav. 2017;71(Pt A):73–8.
Shorvon SD, Trinka E, Steinhoff BJ, Holtkamp M, Villanueva VJP, et al. Eslicarbazepine acetate: its effectiveness as adjunctive therapy in clinical trials and open studies. J Neurol. 2017;264(3):421–31.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Bial–Portela & Ca, S.A., Coronado, Portugal and Sunovion Pharmaceuticals Inc., Marlborough, MA, USA funded the phase III clinical trials reported in this manuscript. The sponsors were involved in the study design, in the collection, analysis, and interpretation of data, writing of the report, and the decision to submit the article for publication.
Conflict of interest
Helena Gama, Mariana Vieira, Raquel Costa, Joana Graça, Luís M. Magalhães, and Patrício Soares-da-Silva are employees of Bial–Portela & Cª, S.A.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gama, H., Vieira, M., Costa, R. et al. Safety Profile of Eslicarbazepine Acetate as Add-On Therapy in Adults with Refractory Focal-Onset Seizures: From Clinical Studies to 6 Years of Post-Marketing Experience. Drug Saf 40, 1231–1240 (2017). https://doi.org/10.1007/s40264-017-0576-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-017-0576-4