
Abstract
Multiple sclerosis treatment made substantial headway during the last two decades with the implementation of therapeutics with new modes of action and routes of application. We are now in the situation that second-generation molecules, approved since 2018, are on the market, characterized by reduced side effects using a more tailored therapeutic approach. Diroximel fumarate is a second-generation fumarate with reduced gastrointestinal side effects. Moreover, several novel, selective, sphingosine-1-phosphate receptor modulators with reduced off-target effects have been developed; namely siponimod, ozanimod, and ponesimod; all oral formulations. B-cell-targeted therapies such as ocrelizumab, given intravenously, and since 2021 ofatumumab, applied subcutaneously, complement the spectrum of novel therapies. The glycoengineered antibody ublituximab is the next anti-CD20 therapy about to be approved. Within the next years, oral inhibitors of Bruton’s tyrosine kinase, currently under investigation in several phase III trials, may be licensed for multiple sclerosis. Those developments currently offer an individualized multiple sclerosis therapy, targeting patient needs with substantial effects on relapses, disability progression, and implications for daily life. In this up-to-date review, we provide a holistic overview about novel developments of the therapeutic landscape and upcoming approaches for multiple sclerosis treatment.
Similar content being viewed by others
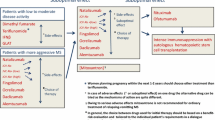

We critically reviewed (second-generation) multiple sclerosis therapeutics licensed since 2018 until 2022 and the outlook regarding new substance classes. |
Differing modes of action, routes of application, and side-effect profiles allow a personalized treatment approach. |
Substances to come include a new anti-CD20 therapeutic, ublituximab, and inhibitors of Bruton’s tyrosine kinase as a new class of medications acting both on B cells and myeloid cells. |
1 Introduction
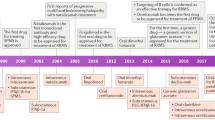
Multiple sclerosis (MS) treatment made tremendous headway during the last decades owing to the development of effective substances for both differing inflammatory activity of the disease as well as different courses [1]. The availability of substances applied subcutaneously, orally, or intravenously as well as differing modes of action and application schedules from day-to-day intake over weekly or monthly therapies to sequenced or pulsed medications has led to a situation allowing an individualized and precision MS therapy in 2022. Modern MS medications not only offer advantages regarding relapse activity and short-term disability, but also regarding the risk reduction in long-term disability. In certain situations, the implementation of highly effective medications is of clear advantage as it reduces long-term disability, as shown in real-world studies comparing initial treatment with highly effective medications over up to 10 years [2] and is associated with a later conversion to secondary progressive MS compared with injectables [3]. In 2018, we described the development of the MS landscape from 2010 to 2018 [1]; in this review, we summarize the development from 2018 until 2022. We focus on the second-generation fumarate diroximel fumarate, associated with an improved gastrointestinal side-effect profile and the second-generation sphingosine-1 (S1P) phosphate receptor modulators siponimod, ozanimod, and ponesimod. Moreover, we provide long-term data about ocrelizumab and review data of ofatumumab as a subcutaneous B-cell-depleting agent. The section about anti-CD20-directed therapies is complemented with an overview about ublituximab. We finish with an outlook regarding new therapeutic approaches on the horizon, namely the inhibition of Bruton’s tyrosine kinase probably coming up during the next years. We searched PubMed, MEDLINE, and clinicaltrials.gov regarding MS trials, long-term extension data, and new medications.
2 Second-Generation Fumarate Drugs for Multiple Sclerosis
2.1 Diroximel Fumarate
Dimethyl fumarate (DMF) has been on the market since 2013 (US Food and Drug Administration [FDA]) and 2014 (European Medicines Agency [EMA]) and with proven long-term effectiveness over up to 13 years as shown with the final results of the ENDORSE study [4]. It is currently the most frequently used oral drug, with 562,123 patients with MS worldwide having been treated as of 31 December, 2021 (Biogen). One of the most important short-term side effects are gastrointestinal side effects, leading to withdrawal from the medication in a considerable percentage of patients. This led to the development of a formulation with better gastrointestinal tolerability, diroximel fumarate (DRF). The active metabolite of both DMF and DRF is monomethyl fumarate, which is generated following processing by an esterase. A byproduct of DMF following cleavage of a methyl group is methanol, leading to gastrointestinal side effects, which, however, is produced nine times less with DRF [5]. As both compounds have the same active metabolite, bioequivalence was postulated; thus, DRF was tested in one head-to-head trial against DMF [6] and one open-label study [7] regarding safety and efficacy.
2.1.1 Efficacy and Safety in Clinical Trials
Diroximel fumarate was tested versus DMF in the randomized, blinded, 5-week, phase III EVOLVE-MS-2 study (NCT03093324) [8]. In total, 504 patients were enrolled, of which 253 patients were randomized to DRF and 251 to DMF. Endpoints consisted of gastrointestinal side effects, investigated using the Individual Gastrointestinal Symptom and Impact Scale and Global Gastrointestinal Symptom and Impact Scale eDiary questionnaires as well as the influence on daily activities and work. Gastrointestinal tolerability adverse events (AEs) were less pronounced in DRF-treated patients compared with DMF-treated patients (DRF, 34.8% [88/253]; DMF, 48.2% [121/251]). Moreover, patients with DRF were less likely to discontinue treatment (DRF, 0.8% vs DMF, 4.8%). The positive effect on gastrointestinal tolerability was also mirrored in positive effects on daily activities (interferences with activities “quite a bit” or “extremely” Individual Gastrointestinal Symptom and Impact Scale: DRF, 9.5% [24/253] vs DMF, 28.9% [72/249]) and work productivity (Global Gastrointestinal Symptom and Impact Scale: DRF, 6.1% [10/165)] vs DMF, 11.3% [18/159] [8]) (for an overview of pivotal trial data, see Table 1).
Diroximel fumarate was also tested in the open-label, 96-week, phase III, EVOLVE-MS-1 study, assessing DRF safety, tolerability, and efficacy in patients with relapsing-remitting MS (RRMS) [NCT02634307] [7]. In an interim analysis from March 2018, 696 patients were enrolled with a median exposure of 59.9 (range: 0.1–98.9) weeks. Treatment with DRF led to a reduction in the mean number of gadolinium-enhancing (Gd+) lesions of 77% from baseline (p < 0.0001). The adjusted annualized relapse rate (ARR) was 0.16 (95% confidence interval [CI] 0.13–0.20). Adverse events were documented in 84.6% (589/696) of patients; the majority of AEs were mild (31.2%; 217/696) or moderate (46.8%; 326/696). Treatment discontinuation because of gastrointestinal AEs was <1%. In an analysis regarding rollovers from glatiramer acetate or interferons, patients with DRF had improved clinical and radiological efficacy outcomes with 74.0% (95% CI 62.0–82.1; p < 0.0001) and 78.8% (95% CI 68.5–85.6; p < 0.0001) reduced ARRs compared with treatment with glatiramer acetate and interferons, respectively [9]. Interestingly, we noted that flushing also seems to be clearly reduced with DRF as compared with DMF (Gold, Faissner personal observation), which has also been shown in a post-hoc analysis from the EVOLVE-MS2 study [10].
2.1.2 Outlook
Diroximel fumarate as a second-generation fumarate is an improvement owing to reduced gastrointestinal side effects with better overall tolerance and should therefore be prioritized over DMF in the case where a fumarate should be started. Moreover, patients experiencing gastrointestinal side effects while taking DMF could be switched. Diroximel fumarate has been available in the USA since December 2020 and in the European Union and UK since 2022. The phase IV EXPERIENCE study will evaluate long-term safety and efficacy (NCT04746976).
Apart from DMF and DRF, monomethyl fumarate is approved in the USA for relapsing MS (RMS) since May 2020. The FDA approval was mainly based on the pivotal DMF trial data and a bioequivalence study, which showed that intake of a single oral dose of two delayed-release 95-mg capsules was bioequivalent to a single oral dose of one DMF 240-mg capsule regarding the plasma monomethyl fumarate concentration [11].
3 Sphingosin-1-Phosphate Receptor Modulators
Sphingosin-1-phosphate receptor modulation has been used as a therapeutic approach since 2010 after fingolimod received market authorization. While fingolimod targets S1P sub-receptors 1, 3, 4, and 5, hence inducing especially cardiac side effects, there was an effort to generate more selective second-generation molecules. This led to the development and approval of siponimod, ozanimod, and ponesimod; all of which are now available.
3.1 Siponimod
3.1.1 Mechanism of Action
Siponimod (BAF312) is a selective oral S1P receptor 1,5 modulator and is authorized as a medication for active secondary progressive MS (SPMS). Siponimod has a mean half-life in plasma of 56.6 hours (human) and is excreted in feces as oxidative metabolites after hepatic biotransformation, mostly mediated by cytochrome P450 (CYP) 2C9 [12]. Siponimod has pleiotropic effects on immune and central nervous system (CNS) cells. Siponimod has lipophilic properties and therefore crosses the blood–brain barrier, thus influencing CNS cells directly [13]. First studies about the mechanism of action in experimental autoimmune encephalomyelitis showed that siponimod antagonizes S1P 1 and 5 receptors, leading to an inhibition of lymphocyte egress from the thymus and secondary lymphoid organs [14]. Siponimod does not influence the S1P 3 receptor such as the first-generation substance fingolimod, which is mainly responsible for adverse cardiac effects such as bradycardia [15]. In a model of demyelination in Xenopus tadpoles, treatment with siponimod led to strong remyelination [16], suggesting that the medication might be effective in progressive forms of MS. Siponimod reduces the release of interleukin-6 in tumor necrosis factor-alpha/interleukin-17-activated microglia in vitro and has protective effects on demyelination in lysophosphatidylcholine-mediated demyelination in organotypic slice cultures [17]. Siponimod has positive effects on the clinical course of encephalomyelitis in C57BL6 mice in accordance with reduced astrogliosis and microgliosis [18]. Moreover, it rescues the loss of parvalbumin-positive GABAergic interneurons in vivo and reduces the release of cytokines RANTES and interleukin-6 in microglia in vitro [18]. Those data altogether are convincing regarding effectiveness in SPMS; especially because therapeutic approaches for progression remain scarce.
3.1.2 Efficacy
Siponimod was first investigated in the phase II BOLD study in patients with RRMS (NCT00879658). The BOLD study was a double-blind, adaptive, dose-ranging phase II study that included 188 and 109 patients in two cohorts. Patients randomly received once-daily siponimod 10 mg, 2 mg, or 0.5 mg or placebo over a period of 6 months. The primary endpoint consisted of a percentage reduction in the monthly number of combined unique active lesions at 3 months of siponimod compared with placebo. In this dose-response study, the medication led to a dose-dependent reduction in combined unique active lesions at 3 months compared with placebo with an effect of 82% (70–90) for siponimod 10 mg [19]. Subsequently, the medication was investigated in the phase III clinical trial EXPAND in 1651 patients, which was the largest SPMS trial performed so far (NCT01665144). The primary endpoint consisted of 3-month confirmed disability progression. Siponimod treatment led to a 21% relative risk reduction compared with placebo (p = 0.013) [20]. Secondary endpoints were also positively attenuated: the ARR was lower in siponimod-treated patients compared with placebo-treated patients with a risk reduction of 55% (rate ratio 0.45, 95% CI 0.34–0.59; p < 0.0001). An increase in T2 lesion load was lower in siponimod-treated patients; brain volume decreased at a lower rate compared with placebo (− 0.50% vs − 0.65%; between-group difference 0.15%, 95% CI 0.07–0.23; p = 0.0002) [20]. Secondary analyses of EXPAND documented significant positive effects on cognitive function. The SDMT was significantly better with a difference of 1.08 at month 12 (95% CI 0.23–1.94; p = 0.0132), 1.23 (95% CI 0.25–2.21; p = 0.0135) at month 18, and 2.30 (95% CI 1.11–3.50; p = 0.0002) at month 24 [21]. Moreover, the risk to have a 4-point decrease was reduced with a hazard ratio of 0.79 (95% CI 0.65–0.96; p = 0.0157) and the likelihood of having a 4-point increase was increased with a hazard ratio of 1.28 (95% CI 1.05–1.55; p = 0.0131). Those data altogether also support a neuroprotective effect of siponimod.
The data of the pivotal trial EXPAND led to the application as therapy in SPMS. Both the FDA and EMA accepted the New Drug Application and Marketing Authorization Application in October 2018. In March 2019, siponimod was authorized as therapy for RRMS and SPMS by the FDA and received market authorization by the EMA in late 2019 for active SPMS. Of note, siponimod did not show efficacy in patients with non-active SPMS [22]; therefore, clinical or radiological activity must be proven prior to therapy initiation.
3.1.3 Safety
Siponimod undergoes hepatic metabolization; therefore, the CYP2C9 genotype has to be tested to choose the correct dosage. In Caucasian patients, 82–89% are fast metabolizers and can receive the full daily dosage of 2 mg (CYP2C9*1*1, *1*2, *2*2), 10–14% have a CYP2C9*2*3 or *1*3 genotype (intermediate metabolizer) and are treated with 1 mg, and 1.7–2.1% of patients cannot receive siponimod (CYP2C9*3*3, weak metabolizer) [23].
The most common AEs reported in more than 10% of treated patients in both treatment arms were headache, nasopharyngitis, urinary tract infection, and falls [20]. Side effects, which occurred more often in siponimod-treated patients, included bradycardia at treatment initiation (4% vs 3%), hypertension (12% vs 9%), lymphopenia (1% vs 0%), macular edema (2% vs < 1%), and convulsions (2% vs < 1%). Moreover, siponimod-treated patients more frequently had herpes zoster reactivation (2% vs 1%).
3.2 Ozanimod
Ozanimod is an oral, selective S1P 1,5 receptor modulator, licensed for RRMS by the EMA and for clinically isolated syndrome, RRMS, and active SPMS by the FDA. Moreover, the medication has received approval for moderate-to-severe active ulcerative colitis (FDA). Ozanimod reduces the severity of encephalomyelitis and penetrates the CNS [24].
3.2.1 Efficacy
Ozanimod was investigated in the phase II clinical trial RADIANCE and the two phase III clinical trials RADIANCE (24 months) and SUNBEAM (at least 12 months). RADIANCE was a 24-month, multicenter, double-blind, double-dummy phase III trial [25]. Patients were randomized to ozanimod 0.5 mg (n = 439) or 1.0 mg (n = 433) or weekly intramuscular interferon-beta-1a 30 μg (n = 441) in a ratio of 1:1:1. The ARR was significantly lower in both ozanimod dosages [1.0 mg: 0.17 (95% CI 0.14–0.21); 0.5 mg: 0.22 (95% CI 0.18–0.26)] compared with interferon beta-1a [0.28 (95% CI 0.23–0.32)] with a rate ratio of 0.62 for the 1.0-mg dosage and 0.79 for the 0.5-mg dosage compared with interferon.
SUNBEAM was a randomized, double-blind, double-dummy, active-controlled phase III trial with 1346 patients enrolled [26]. Patients were randomized to ozanimod 0.5 mg (n = 451) or 1.0 mg (n = 447) or weekly intramuscular interferon-beta-1a 30 μg (n = 448) in a ratio of 1:1:1. The primary endpoint was the ARR. The ARR was significantly lower in ozanimod-treated patients. In the interferon-beta-1a group, the ARR was 0.35 (0.28–0.44) compared with 0.18 (0.14–0.24) in the ozanimod 1.0-mg group and 0.24 (0.19–0.31) in the ozanimod 0.5-mg group. The rate ratio was 0.52 for ozanimod 1.0 mg versus interferon (p < 0.0001) and 0.69 for ozanimod 0.5 mg (p = 0.0013). The number of Gd+ lesions and the cumulative number of T2 lesions were also reduced in the ozanimod groups. There were also (indirect) neuroprotective effects of ozanimod as assessed with neurofilament (Nfl) measurements in a post-hoc analysis of SUNBEAM. In ozanimod-treated patients, Nfl levels were reduced by 20–27% at month 12 and 24, respectively, compared with a reduction of 13–16% in interferon-β-1a-treated patients (p < 0.01) [27]. Greater Nfl reductions were also associated with fewer Gd+ lesions or new/enlarging T2 lesions, less brain volume loss, lower ARR, and a higher percentage of patients with no evidence of disease activity.
3.2.2 Safety
The first generation of S1P receptor modulators had cardiac side effects owing to an alteration of the S1P 3 receptor. Recent data suggest that the selective second-generation substances also elicit effects on heart rate. In a cardiac safety study, ozanimod reduced the heart rate by up to 13.8 bpm compared with placebo [28]. Of note, in ozanimod-treated patients followed in SUNBEAM, there was no significant bradycardia as well as no second-degree or third-degree atrioventricular block. One reason for reduced cardiac side effects may therefore be related to an altered titration schedule. In RADIANCE, AEs leading to treatment discontinuation occurred more often in interferon-beta-1a-treated patients compared with ozanimod, while infections and serious AEs were similar between ozanimod-treated patients and interferon-beta-1a-treated patients [27]. Until now, there was one case of progressive multifocal leukoencephalopathy in the open-label extension of DAYBREAK. Regarding daily clinical practice, patients should be informed that intake of ozanimod together with food containing high levels of tyramine such as cheese, alcohol, or cured meat could potentially lead to hypertension. The “Tyramine and ZEPOSIA food and drug interaction information” suggests that patients taking ozanimod avoid foods and beverages that have more than 150 mg of tyramine [29]. A trial that investigated the interaction has completed recruitment, results are pending (NCT03694119).
3.3 Ponesimod
Ponesimod is a selective S1P 1 receptor modulator licensed for the treatment of clinically isolated syndrome, RRMS, and active SPMS (FDA), and active RMS (EMA). It has a half-life of 32 hours [30]. Preclinical studies suggest that ponesimod penetrates the CNS [30].
3.3.1 Efficacy
Ponesimod was first tested in a double-blind, placebo-controlled, dose-finding phase IIb study in RRMS (NCT01006265). In total, 464 patients were randomized to once-daily oral ponesimod in a dosage of 10, 20, or 40 mg or placebo [31]. The primary endpoint evaluated the cumulative number of new T1 Gd+ lesions, secondary endpoints were ARR and time to first relapse as well as safety and tolerability. The primary endpoint was met with a significant reduction in Gd+ lesions in all dosages (rate ratio 10 mg: 0.57; 20 mg: 0.17; 40 mg: 0.23). The ARR was reduced by up to 52% with 40 mg versus placebo.
Next, ponesimod was investigated in the multicenter, double-blind, active-comparator, superiority, randomized phase III clinical trial OPTIMUM against teriflunomide (NCT02425644) [32]. Patients were randomized to ponesimod 20 mg or teriflunomide 14 mg in a 1:1 ratio (n = 567 patients and n = 566 patients, respectively). The primary endpoint was the ARR. Secondary endpoints assessed the change in fatigue using the Fatigue Symptom and Impact Questionnaire-Relapsing Multiple Sclerosis as well as magnetic resonance imaging endpoints, namely the number of combined unique active lesions, and the time to 12-week and 24-week confirmed disability accumulation. The primary endpoint was met: the relative reduction in the ARR was 30.5 (0.202 vs 0.290; p < 0.001). Secondary endpoints also improved in the ponesimod group with a difference in the fatigue scale of −3.57 (−0.01 vs 3.56; p < 0.001) and a reduction in combined unique active lesions per year by 56% (1.405 vs 3.164; p < 0.001). Confirmed disability accumulation did not differ (17% vs 16%). However, there was a positive effect on brain volume loss: brain volume loss was lower by 0.34% (− 0.91% vs − 1.25%; p < 0.001).
3.3.2 Safety
Adverse events and SAEs were similar between both groups, while treatment discontinuation occurred more often in the ponesimod group compared with teriflunomide-treated patients. Analyses from up to an 8-year follow-up of the phase IIb core and extension studies showed no new safety issues. The most common AEs were nasopharyngitis (30%), headache (24%), and upper respiratory tract infection (21%) [33].
3.4 General Considerations About S1P Receptor Modulators
One important consideration for all S1P receptor modulators is the half-life, which differs quite substantially with potential implications regarding the risk of rebound and sequencing of medication. Fingolimod has a half-life of 6–9 days, siponimod 22–38 h, ozanimod 19–22 h, and ponesimod 32 h [30]. This is of interest both regarding therapeutic considerations for therapy adherence and rebound activity. The risk of rebound is known since the first reports in fingolimod-treated patients, for example, with the development of tumefactive lesions [34]. Disease exacerbation following cessation has also been described for siponimod [35], not yet for ozanimod and ponesimod, either owing to the shorter approval or indeed reflecting the absence of these effects because of the shorter half-life. It is theoretically plausible that all S1P receptor modulators might be at risk of rebound activity following cessation. However, differing pharmacological profiles might shape this risk depending on the half-lives. Another discussion concerns the risk of cancer under S1PR modulation. Data from the FDA Adverse Event Reporting System from 2004 to 2020 showed an increased risk for skin cancer with the strongest association for basal cell carcinoma [36]. Patients should therefore be monitored dermatologically. Another aspect of clinical relevance is that with a more conservative and cautious titration used by the second-generation molecules, a first dose observation over 6 hours is generally not required. This, however, should be performed where patients are at a higher risk regarding cardiac side effects because of comorbidities. In summary, therapy cessations and switches should be planned according to half-lives and pharmacodynamics, especially in highly active patients.
4 Anti-CD20-Directed Monoclonal Antibodies
4.1 Ocrelizumab
4.1.1 Long-Term Data of Ocrelizumab
Ocrelizumab is a humanized monoclonal antibody targeting CD20+ B cells. Ocrelizumab was investigated in the two identical phase III trials OPERA I and II in RMS versus interferon-beta-1a 44 μg three times weekly for 96 weeks [37] and in primary progressive multiple sclerosis (PPMS) in the ORATORIO trial [38], reviewed by us previously in detail [1].
Long-term data of ocrelizumab are now available. The open-label-extension of OPERA I and II, assessing the effect of ocrelizumab over the course of 6.5 years (336 weeks) in the double-blind period and open-label extension of the OPERA I (NCT01247324) and OPERA II (NCT01412333) studies on the time to the Expanded Disability Status Scale score of ≥6.0 over 6.5 years was significantly delayed in ocrelizumab-treated patients compared with interferon-treated patients [39]. The risk of requiring a walking aid, confirmed for ≥24 weeks, was 34% lower (p = 0.024) in early ocrelizumab-treated patients and 48% lower in patients confirmed for ≥48 weeks (p = 0.004), supporting positive long-term effects of ocrelizumab in RRMS.
There are also positive long-term data of the extension in PPMS (ORATORIO) [40]. Five hundred and twenty-seven patients (97%) entered the open-label-extension phase with 86% of the patients ongoing. After 6.5 years, the proportion of patients with disability progression was lower following early treatment compared with patients initially having received placebo. This was assessed with several measures of progression such as the Expanded Disability Status Scale with a difference of 13.1% (95% CI 4.9–21.3; p = 0.0018), the 9-hole peg test with a difference of 12.5% (95% CI 4.1–20.9; p = 0.0035), the Timed-25 Foot Walk with a difference of 7.5% (95% CI − 0.3 to 15.2; p = 0.058), and the composite progression score with a difference of 10.1% (95% CI 3.6–16.6; p = 0.0023). In post-hoc analyses, there were numerical differences regarding benefit according to sex, baseline T1, Gd+ lesions, and age (interaction p < 0.3) with male individuals seeming to benefit more regarding 12-week confirmed disability progression [41].
4.1.2 Safety
Safety data of ocrelizumab, derived from a systematic review of four randomized controlled trials, four open-label trials, 29 observational studies, and 27 case reports, showed that most common AEs were infections (39.2%) and infusion-related reactions (26.2%) [42]. Compared with interferons, the risk of infections was slightly higher (risk ratio [RR] = 1.10; 95% CI 1.01–1.19). Of those, herpes-related infections (RR = 1.75; 95% CI 1.11–2.76), respiratory tract-related infections (RR = 1.42; 95% CI 1.10–1.84 and RR = 1.61; 95% CI 1.10–2.35), nasopharyngitis (RR = 1.47; 95% CI 1.13–1.90), and rhinitis (RR = 4.00; 95% CI 1.13–14.14) were more likely to occur in ocrelizumab-treated patients. Although ORATORIO seemed to have a slightly higher risk of female cancer, this was never confirmed in post-marketing observations. As of October 2021, there have been ten confirmed confounded cases of progressive multifocal leukoencephalopathy in patients with MS who were treated with ocrelizumab, of which nine were carry-over cases from prior disease-modifying therapies (DMTs). The patient who had no carry-over progressive multifocal leukoencephalopathy was aged 78 years, had not been treated with a DMT before, and had a low lymphocyte count grade of 1 prior to ocrelizumab initiation [43].
As a result of the coronavirus disease 2019 (COVID-19) pandemic, there is currently a focus on the risk of infections and vaccination management of patients with MS receiving immunosuppressive therapies. Anti-CD20-directed B-cell-depleting antibodies lead to a depletion of pre-B cells, mature B cells, and memory B cells, while long-living plasma cells, plasmablasts, and lymphoid stem cells are spared [37]. In summary, this leads to an impairment of crucial B-cell functions such as antigen presentation, antibody production, and antibody class switches [44]. Using data from patients with MS from 12 data sources in 28 countries with 657 (28.1%) patients with suspected COVID-19 and 1683 (61.9%) with confirmed COVID-19, it could be revealed that ocrelizumab treatment is associated with higher rates of hospitalization (adjusted odds ratio [aOR] 1.75, 95% CI 1.29–2.38) and intensive care unit admission (aOR 2.55, 95% CI 1.49–4.36) compared with pooled other DMTs including alemtuzumab, cladribine, dimethyl fumarate, glatiramer acetate, interferon, natalizumab, siponimod, and other DMTs [45]. The risk for rituximab compared with pooled other DMTs, however, was higher (hospitalization: aOR 2.76, 95% CI 1.87–4.07; intensive care unit admission: aOR 4.32, 95% CI 2.27–8.23; artificial ventilation: aOR 6.15, 95% CI 3.09–12.27) [45], presumably because of a longer treatment duration. B-cell depletion is also associated with an impaired humoral immune response [46]. Data from Israel showed that patients receiving B-cell depleting therapy develop only in 22.7% a humoral immune response against a severe acute respiratory syndrome–related coronavirus spike 29.5–55 days after the second vaccine dose [46]. In the meantime, we [47] and others [48] have shown that despite an impaired humoral response, B-cell-depleted patients elicit a strong cellular immune response, in part even stronger than in untreated healthy controls [47]. Currently, high-dose ocrelizumab in dosages of 1200 or 1800 mg based on body weight is being investigated in a phase IIIb study (NCT04544436).
4.2 Ofatumumab
4.2.1 Mechanism of Action
Ofatumumab is an anti-CD20-directed monoclonal immunoglobulin-1-k antibody. Ofatumumab binds the small extracellular loop of CD20 and leads to antibody-mediated lysis of the target cell [49] via a complement mediated effect, leading to the activation of the classical pathway of the complement system by binding C1q [50]. This effect is more pronounced than with the chimeric antibody rituximab, presumably owing to binding of the small extracellular loop of CD20, which is closer at the cellular membrane [50]. Ofatumumab is applied subcutaneously. In an animal model, a subcutaneous antibody application leads to a fast antibody delivery to draining lymph nodes via transcytosis [51]. Indeed, it has been shown in experimental models that subcutaneous ofatumumab accumulates in axillary and inguinal lymph nodes [52], which might positively attenuate antigen-presenting cells. Another advantage of this human antibody is the reduced immunogenicity. While 48 weeks after rituximab treatment 24.6% of patients developed anti-chimeric antibodies [53], there were no human anti-human antibodies in the phase II MIRROR study [54].
4.2.2 Efficacy
Ofatumumab was initially developed for chronic lymphatic leukemia. In MS, ofatumumab was first investigated in a small, randomized, placebo-controlled phase II study of 38 patients [54]. Patients received ofatumumab every 2 weeks in a dosage of 100 mg, 300 mg, or 700 mg or placebo via an infusion. After 24 weeks, the treatment arms were swapped. Hence, first, 26 patients received placebo followed by ofatumumab, then 12 patients received ofatumumab followed by placebo. Treatment with ofatumumab led to complete suppression of CD19+ B cells within 1 week following treatment. The repopulation of CD19+ B cells was dose dependent (100-mg group 12–16 weeks, 300-mg group 20 weeks, 700-mg group no repopulation within the observational period). Magnetic resonance imaging activity was also improved with reduced old and new T1 Gd+ lesions and reduced new/enhancing T2 lesions [54]. Apart from infusion reactions on the day of infusion, there were no unexpected safety signals. Of note, none of the patients investigated developed neutralizing antibodies.
To optimize the application, subcutaneous application was implemented in MS, which had successfully been investigated in a small combined phase I/II study in rheumatoid arthritis [55]. The subcutaneous application was also investigated in MS; here, the administration of ofatumumab via a subcutaneous autoinjector was bioequivalent to a pre-filled syringe regarding the dynamics of B-cell depletion as shown in the 12-week, open-label, parallel-group, phase II APLIOS study (NCT03560739) in RMS [56].
In the phase II MIRROR dose-efficacy study, ofatumumab was investigated in dosages of 3, 30, and 60 mg and placebo every 4 weeks. Two hundred and thirty-two patients with RRMS with an Expanded Disability Status Scale score of 0–5.5 were included. The primary endpoint was the cumulative number of new or contrast-enhancing lesions after 12 weeks. The primary endpoint was met, there was a reduction of at least 65% of all dosages compared with placebo (p < 0.001) [57]. After an exclusion period of weeks 1–4, there was a reduction of at least 90% in all dosages from 30 mg and above. There was a dose-dependent reduction in CD19+ B cells. Side effects included injection-site irritation (52% ofatumumab vs 15% placebo), which were of mild or moderate severity.
The pivotal trials were the identical double-blind, double-dummy, phase III trials ASCLEPIOS I and II, which tested ofatumumab 20 mg against the active comparator teriflunomide (n = 946 ofatumumab, n = 936 placebo) [58]. Patients were followed a median of 1.6 years. Patients received a loading dose ofatumumab on days 1, 7, and 14 followed by ofatumumab 20 mg every 4 weeks subcutaneously. The primary endpoint was the ARR, secondary endpoints included confirmed disability progression after 3 or 6 months, the number of Gd+ lesions, the increase of T2 lesions, release of NfL, and changes in brain volume. The ARRs were 0.11 in the ofatumumab group and 0.22 in the teriflunomide group in trial 1 (difference, − 0.11; 95% CI − 0.16 to − 0.06; p < 0.001) and 0.10 and 0.25 in trial 2 (difference, − 0.15; 95% CI − 0.20 to − 0.09; p < 0.001). In the pooled trials, the disability worsening was reduced by 34% in the ofatumumab group compared with the teriflunomide group after 3 months (10.9% ofatumumab, 15.0% teriflunomide, hazard ratio, 0.66; p = 0.002). This effect was confirmed at 6 months (8.1% ofatumumab, 12.0% teriflunomide, hazard ratio 0.68; p = 0.01). In addition, there was also a positive effect on paraclinical endpoints. T1 Gd+ lesions were reduced by at least 94% (p < 0.001), new or enlarging T2 lesions were reduced by at least 85% in ofatumumab-treated patients compared with teriflunomide (p < 0.001). Brain volume loss did not differ. Neurofilament levels, measured in serum, were significantly lower at all timepoints (3, 12, and 24 months) compared with teriflunomide (p < 0.001).
4.2.3 Safety
The overall frequency of side effects in the pivotal trial was similar between both groups (83.6% ofatumumab, 84.2% teriflunomide) [58]. Injection-site reactions occurred in 20.2% of ofatumumab-treated patients compared with 15.0% in teriflunomide-treated patients, who received placebo injections. Serious infections were rare and comparable between both groups (2.5% vs 1.8%). An interesting aspect concerns the dynamics of B-cell reconstitution using this low-dose regimen. The repopulation of B cells is also impaired in ofatumumab-treated patients and will last approximately 40 weeks in a dosage of 20 mg [57]. One case of a patient from the ASCLEPIOS-extension study 42 months after ofatumumab treatment showed effective IgM and IgG responses against the spike protein following COVID-19 infection although B cells were depleted completely [59]. A case series of four patients receiving ofatumumab with COVID-19 infection documented strong T-cell responses also in the absence of a titer [60]. Data regarding vaccination response against severe acute respiratory syndrome-related coronavirus while receiving ofatumumab are, to the best to our knowledge, not yet published. A phase IV trial is currently ongoing (COMB157GDE01).
5 Outlook
5.1 Novel Anti-CD20-Directed Antibodies: Ublituximab
Apart from the established and above-reviewed anti-CD20-directed monoclonal antibodies ocrelizumab, ofatumumab, and off-label rituximab, the glycoengineered antibody ublituximab is at the next stage of anti-CD20 therapy. Ublituximab targets a unique epitope on the CD20 antigen and is glycoengineered for enhanced B-cell targeting through antibody-dependent cellular cytotoxicity [61]. Ublituximab was investigated in a phase II, placebo-controlled, dose-finding study. Patients were treated with three ublituximab infusions (150 mg over 1–4 h on day 1 and 450–600 mg over 1–3 h on day 15 and week 24) in six dosing cohorts (n = 48). B-cell depletion was > 99% by week 4, maintained at weeks 24 and 48 [61]. Adverse events consisted of grade 1–2 infusion-related reactions. Magnetic resonance imaging at weeks 24 and 48 showed no T1 Gd+ lesions (p = 0.003) and a 10.6% decrease in the T2 lesion volume (p = 0.002). This was associated with a low ARR of 0.07 and 93% of relapse-free patients. Of note for clinical practice is that there was no higher risk of infusion-related reactions with a fast infusion time of 1 h, a potential improvement that needs to be confirmed in larger trials and real-world settings. By now, data from the phase III trials ULTIMATE I and II have been released. Ublituximab was investigated against teriflunomide and showed a reduction in the ARR of about 49–59% in both trials [ULTIMATE I, n = 549, ARR ublituximab 0.076 (0.042–0.138); teriflunomide 0.188 (0.124–0.283), NCT03277261; last update available from 6 December, 2021]. The final publication is pending.
5.2 Inhibition of Bruton’s Tyrosine Kinase
Bruton’s tyrosine kinase (BTK) is a cytoplasmic protein-tyrosine kinase expressed in all hematopoietic cells such as macrophages, neutrophils, and mast cells except for T and plasma cells [62]. Moreover, BTK is expressed in microglia, the resident immune cells of the CNS. Bruton’s tyrosine kinase inhibition leads to improved remyelination in a model of organotypic slice cultures [63]. Therefore, inhibition of BTK is considered a promising therapeutic target both for RMS but also for progressive MS owing to the involvement of B cells and cells of innate immunity in the pathogenesis of progression [64, 65].
Initially, BTK inhibitors were developed for the treatment of hematologic conditions such as lymphoproliferative disorders, leukemia, and lymphoma [66]. This was followed by the investigation of BTK inhibitors in autoimmune conditions such as MS, rheumatoid arthritis, pemphigus, and systemic lupus erythematosus. Currently, several BTK inhibitors are under development in MS, such as the irreversible BTK inhibitors tolebrutinib (Sanofi) or evobrutinib (Merck) or remibrutinib (Novartis), and the reversible BTK inhibitor fenebrutinib (Genentech).
Tolebrutinib was investigated in a 16-week, phase IIb, randomized, double-blind, placebo-controlled, crossover, dose-finding trial investigating dosages of 5–60 mg versus placebo [67]. In total, 130 patients were enrolled. Tolebrutinib led to a dose-dependent reduction in the number of new Gd+ lesions (placebo 1.03 mean lesions per patient; 5 mg, 1.39 (3.20); 15 mg, 0.77 (1.48); 30 mg, 0.76 (3.31); 60 mg, 0.13 (0.43); p = 0.03). The most common side effect was headache. There was no safety-related discontinuation or death. Tolebrutinib is currently investigated in three MS phase III clinical trials in RRMS (GEMINI I and II, NCT04410978), SPMS (HERCULES, NCT04411641), and PPMS (PERSEUS, NCT04458051). Moreover, there is a trial in generalized myasthenia gravis (NCT05132569).
Evobrutinib was investigated in a double-blind, randomized, phase II trial in RMS [68]. Evobrutinib was tested against placebo in dosages from 25 mg once daily, 75 mg once daily, 75 mg twice daily, or DMF as the reference. The primary endpoint was a magnetic resonance imaging endpoint, assessing the cumulative number of Gd+ lesions on magnetic resonance imaging. The rate ratio compared with placebo was 0.30 in the evobrutinib 75-mg once-daily group (p = 0.005). The unadjusted ARR at week 24 was 0.13 in the evobrutinib 75-mg once-daily group compared with 0.37 in the placebo group. Side effects included elevations of liver aminotransferases. Currently, evobrutinib is investigated against teriflunomide in two phase III clinical trials in RMS (evolutionRMS 1 and 2, NCT04338061).
Fenebrutinib is investigated in a phase II study against placebo, assessing the number of Gd+ leions after 12 weeks (FENopta; NCT05119569). Furthermore, there are two parallel phase III trials ongoing, assessing fenebrutinib against teriflunomide in RMS (FENhance, NCT04586010, NCT04586023). Of particular note, the phase III trial FENtrepid investigates fenebrutinib against ocrelizumab in PPMS (primary endpoint time to onset of composite 12-week confirmed disability progression; NCT04544449).
Hence, the development of BTK inhibitors is an auspicious new approach to target MS in different phases of the disease. Because of positive effects on the cells of innate immunity and direct effects in the CNS, BTK inhibitors are especially of interest for patients with progressive disease or at a high risk of progression.
6 Conclusions
In summary, the therapeutic MS landscape considerably changed during the last 4 years with the development of second-generation substances such as a second-generation fumarate and more specific S1P receptor modulators with reduced side effects and altered half-lives. B-cell depletion using ofatumumab can now be performed with a fully human antibody with less risk of developing neutralizing antibodies and the advantage of monthly self-administration. Ublituximab might potentially elicit less immunogenicity because of its glycoengineered design. Regarding progression, the advantages of the therapeutic landscape with the approval of siponimod for active SPMS and ocrelizumab for PPMS are a glimpse of hope as progression remains difficult to target (for a further review, see our publication [65]). The development of BTK inhibitors will bring a whole new class of therapeutics to MS within the next years, provided the phase III trials confirm promising results from the phase II trials.
While safety data derived from studies about second-generation molecules are promising, it needs to be taken into consideration that the true safety profile of a new medication is usually not totally evident from clinical trial data and takes time to accumulate following approval. Therefore, long-term data and especially real-word studies are needed for those new agents to understand the safety spectrum, especially over a long treatment period.
Apart from new therapeutic developments regarding those new substances, another urgent and ongoing discussion is the risk/benefit assessment in patients with MS. This holds especially true in aging patients aged older than 60 or 70 years who are more vulnerable because of immunosenescence. This is currently addressed in a discontinuation study in patients aged older than 55 years [“Discontinuation of Disease Modifying Therapies (DMTs) in Multiple Sclerosis (MS)”]. The second burning debate is about sequencing of MS medications. Here, as stated above, data from several registries and studies provided evidence that initial highly active therapeutics are associated with a reduced risk of progression and conversion to SPMS. The question whether highly active therapeutic approaches are indeed superior to moderate DMTs is currently being investigated in two trials, “Traditional Versus Early Aggressive Therapy for Multiple Sclerosis Trial (TREAT-MS)”, which will enroll 900 participants and “Determining the Effectiveness of earLy Intensive Versus Escalation Approaches for RRMS (DELIVER-MS)” [800 participants]. Thus, we are now in an era of MS therapy developing towards a more tailored and individualized therapeutic approach, but important open questions remain.
References
Faissner S, Gold R. Efficacy and safety of the newer multiple sclerosis drugs approved since 2010. CNS Drugs. 2018;32(3):269–87.
He A, Merkel B, Brown JWL, Zhovits Ryerson L, Kister I, Malpas CB, et al. Timing of high-efficacy therapy for multiple sclerosis: a retrospective observational cohort study. Lancet Neurol. 2020;19(4):307–16.
Brown JWL, Coles A, Horakova D, Havrdova E, Izquierdo G, Prat A, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA. 2019;321(2):175–87.
Gold R, Arnold DL, Bar-Or A, Fox RJ, Kappos L, Mokliatchouk O, et al. Long-term safety and efficacy of dimethyl fumarate for up to 13 years in patients with relapsing-remitting multiple sclerosis: final ENDORSE study results. Mult Scler. 2022;28(5):801–16.
Palte MJ, Wehr A, Tawa M, Perkin K, Leigh-Pemberton R, Hanna J, et al. Improving the gastrointestinal tolerability of fumaric acid esters: early findings on gastrointestinal events with diroximel fumarate in patients with relapsing-remitting multiple sclerosis from the phase 3, open-label EVOLVE-MS-1 study. Adv Ther. 2019;36(11):3154–65.
Wundes A, Wray S, Gold R, Singer BA, Jasinska E, Ziemssen T, et al. Improved gastrointestinal profile with diroximel fumarate is associated with a positive impact on quality of life compared with dimethyl fumarate: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. Ther Adv Neurol Disord. 2021;14:1756286421993999.
Naismith RT, Wolinsky JS, Wundes A, LaGanke C, Arnold DL, Obradovic D, et al. Diroximel fumarate (DRF) in patients with relapsing-remitting multiple sclerosis: interim safety and efficacy results from the phase 3 EVOLVE-MS-1 study. Mult Scler. 2020;26(13):1729–39.
Naismith RT, Wundes A, Ziemssen T, Jasinska E, Freedman MS, Lembo AJ, et al. Diroximel fumarate demonstrates an improved gastrointestinal tolerability profile compared with dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: results from the randomized, double-blind, phase III EVOLVE-MS-2 study. CNS Drugs. 2020;34(2):185–96.
Wray S, Then Bergh F, Wundes A, Arnold DL, Drulovic J, Jasinska E, et al. Efficacy and safety outcomes with diroximel fumarate after switching from prior therapies or continuing on DRF: results from the phase 3 EVOLVE-MS-1 Study. Adv Ther. 2022;39(4):1810–31.
Singer BA, Faissner S, Lyons J, Scaramozza M, Woodward C, Chen H, Branco F, Vosoughi R. Flushing and flushing-related adverse events with diroximel fumarate in patients with relapsing-remitting multiple sclerosis: results from the phase 3 EVOLVE-MS-2 study. Presented at the 37th congress of the European committee for treatment and research in multiple sclerosis; October 13–15, 2021, p. 673.
Lategan TW, Wang L, Sprague TN, Rousseau FS. Pharmacokinetics and bioavailability of monomethyl fumarate following a single oral dose of Bafiertam™ (monomethyl fumarate) or Tecfidera® (dimethyl fumarate). CNS Drugs. 2021;35(5):567–74.
Glaenzel U, Jin Y, Nufer R, Li W, Schroer K, Adam-Stitah S, et al. Metabolism and disposition of siponimod, a novel selective S1P1/S1P5 agonist, in healthy volunteers and in vitro identification of human cytochrome P450 enzymes involved in its oxidative metabolism. Drug Metab Dispos. 2018;46(7):1001–13.
Pan S, Gray NS, Gao W, Mi Y, Fan Y, Wang X, et al. Discovery of BAF312 (siponimod), a potent and selective S1P receptor modulator. ACS Med Chem Lett. 2013;4(3):333–7.
Gergely P, Nuesslein-Hildesheim B, Guerini D, Brinkmann V, Traebert M, Bruns C, et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol. 2012;167(5):1035–47.
Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279(14):13839–48.
Mannioui A, Vauzanges Q, Fini JB, Henriet E, Sekizar S, Azoyan L, et al. The Xenopus tadpole: an in vivo model to screen drugs favoring remyelination. Mult Scler. 2018;24(11):1421–32.
O’Sullivan C, Schubart A, Mir AK, Dev KK. The dual S1PR1/S1PR5 drug BAF312 (siponimod) attenuates demyelination in organotypic slice cultures. J Neuroinflamm. 2016;8(13):31.
Gentile A, Musella A, Bullitta S, Fresegna D, De Vito F, Fantozzi R, et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J Neuroinflamm. 2016;13(1):207.
Selmaj K, Li DK, Hartung HP, Hemmer B, Kappos L, Freedman MS, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013;12(8):756–67.
Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263–73.
Benedict RHB, Tomic D, Cree BA, Fox R, Giovannoni G, Bar-Or A, et al. Siponimod and cognition in secondary progressive multiple sclerosis: EXPAND secondary analyses. Neurology. 2021;96(3):e376–86.
FDA NEWS RELEASE: FDA approves new oral drug to treat multiple sclerosis. https://www.fda.gov/news-events/press-announcements/fda-approves-new-oral-drug-treat-multiple-sclerosis. Accessed 20 June 2022.
European Medicines Agency. Siponimod product information. 22 June 2022. https://www.ema.europa.eu/en/documents/product-information/mayzent-epar-product-information_de.pdf. Accessed 14 Jul 2022.
Scott FL, Clemons B, Brooks J, Brahmachary E, Powell R, Dedman H, et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br J Pharmacol. 2016;173(11):1778–92.
Cohen JA, Comi G, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019;18(11):1021–33.
Comi G, Kappos L, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019;18(11):1009–20.
Harris S, Comi G, Cree BAC, Arnold DL, Steinman L, Sheffield JK, et al. Plasma neurofilament light chain concentrations as a biomarker of clinical and radiologic outcomes in relapsing multiple sclerosis: post hoc analysis of phase 3 ozanimod trials. Eur J Neurol. 2021;28(11):3722–30.
Tran JQ, Hartung JP, Olson AD, Mendzelevski B, Timony GA, Boehm MF, et al. Cardiac safety of ozanimod, a novel sphingosine-1-phosphate receptor modulator: results of a thorough QT/QTc study. Clin Pharmacol Drug Dev. 2018;7(3):263–76.
Company B-MS. Zeposia. Prescribing information. Bristol-Myers Squibb; 2021.
Roy R, Alotaibi AA, Freedman MS. Sphingosine 1-phosphate receptor modulators for multiple sclerosis. CNS Drugs. 2021;35(4):385–402.
Olsson T, Boster A, Fernández Ó, Freedman MS, Pozzilli C, Bach D, et al. Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial. J Neurol Neurosurg Psychiatry. 2014;85(11):1198–208.
Kappos L, Fox RJ, Burcklen M, Freedman MS, Havrdová EK, Hennessy B, et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study: a randomized clinical trial. JAMA Neurol. 2021;78(5):558–67.
Freedman MS, Pozzilli C, Havrdova EK, Lemle A, Burcklen M, Larbalestier A, et al. Long-term treatment with ponesimod in relapsing-remitting multiple sclerosis: results from randomized phase 2b core and extension studies. Neurology. 2022 Jun 6.
Faissner S, Hoepner R, Lukas C, Chan A, Gold R, Ellrichmann G. Tumefactive multiple sclerosis lesions in two patients after cessation of fingolimod treatment. Ther Adv Neurol Disord. 2015;8(5):233–8.
Litwin T, Smoliński Ł, Członkowka A. Substantial disease exacerbation in a patient with relapsing-remitting multiple sclerosis after withdrawal from siponimod. Neurol Neurochir Pol. 2018;52(1):98–101.
Stamatellos VP, Rigas A, Stamoula E, Lallas A, Papadopoulou A, Papazisis G. S1P receptor modulators in multiple sclerosis: detecting a potential skin cancer safety signal. Mult Scler Relat Disord. 2022;59: 103681.
Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221–34.
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–20.
Giovannoni G, Kappos L, de Seze J, Hauser SL, Overell J, Koendgen H, et al. Risk of requiring a walking aid after 6.5 years of ocrelizumab treatment in patients with relapsing multiple sclerosis: data from the OPERA I and OPERA II trials. Eur J Neurol. 2022;29(4):1238–42 .
Wolinsky JS, Arnold DL, Brochet B, Hartung HP, Montalban X, Naismith RT, et al. Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post-hoc analysis from the ongoing open-label extension of the randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19(12):998–1009.
Kappos LM, Hauser X, Julian SL, Manfrini L, Belachew M, Model S, et al. Prespecified subgroup analyses of ocrelizumab efficacy in patients with primary progressive multiple sclerosis from the phase III ORATORIO study. Eur J Neurol Supplement. 2018. 4th Congress of the European Academy of Neurology; 16–19 June 2018; Lisbon.
Ng HS, Rosenbult CL, Tremlett H. Safety profile of ocrelizumab for the treatment of multiple sclerosis: a systematic review. Expert Opin Drug Saf. 2020;19(9):1069–94.
Roche. Ocrelizumab and PML. 2021. wwwocrelizumabinfoglobal. Accessed 15 Jul 2022.
Smets I, Reyes S, Baker D, Giovannoni G. Blunted vaccines responses after ocrelizumab highlight need for immunizations prior to treatment. Mult Scler Relat Disord. 2021;21(50): 102851.
Simpson-Yap S, De Brouwer E, Kalincik T, Rijke N, Hillert JA, Walton C, et al. Associations of disease-modifying therapies with COVID-19 severity in multiple sclerosis. Neurology. 2021;97(19):e1870–85.
Achiron A, Mandel M, Dreyer-Alster S, Harari G, Magalashvili D, Sonis P, et al. Humoral immune response to COVID-19 mRNA vaccine in patients with multiple sclerosis treated with high-efficacy disease-modifying therapies. Ther Adv Neurol Disord. 2021;14:17562864211012836.
Faissner S, Heitmann N, Rohling R et al. Preserved T-cell response in anti-CD20 treated multiple sclerosis patients following SARS-CoV-2 vaccination, 09 December 2021, PREPRINT (Version 1) available at Research Square. https://doi.org/10.21203/rs.3.rs-1109886/v1
Apostolidis SA, Kakara M, Painter MM, Goel RR, Mathew D, Lenzi K, et al. Cellular and humoral immune responses following SARS-CoV-2 mRNA vaccination in patients with multiple sclerosis on anti-CD20 therapy. Nat Med. 2021;27(11):1990–2001.
Masoud S, McAdoo SP, Bedi R, Cairns TD, Lightstone L. Ofatumumab for B cell depletion in patients with systemic lupus erythematosus who are allergic to rituximab. Rheumatology (Oxford). 2018;57(7):1156–61.
Pawluczkowycz AW, Beurskens FJ, Beum PV, Lindorfer MA, van de Winkel JG, Parren PW, et al. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol. 2009;183(1):749–58.
Kähäri L, Fair-Mäkelä R, Auvinen K, Rantakari P, Jalkanen S, Ivaska J, et al. Transcytosis route mediates rapid delivery of intact antibodies to draining lymph nodes. J Clin Invest. 2019;129(8):3086–102.
Torres J et al. Distribution and efficacy of Ofatumumab and Ocrelizumab in humanized-CD20 mice following subcutaneous or intravenous administration. Poster presentation. 2019 AAN.
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88.
Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology. 2014;82(7):573–81.
Kurrasch R, Brown JC, Chu M, Craigen J, Overend P, Patel B, et al. Subcutaneously administered ofatumumab in rheumatoid arthritis: a phase I/II study of safety, tolerability, pharmacokinetics, and pharmacodynamics. J Rheumatol. 2013;40(7):1089–96.
Bar-Or A, Wiendl H, Montalban X, Alvarez E, Davydovskaya M, Delgado SR, et al. Rapid and sustained B-cell depletion with subcutaneous ofatumumab in relapsing multiple sclerosis: APLIOS, a randomized phase-2 study. Mult Scler. 2021;4:13524585211044480.
Bar-Or A, Grove RA, Austin DJ, Tolson JM, VanMeter SA, Lewis EW, et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: the MIRROR study. Neurology. 2018;90(20):e1805–14.
Hauser SL, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–57.
Flores-Gonzalez RE, Hernandez J, Tornes L, Rammohan K, Delgado S. Development of SARS-CoV-2 IgM and IgG antibodies in a relapsing multiple sclerosis patient on ofatumumab. Mult Scler Relat Disord. 2021;19(49): 102777.
Adamec I, Rogić D, Penz MG, Braun C, Habek M. Humoral and cellular immunity in convalescent COVID-19 people with multiple sclerosis treated with ofatumumab. J Neuroimmunol. 2022;15(362): 577788.
Fox E, Lovett-Racke AE, Gormley M, Liu Y, Petracca M, Cocozza S, et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti-CD20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mul Scler. 2021;27(3):420–9.
de Weers M, Verschuren MC, Kraakman ME, Mensink RG, Schuurman RK, van Dongen JJ, et al. The Bruton’s tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol. 1993;23(12):3109–14.
Martin E, Aigrot MS, Grenningloh R, Stankoff B, Lubetzki C, Boschert U, et al. Bruton’s tyrosine kinase inhibition promotes myelin repair. Brain Plast. 2020;5(2):123–33.
Faissner S, Gold R. Progressive multiple sclerosis: latest therapeutic developments and future directions. Ther Adv Neurol Disord. 2019;12:1756286419878323.
Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019;18(12):905–22.
Estupiñán HY, Berglöf A, Zain R, Smith CIE. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front Cell Dev Biol. 2021;9: 630942.
Reich DS, Arnold DL, Vermersch P, Bar-Or A, Fox RJ, Matta A, et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021;20(9):729–38.
Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380(25):2406–17.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this article. Open Access funding enabled and organized by Projekt DEAL.
Conflicts of Interest/Competing Interests
Simon Faissner and Ralf Gold have no conflicts of interest that are directly relevant to the content of this article. Simon Faissner has received speaker’s and/or scientific board honoraria from Biogen, BMS, Celgene, Genesis Pharma, Novartis, and Roche and grant support from Ruhr-University Bochum, DMSG, Stiftung für therapeutische Forschung, Lead Discovery Center GmbH, and Novartis. Ralf Gold serves on scientific advisory boards for Teva Pharmaceutical Industries Ltd., Biogen, Bayer Schering Pharma, and Novartis; has received speaker honoraria from Biogen, Teva Pharmaceutical Industries Ltd., Bayer Schering Pharma, and Novartis; serves as an editor for Therapeutic Advances in Neurological Diseases and on the editorial boards of Experimental Neurology and the Journal of Neuroimmunology; and receives research support from Teva Pharmaceutical Industries Ltd., Biogen Idec, Bayer Schering Pharma, Genzyme, Merck Serono, and Novartis.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code Availability
Not applicable.
Authors’ Contributions
SF and RG discussed data and performed the literature review. SF wrote the first draft of the manuscript. RG critically revised the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Faissner, S., Gold, R. Efficacy and Safety of Multiple Sclerosis Drugs Approved Since 2018 and Future Developments. CNS Drugs 36, 803–817 (2022). https://doi.org/10.1007/s40263-022-00939-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-022-00939-9