
Abstract
Recombinant interferon (IFN) β-1b was approved by the US Food and Drug Administration as the first disease-modifying therapy (DMT) for multiple sclerosis (MS) in 1993. Since that time, clinical trials and real-world observational studies have demonstrated the effectiveness of IFN therapies. The pivotal intramuscular IFN β-1a phase III trial published in 1996 was the first to demonstrate that a DMT could reduce accumulation of sustained disability in MS. Patient adherence to treatment is higher with intramuscular IFN β-1a, given once weekly, than with subcutaneous formulations requiring multiple injections per week. Moreover, subcutaneous IFN β-1a is associated with an increased incidence of injection-site reactions and neutralizing antibodies compared with intramuscular administration. In recent years, revisions to MS diagnostic criteria have improved clinicians’ ability to identify patients with MS and have promoted the use of magnetic resonance imaging (MRI) for diagnosis and disease monitoring. MRI studies show that treatment with IFN β-1a, relative to placebo, reduces T2 and gadolinium-enhancing lesions and gray matter atrophy. Since the approval of intramuscular IFN β-1a, a number of high-efficacy therapies have been approved for MS, though the benefit of these high-efficacy therapies should be balanced against the increased risk of serious adverse events associated with their long-term use. For some subpopulations of patients, including pregnant women, the safety profile of IFN β formulations may provide a particular benefit. In addition, the antiviral properties of IFNs may indicate potential therapeutic opportunities for IFN β in reducing the risk of viral infections such as COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Since 1981, clinical and real-world observational studies have demonstrated the effectiveness of interferon therapies in reducing relapse rate, disability worsening/progression, and the number of new or newly enlarging lesions in patients with multiple sclerosis (MS). |
The pivotal intramuscular interferon β-1a phase III trial in 1996 (Multiple Sclerosis Collaborative Research Group [MSCRG]) was the first to demonstrate that disease-modifying therapy could reduce the accumulation of sustained disability in MS. |
Other studies suggest that interferon treatment may improve cognition and benefit patients’ quality of life. |
1 Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating and neurodegenerative disease of the central nervous system (CNS) of unknown etiology with a projected 2020 prevalence of > 0.9 million cases in the United States [1] and an estimated 2020 global prevalence of > 2.8 million cases [2]. Symptoms of MS usually appear in adults between 20 and 50 years of age [3,4,5] and may include fatigue, visual impairment, motor weakness, ataxia, reduced mobility, tremor, sensory loss, pain, impaired genitourinary function, depression, and cognitive impairment [6]. These symptoms have a negative impact on patients’ quality of life (QoL) by interfering with gainful employment, interpersonal relationships, intimate and leisure activities, and other daily activities [7, 8]. Most people with MS (> 85%) present with relapsing-remitting MS (RRMS) [4], characterized by an initial phase of recurrent neurologic episodes (relapses) followed by remission and a second phase consisting of the progressive accrual of neurologic disability [9, 10].
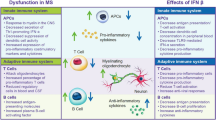
The pathological mechanisms underlying the initiation and progression of MS are multifactorial and not completely known (reviewed in [11,12,13,14,15]). The autoimmune inflammatory state is generally believed to be initiated by myelin-reactive CD4+ T helper cells [15,16,17], although other immune cells, including CD8+ T cells and B cells, are also significantly involved [18, 19]. Epidemiological studies have identified geographic [20], environmental [21,22,23], commensal [24,25,26], and genetic [27] risk factors for MS. Genome-wide association studies show over 200 loci that may contribute to the genetic risk for MS, including major histocompatibility complex alleles and genes regulating interferon (IFN) responses (reviewed in [28,29,30,31]). While some of the genetic loci and most of the identified environmental risk factors affect adaptive and/or innate immunity, no one risk factor is able to explain all cases of MS. This suggests that one or more risk factors may help to ‘trigger’ MS in genetically susceptible persons [32, 33].
Twenty-five years ago, intramuscular (IM) IFN β-1a (Avonex®) was shown to be the first treatment for patients with MS that could impact the course of the disease by reducing the accrual of sustained disability in addition to reducing relapses. Since the approval of IM IFN β-1a, the MS therapeutic landscape has changed considerably with the addition of new therapeutic agents. In the following, we present a narrative review of the history, mechanism of action, and clinical and real-world experience with IM IFN β-1a for the treatment of MS.
2 The Interferons (IFNs): Discovery and Mechanism of Action in Multiple Sclerosis Therapy
The IFNs were discovered in 1957 during investigations into mechanisms of viral interference [34]. In humans, IFNs form a family of secreted autocrine and paracrine cytokines [35] with diverse and essential roles in mediating antiviral activity, regulating cell growth and proliferation, and modulating immune responses [36, 37].
Based on sequence homology and signaling properties, IFNs can be grouped into three types. All type I IFNs bind to and signal through the same heterodimeric IFNAR1/IFNAR2 receptor [38, 39]. In addition to the type I IFNs, a single type II IFN, IFN γ, signals through different receptors and elicits different cellular responses [40], though both types regulate inflammatory responses, primarily by signaling through the JAK-STAT family of signal transducers [41]. The type III IFNs, which include IFN λ1-4, primarily target epithelial cells and have antiviral and immunomodulatory functions [42].
Epidemiological evidence for a viral trigger of MS, coupled with the known antiviral activity of type I IFNs [34, 43], formed the rationale for a small study in patients with MS randomly assigned to a group receiving intrathecal IFN β (n = 10) administered over 6 months or a non-treated control group (n = 10), which was reported in 1981 [44, 45] (Table 1). Over 1.5–2.0 years of observation, patients who received repeated intrathecal IFN β injections had fewer relapses and were more likely to show improvement in their clinical condition than control patients [44, 45]. Approximately 90% of the treated population required no additional injections nearly 4 years after the last intrathecal injection [46]. In 1984, a clinical trial of subcutaneous (SC) IFN α administration reported a trend toward reduced relapse rates [47]. These advances, coupled with the availability of biologically active recombinant IFNs [48], led to the pivotal clinical trials that resulted in US Food and Drug Administration (FDA) approval of recombinant SC IFN β-1b (Betaseron®/Betaferon® and Extavia®) in 1993 [49,50,51] and recombinant IM IFN β-1a (Avonex®) in 1996 [52] for treatment of relapsing MS (Fig. 1).

Timeline of key developments in the history of interferon beta-1 (IFN β-1) for MS. CHAMPIONS Controlled High-Risk Avonex Multiple Sclerosis Prevention Study in Ongoing Neurological Surveillance, CHAMPS Controlled High Risk Avonex Multiple Sclerosis Study, FDA Food and Drug Administration, IFN interferon, IM intramuscular, IT intrathecal, MSCRG MS Collaborative Research Group, SC subcutaneous; aKrause and Petska (2005) [199]; bIsaacs et al. (1957) [34]; cJacobs et al. (1981, 1982) [44, 45]; dSC IFN β-1b (Betaseron® and Extavia®) (2020) [49, 51]; eJacobs et al. (1996) [78]; fIM IFN β-1a (Avonex®) (2020) [52]; gJacobs et al. (2000) [79]; hSC IFN β-1a (Rebif®) (2020) [53]; iKinkel et al. (2012) [85]; jpegylated IFN β-1a (Plegridy®) (2021) [54]; kCohan et al. (2018) [116]; Fox et al. (2013) [119]; Gajofatto et al. (2009) [195]; Einarson et al. (2017) [127]
IFN β-1b and IFN β-1a were among the first disease-modifying therapies (DMTs) approved for treatment of relapsing forms of MS, ushering in the modern era of DMTs for MS. A third IFN, SC-administered IFN β-1a (Rebif®), was approved in 2002 [53]. SC and IM formulations of pegylated IFN β-1a (Plegridy®), which has a longer half-life and thus requires less frequent dosing, were approved in 2014 and 2021, respectively [54] (Fig. 1). The beta IFNs are among the most widely prescribed DMTs for MS worldwide [55]. As of March 2020, > 580,000 patients have been prescribed IM IFN β-1a since its approval, representing > 2,600,000 patient-years of exposure [56].
The complex effects of IFN β on immunomodulatory signaling that contribute to its effectiveness in treating MS have been reviewed elsewhere [57,58,59,60,61,62] and will only be summarized here. IFN β inhibits production of the inflammatory Th1 cytokines, IFN γ and interleukin 12 (IL-12), by human dendritic cells [63, 64]. In patients with stable MS, long-term treatment with IFN β (over an average of 8 years) suppresses expression of Th1/Th2/Th17 inflammatory cytokines and costimulatory proteins, reducing the ‘cytokine storm’ observed in untreated MS patients [65].
Deficiencies or dysfunction in the maintenance of immunological self-tolerance by regulatory T cells can contribute to autoimmune disease [66], and expression of FOXP3, a master regulator of transcription in regulatory T cells, is reduced in regulatory CD4+CD25+ T cells isolated from patients with MS [67]. Within 6 months after starting therapy with IFN β, including IM IFN β-1a, naive CD4+ regulatory T cells increase, memory regulatory T cells decrease, and CD4 regulatory T-cell function is at least partially restored [68].
IFN β treatment improves the subnormal function seen in nonspecific CD8+ suppressor cells isolated from untreated MS patients. CD8+ T cells play an important role in the resolution of inflammation. Patients in relapse have fewer CD8+CD25+FOXP3+ regulatory T cells in their cerebrospinal fluid (CSF) than patients in remission [69]. CD8+ regulatory T cells isolated from clinically quiescent treatment-naive patients with MS or healthy subjects similarly suppress the proliferation of autoreactive CD4+ T cells, but CD8+ cells isolated from patients during relapse are deficient in T-cell suppressor function [70]. CD4+ and CD8+ T cells from IFN β-treated patients also demonstrate reduced proliferation and autoreactivity and decreased production of IFN γ in response to challenge with a myelin-derived antigen compared with cells from untreated MS patients [71].
IFN β also promotes the production of the anti-inflammatory and tolerogenic cytokine interleukin-10 (IL-10). Low levels of IL-10 are associated with an elevated risk of developing autoimmune diseases [72]. IFN β-1a induces expression of IL-10 in cultured peripheral blood mononuclear cells from patients with MS and healthy controls [73, 74]. IL-10 levels in CSF decrease during MS relapse and increase during remission, and IFN β-1a therapy increases serum IL-10 levels in patients with relapsing MS [75].
Finally, in animal models of MS, IFN β reduces the expression of vascular and intercellular adhesion molecules [76] and decreases both the inflammatory activity of dendritic antigen-presenting cells and their migration into the CNS [77].
Thus, type I IFNs have broad effects in reducing inflammatory cytokines, correcting defective regulatory cell function in MS, and reducing expression of molecules that allow penetration of the blood–brain barrier.
3 The Efficacy of Intramuscular (IM) IFN β-1a: Clinical Studies
3.1 Pivotal Clinical Trials: Multiple Sclerosis Collaborative Research Group (MSCRG) and Controlled High-Risk Subjects Avonex Multiple Sclerosis Prevention Study (CHAMPS)
The pivotal phase III MSCRG randomized clinical trial of recombinant IM IFN β-1a versus placebo in patients with relapsing MS was the first large study designed to assess the effect of IFN treatment on physical disability [78]. Kaplan-Meier–estimated time to confirmed disability worsening (CDW) over 104 weeks of treatment (the primary study endpoint, defined as a 1.0-point increase from baseline Expanded Disability Status Scale [EDSS] score, confirmed after 6 months) was greater for patients treated with IM IFN β-1a than with placebo (p = 0.02).
CHAMPS was a 3-year randomized controlled study that examined the effect of IM IFN β-1a versus placebo in 383 patients after a first demyelinating episode involving either the optic nerve (optic neuritis), the brainstem/cerebellum, or the spinal cord (incomplete transverse myelitis) [79]. The primary study endpoint was the development of clinically definite MS (CDMS), defined as the occurrence of either a new clinical neurologic event or an increase after ≥ 1 month of treatment of ≥ 1.5 points in EDSS score. Evidence of new radiological activity (new or enlarging T2 or new gadolinium-enhancing [Gd+] lesions) was assessed as a prespecified secondary endpoint. After 3 years of treatment, the estimated cumulative probability of disease worsening was significantly lower in the IM IFN β-1a group than the placebo group (p = 0.002) [79]. These results were confirmed in a subgroup analysis of CHAMPS [80]. Based on a composite measure consisting of either the development of CDMS (as specified above) or more than one new or enlarging T2 lesion on magnetic resonance imaging (MRI), IM IFN β-1a demonstrated benefit compared with placebo regardless of the initial demyelinating event. Table 1 provides more complete details of the MSCRG and CHAMPS studies.
While an earlier study of IM IFN β-1b did show a reduction in relapses in IFN-treated patients compared with a placebo group [81], that trial was not designed to determine the effect of treatment on physical disability. The MSCRG results for IM IFN β-1a were the first demonstration that DMT treatment of patients with relapsing MS could alter the course of the disease by reducing the accumulation of confirmed/sustained disability. The CHAMPS trial, in addition to providing evidence for the value of early DMT initiation, also demonstrated the value of early MRI scans in MS diagnosis and treatment monitoring, and both concepts continue to inform clinical studies of treatments for MS.
3.2 Long-Term Efficacy of IM IFN β-1a in the MSCRG and CHAMPS Extension Studies
Of the 301 patients originally randomized in the MSCRG pivotal study, 172 (57.1%) completed the full 2 years, 85 of whom were originally randomized to receive IM IFN β-1a. Follow-up studies evaluated the long-term efficacy of IM IFN β-1a based on assessments performed 8 years [82] and 15 years (ASSURANCE) [83] after original entry into the MSCRG trial. At 8 years, a lower proportion of patients initially randomized to IM IFN β-1a rather than to placebo had progressed to an EDSS score ≥ 4.0 (44.3% vs 65.4%; relative risk reduction 32.3%; p = 0.007) or ≥ 5.0 (34.2% vs 54.3%; relative risk reduction 36.9%; p = 0.01), demonstrating that early treatment with IM IFN β-1a reduced the proportion of patients with EDSS progression (Table 2). At the year-15 ASSURANCE follow-up assessment, in 122 eligible patients (63 and 59 patients originally randomized to IM IFN β-1a or placebo, respectively; 14 patients were deceased), those randomized to IM IFN β-1a had better disability outcomes than those randomized to placebo on metrics including mean EDSS score (5.1 vs 5.7), increase from baseline in EDSS score (2.9 vs 3.3 points), and progression to EDSS score ≥ 4.0 (73.9% vs 79.1%), ≥ 6.0 (47.8% vs 58.2%), or ≥ 7.0 (24.6% vs 31.3%) [83]. At 15 years, patients with ongoing IM IFN β-1a therapy had a lower mean EDSS score (4.4 vs 5.7; p = 0.011) and a smaller mean increase in EDSS score from baseline (2.3 vs 3.3; p = 0.011) than patients who had discontinued IM IFN β-1a [83] (Table 2).
The open-label extension study, CHAMPIONS, provided long-term efficacy data on patients originally enrolled in CHAMPS and randomized to placebo or IM IFN β-1a treatment [84]. All CHAMPS patients who enrolled in CHAMPIONS (203 of 383 [53%]) were offered IM IFN β-1a, regardless of their original assignment. After 5 years of treatment with IM IFN β-1a, the cumulative probability of developing CDMS (as defined in CHAMPS) was lower in patients randomized to receive IM IFN β-1a in CHAMPS (the immediate-treatment group) than in those who initially received placebo (the delayed-treatment group; mean [standard deviation (SD)] 5-year incidence 36% [9%] vs 49% [10%]; p = 0.03; Table 2).
Similar results were seen in a subset of CHAMPIONS patients at 10 years of follow-up [85]. The cumulative probability of developing CDMS was lower in the immediate-treatment group than in the delayed-treatment group (58% vs 69%; p = 0.004). Patients in the immediate-treatment group also had a significantly lower annualized relapse rate (ARR) than those in the delayed-treatment group during years 5–10 (0.14 vs 0.31; p = 0.03) and over the entire 10-year follow-up period (0.16 vs 0.33; p = 0.02; Table 2). At 10 years, 81% of all patients had an EDSS score < 3.0. There were no significant between-group differences for EDSS worsening, change in T2 lesion volume, number of Gd+ lesions, or number of new or enlarging T2 lesions in years 5–10. The CHAMPIONS 10-year follow-up results suggest that early initiation of treatment has a more beneficial impact on MS disease course in patients than later initiation. Over the 10-year follow-up period, IM IFN β-1a was associated with a low percentage of serious adverse events (SAEs) and no new safety concerns (see also Sect. 6 below).
4 Comparative Efficacy of IM IFN β-1a
The efficacy of IM IFN β-1a has been compared with other in-class DMTs in head-to-head studies, and IM IFN β-1a has been used as a comparator in clinical trials of newer DMTs.
4.1 Head-to-Head Efficacy Comparison in EVIDENCE
EVIDENCE was a multicenter, randomized controlled trial of the efficacy and safety of SC IFN β-1a versus IM IFN β-1a in patients with RRMS [86, 87]. In the primary clinical outcome, more patients receiving SC IFN β-1a than IM IFN β-1a were relapse free after 24 weeks (75% vs 63%; p = 0.0005) and 48 weeks (62% vs 52%; p = 0.009) [86]. Similarly, ARR was significantly lower in the SC IFN β-1a group than the IM IFN β-1a group at 24 weeks (0.29 vs 0.41; p = 0.022) but not at 48 weeks (0.54 vs 0.64; p = 0.094). More patients receiving SC IFN β-1a than IM IFN β-1a had no new MRI activity (no new Gd+ lesions or new or enlarging T2 lesions) at 24 weeks (p < 0.001). However, in an analysis of 6-month CDW (defined as a ≥ 1.0-point increase in EDSS score) over 48 weeks, the proportions of SC IFN β-1a and IM IFN β-1a patients with disability progression did not differ significantly (5.9% vs 8.3%; p = 0.23; Table 3) [86]. Between-group differences were maintained when EVIDENCE patients were followed for 64 weeks [87]. More SC IFN β-1a than IM IFN β-1a patients remained relapse free (56% vs 48%; p = 0.023) and free of new or enlarging T2 lesions (58% vs 38%; p < 0.001). However, as in the 48-week analyses, there was no significant difference in the proportions of SC IFN β-1a and IM IFN β-1a patients with 6-month CDW (16% vs 17%; p = 0.710; Table 3).
An assessment using the composite outcome measure ‘no evidence of disease activity’ (NEDA, defined as no relapses, no 3-month confirmed EDSS worsening, no new Gd+ lesions, and no new or enlarging T2 lesions) showed that more patients treated with SC IFN β-1a than with IM IFN β-1a achieved NEDA (59.5% vs 41.2%; p < 0.001) over 24 weeks in the EVIDENCE study [88].
4.2 Other Comparative Studies
TRANSFORMS was a 12-month, randomized, double-blind, double-dummy, phase III efficacy study of fingolimod, an orally bioavailable functional sphingosine-1-phosphate–receptor modulator, compared with IM IFN β-1 [89]. In the primary study outcome, ARR at 12 months was significantly lower for patients receiving either 1.25 mg or 0.5 mg fingolimod than for those receiving IM IFN β-1 30 µg (both p < 0.001). However, in a key secondary outcome, there was no significant difference in the proportion of patients free of 3-month confirmed disability progression on IM IFN β-1 (92.1%) compared with fingolimod 1.25 mg (93.3%; p = 0.50) or fingolimod 0.5 mg (94.1%; p = 0.25; Table 3) [89].
Similar results were obtained in DECIDE, a randomized, double-blind, active-controlled phase III study of daclizumab, an anti–IL-2 receptor monoclonal antibody (voluntarily withdrawn from the market in March 2018), compared with IM IFN β-1a [90]. ARR over 144 weeks, the primary study outcome, was lower with daclizumab than with IM IFN β-1a, as was the number of new or enlarging T2 lesions over 96 weeks (both p < 0.001). Paralleling the results of EVIDENCE and TRANSFORMS, there was no significant difference in the estimated proportions of patients with 3-month CDW between the daclizumab and IM IFN β-1a treatment arms over 144 weeks of treatment (16% vs 20%; p = 0.16; Table 3) [90].
RADIANCE was a 24-month, multicenter, double-blind, double-dummy, phase III trial that compared the safety and efficacy of ozanimod, a sphingosine 1-phosphate receptor modulator, with IM IFN β-1a [91]. Over 24 months, ARR was significantly lower in patients treated with ozanimod 1.0 mg than with IM IFN β-1a (p < 0.0001). Similarly, the adjusted mean number of new or enlarging T2 lesions per scan was significantly lower with ozanimod than with IM IFN β-1a (1.84 vs 3.18; p < 0.0001), as was the adjusted mean number of Gd+ lesions at 2 years (0.18 vs 0.37; p = 0.0006). However, the proportion of patients with disability progression (defined as an EDSS score increase of ≥1.0 point confirmed after 3 or 6 months) did not differ significantly in the ozanimod and IM IFN β-1a treatment groups (3-month CDW: 12.5% vs 11.3% [p = 0.8224]; 6-month CDW: 9.7% vs 6.6% [p = 0.1353]) [92].
Taken together, these comparative studies suggest that IM IFN β-1a provides substantial long-term benefit to patients, as evidenced by the consistently low proportions of patients with 3- or 6-month CDW, despite the greater proportion of IM IFN β-1a–treated patients with relapse or MRI activity compared with those treated with alternative DMTs. It is possible that the durations of the comparative studies (12–36 months) were not sufficient to detect a difference in 3- or 6-month CDW between IM IFN β-1a and the comparator DMTs, and these results should be interpreted cautiously. However, the results are consistent with the findings from a 10-year, longitudinal prospective study of 480 patients with RRMS or clinically isolated syndrome that brain atrophy, but not relapse activity, is associated with long-term disability progression [93].
5 Brain Atrophy and Pseudoatrophy
Brain atrophy/brain volume loss (BVL) predicts long-term disability in MS [93, 94]. BVL is included in an expanded NEDA outcome measure, NEDA-4 [95]. However, reliable assessment of BVL can be difficult due to the variability and limited reproducibility of imaging methods. In addition, brain volume changes associated with aging in healthy individuals as well as those with MS make it difficult to identify the precise extent of pathological BVL attributable to MS [96]. Nevertheless, longitudinal imaging studies of BVL have found a larger annual percentage brain volume change (PBVC) in patients with MS than in normal controls (PBVC mean [SD] −0.51% [0.27] vs −0.27% [0.15]; p < 0.0001) [97].
Another difficulty in using brain atrophy as a therapeutic endpoint is the loss of brain volume during short-term (approximately 1 year) treatment with anti-inflammatory DMTs [98]. This phenomenon, termed ‘pseudoatrophy,’ presumably results from resolution of edema in brain along with reduction in the number or volume of inflammatory cells, including microglia [99]. For example, in MSCRG, a post-hoc analysis observed that IM IFN β-1a reduced the progression of brain atrophy, but only by the end of the second year of treatment [100]. It is possible that pseudoatrophy in year 1 masked improvements in BVL during this time.
Supporting this hypothesis, a 3-year prospective study of 386 patients treated with IM IFN β-1a reported that the relative decrease from baseline in brain parenchymal fraction (BPF, a normalized measure of whole-brain atrophy) was 0.686% after 1 year of therapy, 0.377% after 2 years, and 0.378% after 3 years [101]. Subgroup analysis revealed that 68% of the decrease in BPF during the first year occurred during the first 4 months of treatment (0.482% decrease at 4 months; 0.710% change at 1 year). The pretreatment annualized rate of atrophy was 1.06%, whereas the yearly rate of atrophy was 0.33% from 4 months to 3 years of active therapy [101].
Gray matter (GM) atrophy may be the most clinically relevant component of overall brain atrophy in MS [102], as it correlates with physical disability [103] and cognitive impairment [104]. The thalamus is a GM region involved in sensorimotor, cognitive, and attentional circuit functions. A subgroup analysis of 39 patients found reduced thalamic volume in patients with MS compared with normal controls (p < 0.0001) and a strong correlation between thalamic volume and cognitive performance [105]. A retrospective study of MSCRG patients with available whole brain atrophy (BPF) data during the second year of treatment showed significantly less GM atrophy with IM IFN β-1a therapy than with placebo [106]. There was no significant difference in white matter (WM) atrophy, though pseudoatrophy was more apparent in WM than in GM.
6 Cognition
Cognitive impairment occurs frequently in MS, affecting up to 65% of patients [107]. Cognitive deficits adversely impact family and social relationships, work, and self-care [108]. Results from MSCRG provided early evidence that IM IFN β-1a treatment could slow cognitive decline in patients with MS. At 2 years, IM IFN β-1a treatment improved information processing speed and learning/memory as assessed by the Comprehensive Neuropsychological Battery compared with placebo [109].
In the CHAMPIONS 10-year follow-up, > 95% of patients treated with IM IFN β-1a showed cognitive stability, as assessed by the Paced Auditory Serial Addition Test (PASAT), over years 5–10 of the study [110]. Patient attrition over 10 years may have introduced selection bias into CHAMPIONS, but these results still seem notable, given that a small longitudinal study of patients with untreated early-onset MS showed cognitive decline in 46% of patients over 10 years [111]. It should also be noted that learning effects may have elevated PASAT scores in CHAMPIONS, and therefore these longitudinal cognitive test results should be interpreted with caution.
The DECIDE study of patients with RRMS treated with IM IFN β-1a or daclizumab demonstrated improvements in processing speed on the Symbol Digit Modalities Test (SDMT) for both treatments over 96 weeks, with significantly greater improvements seen with daclizumab [112]. An analysis of the DECIDE patients treated with IM IFN β-1a (n = 922) showed an association between processing speed on the SDMT and cortical GM (CGM) and thalamic volume. With MRI results re-baselined at 24 weeks (to allow time for full clinical benefit and to adjust for pseudoatrophy), SDMT improvement at 96 weeks was more pronounced in patients with low (no greater than median) CGM loss over weeks 24–96 than in those with high (greater than median) CGM loss (p = 0.0031). Similarly, SDMT improvement at 96 weeks was more pronounced in patients with low thalamic volume loss than in those with high thalamic volume loss (p = 0.0445; Table 3) [113]. A recent systematic review of cognitive test performance outcomes in 44 studies revealed positive effects on cognitive test performance for DMTs in general; no significant differences between the effects were seen with any single DMT, including high-efficacy DMTs, and IFN β [114].
7 Real-World Effectiveness and Adherence
Real-world observational studies provide data on long-term MS treatment outcomes and drug adherence in routine clinical practice settings and complement the information from randomized clinical trials [115]. Real-world data from a US claims database were used to investigate outcome measures in patients with MS who were stable (relapse free for 1 year) on any IFN β therapy and then either switched to a different IFN β therapy or kept on their existing therapy [116]. For the subset of patients who were stable on IM IFN β-1a at baseline, ARR was twice as high in those who switched to a different IFN β therapy (n = 90) than in those who continued on IM IFN β-1a (n = 270) over 1 year of follow-up (0.14 vs 0.07; p = 0.012). Patients who switched to a different IFN β therapy also had lower medication adherence rates, as assessed by medication possession ratio (defined as the sum of all days’ supplies for all fills of the drug in a particular time period divided by the number of days in that time period), than those who remained on IM IFN β-1a (0.76 vs 0.92; p < 0.001).
Adherence can have a major impact on treatment effectiveness, and multiple studies show poor adherence with self-administered medications for chronic diseases [117]. A retrospective claims database study of 6680 patients with MS initiating treatment with an IFN β or glatiramer acetate (GA) found higher adherence in patients initiating IM IFN β-1a than in those initiating the other DMTs (regression-adjusted odds ratio range, 0.656–0.829). Greater adherence with IM IFN β-1a, possibly attributable to less frequent dosing, is consistent with the results of a questionnaire-based discrete-choice survey of 205 patients indicating that patients value a reduction in injection frequency just as much as a decrease in adverse events (AEs) or an improvement in efficacy [118]. A questionnaire-based study of IFN β discontinuation using the North American Research Committee on Multiple Sclerosis (NARCOMS) registry found that treatment-adherent patients reported less disability progression with IM IFN β-1a (44.4%) than with SC IFN β-1a or SC IFN β-1b (59.2% for the combined groups; p = 0.007) [119]. Safety concerns prompted fewer patients to discontinue IM IFN β-1a (22.3%) than SC IFN β-1a (36.3%; p < 0.001) or SC IFN β-1b (37.8%; p < 0.001). Adherence may be improved by patient education, though a non-interventional observational study of 731 patients with MS treated with IM IFN β-1a found no significant difference in treatment adherence between those who participated in a drug patient management program, consisting of injection training, support, and quarterly visits for up to 12 months after initiation of therapy, and those who did not participate (84% vs 80%; p = 0.3058) [120]. However, treatment cost savings were twice as great for treated patients who participated in the patient management program for 6 months as for non-participants [120].
8 Treatment Non-Responders and Immunogenicity
A post-hoc analysis of MSCRG suggested that the quartile of patients with the greatest MRI activity while taking IM IFN β-1a could be categorized as ‘treatment non-responders’ [121]. A single-nucleotide polymorphism study performed in 830 patients with MS treated with IFN β, including 416 IFN β responders (defined as patients with no relapses and no increase in the EDSS score over ≥ 2 years of treatment) and 414 non-responders (defined as patients with ≥ 1 relapses and an EDSS score increase of ≥ 1.0 point confirmed at 6 months over the same period), failed to demonstrate an association between polymorphisms located in type I IFN-induced genes, toll-like receptor pathway genes, or genes encoding neurotransmitter receptors and IFN β response [122]. Since DNA links are not obvious in response to IFN β-1a, response heterogeneity may be due to variation in expression of IFN β–responsive genes. Short- and long-term transcriptome profiling of peripheral blood mononuclear cells isolated from patients with MS treated with IFN β and followed for an average of 8.14 years (range 1–19 years; complete or partial responders), DMT-naive patients (with stable or active disease), and healthy controls demonstrated that for IFN β–treated patients, there were short-term changes in the expression profile of 1233 coding and 664 non-coding sequences 4 and 24 hours after injection (fold-change ≥ 1.5 or ≤ −1.5; p < 0.05) [65]. Sequences associated with immune regulation, inflammation, antiviral responses, cell cycle progression, and promotion of apoptosis were the most significantly affected. Long-term alterations in 6434 coding and 2362 non-coding sequences were also identified in one or more of the treated and/or untreated MS groups compared with healthy controls. Nearly all (95%) of the differentially expressed sequences were identified only in the therapy-naive groups. In contrast, both the completely and partially IFN β–responsive patients had a long-term expression profile similar to that of the healthy controls [65]. Such large-scale differential gene expression in response to IFN β could account for the observed non-responders to IM IFN β-1a. IFN signature gene expression has been suggested as a candidate biomarker to identify IFN β–responsive patients, and it may also identify novel candidate proteins or target pathways for therapeutic intervention.
The presence of IFN-neutralizing antibodies (NAbs) has been associated with reduced therapeutic effectiveness [123,124,125]. The titer and frequency of developing anti–IFN β NAbs varies with the particular IFN β therapy, as well as with the frequency and route of administration [126]. During the MSCRG phase III trial, 22% of the IFN treatment group developed NAbs after 2 years. However, in the subsequent CHAMPS study of patients at risk of MS after a first demyelinating event, NAbs were detected in the serum of < 1% of treated patients at 18 months and 2% of patients at 30 months [79]. This marked reduction in NAb frequency likely reflects improvements in the methodology of IFN β-1a synthesis. In addition, IM administration may pose a lower risk of NAb development than SC administration. A 2017 meta-analysis of 36 studies that included > 30,000 patients with MS receiving IFN therapy found that after ≥ 1 year of continuous treatment, the proportion (SD) of patients developing NAbs to IFN was lower in those treated with IM IFN β-1a (4.7% [1.5%]; n = 188) than in those treated with SC IFN β-1a (21.4% [2.8%]; n = 716; p < 0.001) or SC IFN β-1b (32.2% [3.3%]; n = 195; p < 0.001) [127]. Neutralization can depend on NAb titers, which also vary among IFN preparations. Over 2 years of treatment with PEGylated IFN β-1a, < 1% of patients developed NAbs, an incidence rate that may be due in part to a reduction in immunogenicity with pegylation [128].
9 Safety and Tolerability of IM IFN β-1a
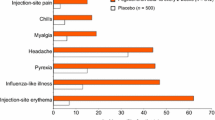
More than 25 years of clinical trial and real-world experience with IFN βs for MS have demonstrated the long-term safety and tolerability of these therapies [129]. In the MSCRG pivotal trial, AEs were reported significantly more frequently (defined as p < 0.1) by IM IFN β-1a than placebo patients and were limited to flu-like symptoms (FLS; 61% vs 40%), muscle aches (34% vs 15%), chills (21% vs 7%), fever (23% vs 13%), and asthenia (21% vs 13%). Seven of 158 IM IFN β-1a patients (4%) and two of 143 placebo patients (1%) discontinued treatment due to AEs [78]. Following completion of the MSCRG trial, long-term safety and tolerability of IM IFN β-1a were evaluated in an open-label extension study over 6 years [130]. Of the 382 patients included in the safety assessment, 218 (115 from the IM IFN β-1a group and 103 from the placebo group) had participated in the MSCRG pivotal trial. Of the 164 patients who did not participate in the MSCRG trial, 140 had received prior treatment with IM IFN β-1b. Overall, 84 patients in the open-label study were naive to IFN β treatment. Over the duration of the study, the type and incidence of AEs were similar to those seen in the MSCRG trial; the most commonly reported AEs were FLS (74%), headache (58%), muscle aches (48%), cold symptoms (46%), and accidental injury (45%). A majority of patients (275 of 382 [72%]) completed the study, with eight participants (2%) withdrawing due to AEs [130].
Safety outcomes in the CHAMPS trial of IM IFN β-1a in patients with a first demyelinating event were similar to those seen in the MSCRG phase III and open-label studies. The AEs reported by more IM IFN β-1a than placebo patients were FLS (54% vs 26%) and depression (20% vs 13%) [79]. One patient in the IM IFN β-1a group (<1%) and seven patients in the placebo group (4%) discontinued treatment due to AEs, and there were no SAEs attributed to study drug. Long-term safety outcomes in patients originally enrolled in CHAMPS were assessed in the open-label CHAMPIONS extension study [84, 85]. Of the 203 patients who enrolled in CHAMPIONS, 103 had originally received IM IFN β-1a and 100 had received placebo in CHAMPS. A majority of patients (155 of 203 [76%]) completed the 10-year follow-up study. During CHAMPIONS, a total of 34 SAEs in 25 patients were reported, none of which were considered likely to be related to treatment, and no new safety signals were identified [84, 85]. Safety outcomes for IM IFN β-1a and SC IFN β-1a were investigated over 1 year of treatment in the EVIDENCE study [86]. Injection-site reactions (ISRs) were more frequent with SC IFN β-1a than with IM IFN β-1a, as were liver enzyme abnormalities, altered leukocyte counts, and the development of NAbs, though patient discontinuation rates were similar. At the last assessment after 16 months of treatment, 21% of SC IFN β-1a patients were persistently NAb-positive, compared with 3% of IM IFN β-1a patients (p < 0.001) [86]. Similarly, a phase IV, multicenter, retrospective and prospective observational study found that while AEs were reported by similar proportions of patients receiving IM IFN β-1a and SC IFN β-1a, fewer patients treated with IM IFN β-1a experienced ISRs [131].
9.1 Flu-Like Symptoms and Injection-Site Reactions
One of the most commonly reported AEs/safety concerns for each of the IFN β therapies during clinical trials and post-marketing surveillance is FLS [49, 51,52,53]. This is not surprising, given that native IFNs are responsible for these symptoms during infection by influenza viruses [132, 133]. AEs such as FLS and ISRs are important factors in lack of adherence to IFN β, typically ranking just below perceived lack of treatment effectiveness in importance. In MSCRG, FLS were the most common AE, reported by 61% of the IM IFN β-1a group and 40% of the placebo group [78]. Similar proportions of patients had FLS during the first 6 months of CHAMPS [79]. A retrospective hospital-chart–based study of 394 patients with MS treated with IFN β for ≤ 8 years found that FLS and ISRs were the most-often cited reasons for discontinuation [134]. Patients receiving IM IFN β-1a are less likely to experience ISRs than patients receiving SC IFN β-1a, so it was anticipated that patients receiving IM treatment would be less likely to discontinue medication as well [131]. Consistent with this, a NARCOMS registry study reported that patients treated with IM IFN β-1a reported fewer discontinuations due to safety concerns than patients treated with SC IFN β-1a or IFN β-1b [119]. Similarly, a recent open-label, randomized, crossover phase I study in healthy subjects found a significantly lower incidence of treatment-emergent ISRs with IM administration of PEGylated IFN β-1a than with SC administration (14.4% vs 32.1%; p = 0.0005) [135].
To reduce the negative impact of FLS and ISRs upon treatment adherence, successful strategies for mitigation and management of these AEs have been developed [136, 137]. Patient education that includes information about the frequency, severity, and management of FLS and ISRs may optimize treatment adherence and clinical outcomes [136, 138,139,140]. The 12-month observational PERSIST study of 232 patients with MS demonstrated that the use of an IM IFN β-1a autoinjector pen was also associated with high levels of persistence, compliance, adherence, and patient satisfaction, with little to no pain and minimal need for caregiver assistance [173].
10 Pregnancy
Women are disproportionately affected by MS and are often diagnosed and treated during childbearing years [141]. While patients can continue to take IFN β before pregnancy, treatment choice during pregnancy planning and pregnancy and postpartum strategies regarding resumption of therapy are essential.
Recent observational data from European and Nordic (Finland and Sweden) healthcare registries of pregnant women with MS treated with IFN β indicate that exposure to IFNs is not associated with adverse pregnancy outcomes. It should be noted that most exposure to IFN β in these cases occurred before or during the first trimester of pregnancy [142, 143]. European MS registry data showed that 82.0% (777 of 948) of pregnancies in women who were treated with IFN β resulted in live births without congenital anomalies. The prevalence of congenital anomalies (1.8% [17 of 948]), spontaneous abortions (10.7% [101 of 948]), and ectopic pregnancies (0.4% [4 of 948]) with live birth were within the ranges reported for the general population [142]. Similar results were obtained from the Nordic registries for pregnant women with MS exposed only to IFN β compared with pregnant women with no exposure to any MS DMT. In the IFN β–exposed and unexposed cohorts, live births with congenital anomaly occurred in 1.8% (12 of 666) and 4.0% (56 of 1397) of cases, respectively, spontaneous abortions in 8.3% (66 of 797) and 12.0% (197 of 1647) of cases, respectively, and ectopic pregnancies in 1.6% (13 of 797) and 3.2% (53 of 1647) of cases, respectively [144].
A separate analysis of Nordic registry data demonstrated that exposure to IFN β did not affect infant outcomes (including birth weight, head circumference, and live birth with congenital malformation) compared with pregnancies with no exposure to any DMT. There was no difference in birth weight or infant head circumference between the exposed and unexposed cohorts [145].
Exposure to IM IFN β-1a while breastfeeding does not appear to be associated with significant infant exposure. A small study of six breastfeeding patients with MS estimated, using the highest measured value of IM IFN β-1a in breastmilk, that the relative infant dose was 0.006% of the maternal dose [146]. Similar results were recently reported for infant exposure to pegylated IFN β-1a in breastmilk [147].
Recently, the FDA and the European Medicines Agency approved updates to the IFN β product information labels indicating that women may continue taking IFN β during pregnancy and while breastfeeding [49,50,51,52,53, 148,149,150].
11 IFN β and Quality of Life
Patient-reported outcomes (PROs) have become increasingly important in therapeutic research and clinical practice in understanding the effects of MS on patients’ daily lives and how DMTs ameliorate those effects. PROs can include overall health status, MS symptom impact, physical and mental functioning, and health-related QoL (HRQoL). PROs are also applicable to comparative effectiveness research and real-world clinical settings [151].
Neurological symptoms experienced by patients with MS may interfere with daily life. Patients with MS rank their HRQoL lower than individuals with other neurological and chronic diseases or healthy individuals [8]. The effect of IM IFN β-1a on HRQoL was evident in a retrospective evaluation of a subgroup of patients originally enrolled in MSCRG. QoL assessed by the Sickness Impact Profile, a validated patient-reported measure of overall health, remained stable with IM IFN β-1a treatment over 2 years [152]. At the ASSURANCE year-15 follow-up assessment of patients who completed the 2-year pivotal phase III MSCRG trial, patients who stayed on IM IFN β-1a, compared with patients who discontinued treatment, had better average scores on the physical component summary (39.3 vs 31.0; p < 0.0001) and the physical functioning (53.0 vs 27.7; p < 0.001), role-physical (46.0 vs 27.8; p < 0.05), and general health (64.0 vs 51.4; p < 0.05) subscales of the Short-Form Health Survey-36 [83].
Observational studies also demonstrate an association between IM IFN β-1a treatment and improvements in QoL. A 2-year, prospective, observational, phase IV study in 284 patients found that IM IFN β-1a treatment was associated with an increase in Multiple Sclerosis Quality of Life-54 (MS54QoL) physical domain score from 56.6 at baseline to 61.0 at 2 years (p < 0.05). There was a trend towards improvement in MS54QoL mental domain score from 57.2 to 61.1 (p = 0.07) over the same period [153]. In another observational retrospective study of 445 patients with RRMS treated with injectable DMTs, the Treatment Satisfaction Questionnaire for Medication showed that patient satisfaction ranged from 62% to 72%; of the patients treated with IM IFN β-1a, 69% expressed overall satisfaction with their treatment [154].
11.1 IFN β and Depression
Therapeutic use of IFNs has been associated with an increased risk of subjective feelings of illness, including fatigue and depression, collectively known as cytokine sickness behavior [155]. Neuropsychiatric side effects, including depression and suicide, were first identified in patients treated with IFNα for viral hepatitis infection [156, 157], and a 2012 meta-analysis confirmed a 25% incidence of major depressive episodes in patients who initiate treatment with IFN α [158]. While the labels for all MS IFN β therapies carry a warning for depression and suicide, much of the evidence from the published literature does not support the association of IFN β therapy with depression. A retrospective review of medical records from 2000 to 2007 of 112 patients with RRMS treated exclusively with IFN β or GA found no change in baseline depression scores over 4 years and no exacerbation of depressive symptoms with either DMT [159]. Similarly, a cross-sectional study of 694 patients with MS found no significant relationship between depression levels and IFN treatment [160]. A systematic review of 10 studies found that while most of the included studies did not suggest an association between depression and IFN β therapy, three studies indicated that patients with a prior history of depression may develop a major depressive episode during the first 6 months of treatment [161]. As noted in section 1.1, MS has a profoundly negative impact on patients’ QoL, and persons with MS report lower QoL than those with other chronic conditions, including rheumatoid arthritis, inflammatory bowel disease, epilepsy, and diabetes mellitus [162, 163]. The prevalence of depression is also greater in patients with MS than with other chronic conditions [164]; depression has been associated with increased neuroinflammatory activity and elevated levels of proinflammatory cytokines [155, 165, 166]. However, treatment of depression in patients with MS is associated with significant increases in QoL and improved adherence to treatment [167,168,169,170].
12 The Role of IM IFN β-1a in the Current MS Therapeutic Landscape
12.1 Escalation Versus High-Efficacy Therapy
As the number of approved MS therapies has expanded, so has the complexity of treating MS. The newer therapies vary in efficacy, safety profile, and route and ease of administration [171]. The benefit of early treatment in MS highlights the importance of the choice of initial therapy [172]. Many factors play a role in treatment decisions, including age, sex, child-bearing potential, disease activity, comorbid conditions, previous DMT use, risk tolerance, and cost.
One common therapeutic approach is to use a first-line DMT that is considered safe and moderately efficacious (relative to higher-efficacy DMTs), followed by a period of monitoring and escalation to more efficacious treatment as disease activity necessitates [173]. Conversely, treating early with high-efficacy therapies may prevent the accumulation of irreversible long-term CNS damage [174]. However, high-efficacy therapies may be associated with greater risk of SAEs [175]. Thus, the decision whether to initiate treatment with high-efficacy medications or switch to them later must be balanced against the benefit-risk profile of each treatment approach and should be the result of a shared decision by healthcare providers and patients. The ongoing Determining the Effectiveness of earLy Intensive Versus Escalation Approaches for RRMS (DELIVER-MS) [176] and TRaditional versus Early Aggressive Therapy for MS (TREAT-MS, ClinicalTrials.gov NCT03500328) studies will compare the benefits and risks of starting treatment with an escalation approach versus initiating treatment with high-efficacy medications in patients with RRMS.
In another approach, after beginning treatment with a high-efficacy oral or infusible DMT as their initial therapy, patients may be switched to a first-line DMT after a period of stable disease or if/when AEs associated with the high-efficacy therapy occur. Initial treatment with immunosuppressive agents that target and ablate B-cell lymphocytes followed by long-term treatment with immunomodulatory DMTs, including IFNs (‘induction therapy’), has been studied in randomized clinical trials (reviewed in [174]). In the randomized, partially placebo-controlled exploratory RESTORE study, patients with relapsing MS who were stable on natalizumab for ≥ 1 year were randomized 1:1:2 to continue natalizumab, switch to placebo, or switch to an alternate immunomodulatory therapy (IM IFN β-1a, GA, or methylprednisolone). After 24 weeks, 40% of patients who switched to an alternate therapy or placebo had disease recurrence as assessed by new Gd+ lesions on MRI, including 1 of 14 patients receiving IM IFN β-1a (7%), 8 of 15 patients receiving GA (53%), 21 of 52 patients receiving methylprednisolone (40%), and 19 of 41 patients receiving placebo (46%) [177].
12.2 Patient Subpopulations Who Might Benefit from IFN β Therapy
As previously noted, IFNs are well tolerated in pregnant and nursing women. Women receiving DMTs that are contraindicated before/during pregnancy (i.e., fingolimod or teriflunomide) may, in consultation with their healthcare provider, consider IFN β treatment for a ‘bridge’ period after discontinuation of these DMTs. In addition, patients at high risk for serious infections, such as older patients with active inflammatory disease and patients with comorbidities that elevate the risks associated with higher-efficacy medications, may benefit from the established safety profile of IFN β.
Given the role of IFNs in native immune responses to viral infections, it is not surprising that the antiviral properties of IFN βs have been investigated in MS. Sera from patients treated with IM IFN β-1 (n = 23) had lower levels of latent neurotropic human herpes virus 6 (HHV6) cell–free DNA and higher levels of anti-HHV6 immunoglobulin M antibodies than sera from untreated patients with MS or healthy individuals [178].
In response to the novel coronavirus first identified in late 2019 (COVID-19), it has been hypothesized that type I IFNs (including IFN β-1a) may, in combination with other antiviral agents, be effective for the treatment of human coronavirus infection, including infection by Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, the causative agent of COVID-19), if administered early in the infection (reviewed in [179]). Recently, a small, prospective randomized study of patients hospitalized with COVID-19 infection found a reduction in the duration of viral shedding and hospital stay and greater alleviation of symptoms in patients treated with a combination of lopinavir, ritonavir, ribavirin, and IFN β-1b compared with those treated with lopinavir and ritonavir alone [180]. A recent observational study of confirmed or suspected COVID-19 cases in 844 patients with MS currently taking a prescribed DMT demonstrated a lower risk of COVID-19 infection in patients currently taking an IFN β therapy than would be expected based on the size of the overall MS population [181]. With respect to management of patients with MS, it is important to note that most DMTs, including IFN β, do not appear to have major short- or long-term impacts on immune protection against COVID-19 [182, 183]. However, adjustments to dosing intervals and monitoring visits may reduce the risk of viral infection and allay other concerns of patients with MS being treated during the COVID-19 pandemic [184].
The age of the MS patient population is increasing due to the increased use of efficacious therapies for MS and better interventions for MS-related comorbidities [185,186,187]. Elderly patients with MS frequently have age- and disease-related comorbidities that can complicate their management [187, 188]. In a cross-sectional study of 2738 individuals with self-identified RRMS or progressive MS over age 60 years (median age approximately 66 years) and a mean disease duration of approximately 23 years, the most common comorbidities (affecting >30% of patients in either group) were pain, indications of cardiac disease, depression, hypertension, dyslipidemia, and obesity [189]. Aging is frequently accompanied by immunosenescence, a reduction in function of the innate and adaptive immune systems [190]. In particular, a paucity of recent thymocyte emigrants, as well as changes in B cells [191] and CD4+ and CD8+ T cells [191, 192] in patients with MS can compound the immunosuppressive effect of some DMTs [193]. The well-established safety profile of IFN β DMTs suggests its potential usefulness in older patients, particularly those with comorbidities, the risks of which might be heightened by certain MS therapies. Additional real-world studies of treatment outcomes are needed to better understand disease characteristics and responses to DMTs in older patients, especially given that this segment of the MS population is anticipated to increase along with the population as a whole.
13 Conclusions
Since the discovery of the antiviral role of IFNs in 1957, research has demonstrated that IFN β is immunomodulatory [194], promotes the production of anti-inflammatory cytokines [73, 74], and participates in multiple immune regulatory pathways [65]. MSCRG, the pivotal IM IFN β-1a phase III trial, was the first study to show that a DMT could improve disability outcomes in MS [78]. Clinical trials and subsequent real-world observational studies have since demonstrated the effectiveness of IM IFN β-1a for treating MS [79, 80, 84, 85, 116, 119, 127, 195]. Treatment with IM IFN β-1a is associated with significant reductions in relapses [78, 85], disability worsening [80, 83, 85, 196], the number and volume of T2 and Gd+ MRI lesions [78, 197], and GM atrophy relative to placebo [106]. Head-to-head comparisons of several newer MS therapies with IM IFN β-1a have consistently found no between-group differences with regard to disability outcomes [89, 90]. Finally, there are cognitive benefits [109, 110] and QoL improvement [83, 152, 153] with ≥ 2 years of treatment with IM IFN β-1a.
In the years since the approval of the IFN βs, the MS therapeutic landscape has changed significantly [198]. This changing landscape presents challenges in terms of balancing efficacy with safety, and opportunities to develop individualized long-term treatment strategies for persons with MS.
References
Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, et al. The prevalence of MS in the United States: a population-based estimate using health claims data. Neurology. 2019;92(10):e1029–40.
Atlas of MS (3rd edition). The Multiple Sclerosis International Federation; 2020.
McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: a review. J Am Med Assoc. 2021;325(8):765–79.
Iwanowski P, Losy J. Immunological differences between classical phenotypes of multiple sclerosis. J Neurol Sci. 2015;349(1):10–4.
Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol. 2019;9:3116.
Williams AE, Vietri JT, Isherwood G, Flor A. Symptoms and association with health outcomes in relapsing-remitting multiple sclerosis: results of a US patient survey. Mult Scler Int. 2014;2014:203183.
Zwibel HL, Smrtka J. Improving quality of life in multiple sclerosis: an unmet need. Am J Manag Care. 2011;17:S139–45.
Benito-León J, Manuel Morales J, Rivera-Navarro J, Mitchell AJ. A review about the impact of multiple sclerosis on health-related quality of life. Disabil Rehabil. 2003;25(23):1291–303.
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86.
Confavreux C, Vukusic S. The clinical course of multiple sclerosis. Handb Clin Neurol. 2014;122:343–69.
Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron. 2018;97(4):742–68.
Lassmann H. Multiple sclerosis pathology. Cold Spring Harb Perspect Med. 2018;8:a028936.
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545–58.
Grigoriadis N, van Pesch V, ParadigMS Group. A basic overview of multiple sclerosis immunopathology. Eur J Neurol. 2015;22(Suppl 2):3–13.
Kunkl M, Frascolla S, Amormino C, Volpe E, Tuosto L. T helper cells: the modulators of inflammation in multiple sclerosis. Cells. 2020;9(2):482.
Chitnis T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol. 2007;79:43–72.
Wagner CA, Roqué PJ, Goverman JM. Pathogenic T cell cytokines in multiple sclerosis. J Exp Med. 2020;217(1):e20190460.
Arneth BM. Impact of B cells to the pathophysiology of multiple sclerosis. J Neuroinflammation. 2019;16(1):128.
Salou M, Nicol B, Garcia A, Laplaud D-A. Involvement of CD8(+) T cells in multiple sclerosis. Front Immunol. 2015;6:604.
Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev. 2010;9(5):A387–94.
Marrie RA. Environmental risk factors in multiple sclerosis aetiology. Lancet Neurol. 2004;3(12):709–18.
O’Gorman C, Lucas R, Taylor B. Environmental risk factors for multiple sclerosis: a review with a focus on molecular mechanisms. Int J Mol Sci. 2012;13(9):11718–52.
Ascherio A. Environmental factors in multiple sclerosis. Expert Rev Neurother. 2013;13(12 Suppl):3–9.
Mirza A, Forbes JD, Zhu F, Bernstein CN, Van Domselaar G, Graham M, et al. The multiple sclerosis gut microbiota: a systematic review. Mult Scler Relat Disord. 2020;37:101427.
Ochoa-Repáraz J, Kirby TO, Kasper LH. The gut microbiome and multiple sclerosis. Cold Spring Harb Perspect Med. 2018;8(6):a029017.
Mowry EM, Glenn JD. The dynamics of the gut microbiome in multiple sclerosis in relation to disease. Neurol Clin. 2018;36(1):185–96.
Sadovnick AD, Dyment D, Ebers GC. Genetic epidemiology of multiple sclerosis. Epidemiol Rev. 1997;19(1):99–106.
Cotsapas C, Mitrovic M. Genome-wide association studies of multiple sclerosis. Clin Transl Immunology. 2018;7(6):e10118.
Patsopoulos NA, De Jager PL. Genetic and gene expression signatures in multiple sclerosis. Mult Scler. 2020;26(5):576–81.
Lincoln MR, Montpetit A, Cader MZ, Saarela J, Dyment DA, Tiislar M, et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet. 2005;37(10):1108–12.
Patsopoulos NA. Genetics of multiple sclerosis: an overview and new directions. Cold Spring Harb Perspect Med. 2018;8(7):a028951.
Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. 2016;13:25.
Waubant E, Lucas R, Mowry E, Graves J, Olsson T, Alfredsson L, et al. Environmental and genetic risk factors for MS: an integrated review. Ann Clin Transl Neurol. 2019;6(9):1905–22.
Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147(927):258–67.
Dinarello CA. Historical insights into cytokines. Eur J Immunol. 2007;37(S1):S34-45.
Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6(12):975–90.
Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem. 2007;282(28):20047–51.
de Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282(28):20053–7.
Uze G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol. 2007;316:71–95.
Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. 2018;9(2061):1–10.
Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–63.
Zanoni I, Odendall C. Editorial: Interferon-λs: new regulators of inflammatory processes. Front Immunol. 2019;10:2117.
Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14(4):778–809.
Jacobs L, O’Malley J, Freeman A, Ekes R. Intrathecal interferon reduces exacerbations of multiple sclerosis. Science. 1981;214(4524):1026–8.
Jacobs L, O’Malley J, Freeman A, Murawski J, Ekes R. Intrathecal interferon in multiple sclerosis. Arch Neurol. 1982;39(10):609–15.
Jacobs L, O’Malley JA, Freeman A, Ekes R, Reese PA. Intrathecal interferon in the treatment of multiple sclerosis. Patient follow-up. Arch Neurol. 1985;42(9):841–7.
Knobler RL, Panitch HS, Braheny SL, Sipe JC, Rice GP, Huddlestone JR, et al. Systemic alpha-interferon therapy of multiple sclerosis. Neurology. 1984;34(10):1273–9.
Jacobs L, Johnson KP. A brief history of the use of interferons as treatment of multiple sclerosis. Arch Neurol. 1994;51(12):1245–52.
Betaseron [prescribing information]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc.; 2020.
Betaferon [summary of product characteristics]. Leverkusen, Germany: Bayer AG; 2019.
Extavia [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corp.; 2020.
Avonex [prescribing information]. Cambridge, MA: Biogen Inc.; 2020.
Rebif [prescribing information]. Rockland, MA: EMD Serono; 2020.
Plegridy [prescribing information]. Cambridge, MA: Biogen Inc.; 2021.
Westad A, Venugopal A, Snyder E. The multiple sclerosis market. Nat Rev Drug Discov. 2017;16(10):675–6.
Hua LH, Liao S, Hersh CM, Fitzgerald K, Altincatal A, Naylor ML, et al. Characteristics and clinical outcomes of older patients with MS treated with peginterferon beta-1a or intramuscular interferon beta-1a in MS PATHS. ECTRIMS [online]; September 11–13, 2020.
Kasper LH, Reder AT. Immunomodulatory activity of interferon-beta. Ann Clin Transl Neurol. 2014;1(8):622–31.
Jakimovski D, Kolb C, Ramanathan M, Zivadinov R, Weinstock-Guttman B. Interferon beta for multiple sclerosis. Cold Spring Harb Perspect Med. 2018;8(11):a032003.
Kieseier BC. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. 2011;25(6):491–502.
Blank T, Prinz M. Type I interferon pathway in CNS homeostasis and neurological disorders. Glia. 2017;65(9):1397–406.
Rommer PS, Milo R, Han MH, Satyanarayan S, Sellner J, Hauer L, et al. Immunological aspects of approved MS therapeutics. Front Immunol. 2019;10:1564.
Scheu S, Ali S, Mann-Nuttel R, Richter L, Arolt V, Dannlowski U, et al. Interferon beta-mediated protective functions of microglia in central nervous system autoimmunity. Int J Mol Sci. 2019;20(1):190.
McRae BL, Semnani RT, Hayes MP, van Seventer GA. Type I IFNs inhibit human dendritic cell IL-12 production and Th1 cell development. J Immunol. 1998;160(9):4298–304.
Nagai T, Devergne O, van Seventer GA, van Seventer JM. Interferon-beta mediates opposing effects on interferon-gamma-dependent interleukin-12 p70 secretion by human monocyte-derived dendritic cells. Scand J Immunol. 2007;65(2):107–17.
Feng X, Bao R, Li L, Deisenhammer F, Arnason BGW, Reder AT. Interferon-beta corrects massive gene dysregulation in multiple sclerosis: short-term and long-term effects on immune regulation and neuroprotection. EBioMedicine. 2019;49:269–83.
Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6(4):345–52.
Huan J, Culbertson N, Spencer L, Bartholomew R, Burrows GG, Chou YK, et al. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res. 2005;81(1):45–52.
Korporal M, Haas J, Balint B, Fritzsching B, Schwarz A, Moeller S, et al. Interferon beta-induced restoration of regulatory T-cell function in multiple sclerosis is prompted by an increase in newly generated naive regulatory T cells. Arch Neurol. 2008;65(11):1434–9.
Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol. 2010;67(5):625–38.
Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, et al. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J Autoimmun. 2011;36(2):115–24.
Zafranskaya M, Oschmann P, Engel R, Weishaupt A, van Noort JM, Jomaa H, et al. Interferon-beta therapy reduces CD4+ and CD8+ T-cell reactivity in multiple sclerosis. Immunology. 2007;121(1):29–39.
Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. 2012;32(1):23–63.
Rudick RA, Ransohoff RM, Peppler R, Medendorp SV, Lehmann P, Alam J. Interferon beta induces interleukin-10 expression: relevance to multiple sclerosis. Ann Neurol. 1996;40(4):618–27.
Feng X, Yau D, Holbrook C, Reder AT. Type I interferons inhibit interleukin-10 production in activated human monocytes and stimulate IL-10 in T cells: implications for Th1-mediated diseases. J Interferon Cytokine Res. 2002;22(3):311–9.
Rudick RA, Ransohoff RM, Lee JC, Peppler R, Yu M, Mathisen PM, et al. In vivo effects of interferon beta-1a on immunosuppressive cytokines in multiple sclerosis. Neurology. 1998;50(5):1294–300.
Floris S, Ruuls SR, Wierinckx A, van der Pol SMA, Döpp E, van der Meide PH, et al. Interferon-β directly influences monocyte infiltration into the central nervous system. J Neuroimmunol. 2002;127(1):69–79.
Pennell LM, Fish EN. Interferon-beta regulates dendritic cell activation and migration in experimental autoimmune encephalomyelitis. Immunology. 2017;152(3):439–50.
Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39(3):285–94.
Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med. 2000;343(13):898–904.
Beck RW, Chandler DL, Cole SR, Simon JH, Jacobs LD, Kinkel RP, et al. Interferon beta-1a for early multiple sclerosis: CHAMPS trial subgroup analyses. Ann Neurol. 2002;51(4):481–90.
The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I/ Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial/ The IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):655–61.
Rudick RA, Cutter GR, Baier M, Weinstock-Guttman B, Mass MK, Fisher E, et al. Estimating long-term effects of disease-modifying drug therapy in multiple sclerosis patients. Mult Scler. 2005;11(6):626–34.
Bermel RA, Weinstock-Guttman B, Bourdette D, Foulds P, You X, Rudick RA. Intramuscular interferon beta-1a therapy in patients with relapsing-remitting multiple sclerosis: a 15-year follow-up study. Mult Scler. 2010;16(5):588–96.
Kinkel RP, Kollman C, O’Connor P, Murray TJ, Simon J, Arnold D, et al. IM interferon beta-1a delays definite multiple sclerosis 5 years after a first demyelinating event. Neurology. 2006;66(5):678–84.
Kinkel RP, Dontchev M, Kollman C, Skaramagas TT, O’Connor PW, Simon JH, et al. Association between immediate initiation of intramuscular interferon beta-1a at the time of a clinically isolated syndrome and long-term outcomes: a 10-year follow-up of the Controlled High-Risk Avonex Multiple Sclerosis Prevention Study in Ongoing Neurological Surveillance. Arch Neurol. 2012;69(2):183–90.
Panitch H, Goodin DS, Francis G, Chang P, Coyle PK, O’Connor P, et al. Randomized, comparative study of interferon beta-1a treatment regimens in MS: the EVIDENCE trial. Neurology. 2002;59(10):1496–506.
Panitch H, Goodin D, Francis G, Chang P, Coyle P, O’Connor P, et al. Benefits of high-dose, high-frequency interferon beta-1a in relapsing-remitting multiple sclerosis are sustained to 16 months: final comparative results of the EVIDENCE trial. J Neurol Sci. 2005;239(1):67–74.
Coyle PK, Reder AT, Freedman MS, Fang J, Dangond F. Early MRI results and odds of attaining ‘no evidence of disease activity’; status in MS patients treated with interferon β-1a in the EVIDENCE study. J Neurol Sci. 2017;379:151–6.
Cohen JA, Barkhof F, Comi G, Hartung H-P, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15.
Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A, et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2015;373(15):1418–28.
Cohen JA, Comi G, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019;18(11):1021–33.
Cohen JA, Comi G, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Ozanimod vs interferon β-1a: clinical and MRI results of RADIANCE part B—a 2-year phase 3 trial in relapsing multiple sclerosis. ECTRIMS; October 25–28, 2017; Paris, France.
Cree BAC, Hollenbach JA, Bove R, Kirkish G, Sacco S, Caverzasi E, et al. Silent progression in disease activity–free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653–66.
Popescu V, Agosta F, Hulst HE, Sluimer IC, Knol DL, Sormani MP, et al. Brain atrophy and lesion load predict long term disability in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2013;84(10):1082–91.
Kappos L, De Stefano N, Freedman MS, Cree BA, Radue EW, Sprenger T, et al. Inclusion of brain volume loss in a revised measure of “no evidence of disease activity” (NEDA-4) in relapsing-remitting multiple sclerosis. Mult Scler. 2016;22(10):1297–305.
Andorra M, Nakamura K, Lampert EJ, Pulido-Valdeolivas I, Zubizarreta I, Llufriu S, et al. Assessing biological and methodological aspects of brain volume loss in multiple sclerosis. JAMA Neurol. 2018;75(10):1246–55.
De Stefano N, Stromillo ML, Giorgio A, Bartolozzi ML, Battaglini M, Baldini M, et al. Establishing pathological cut-offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(1):93–9.
Sastre-Garriga J, Tur C, Pareto D, Vidal-Jordana A, Auger C, Rio J, et al. Brain atrophy in natalizumab-treated patients: a 3-year follow-up. Mult Scler. 2015;21(6):749–56.
De Stefano N, Arnold DL. Towards a better understanding of pseudoatrophy in the brain of multiple sclerosis patients. Mult Scler. 2015;21(6):675–6.
Rudick RA, Fisher E, Lee JC, Simon J, Jacobs L, The Multiple Sclerosis Collaborative Research Group. Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing-remitting MS. Neurology. 1999;53(8):1698–704.
Hardmeier M, Wagenpfeil S, Freitag P, Fisher E, Rudick RA, Kooijmans M, et al. Rate of brain atrophy in relapsing MS decreases during treatment with IFNβ-1a. Neurology. 2005;64(2):236–40.
Bergsland N, Horakova D, Dwyer MG, Uher T, Vaneckova M, Tyblova M, et al. Gray matter atrophy patterns in multiple sclerosis: a 10-year source-based morphometry study. NeuroImage Clin. 2018;17:444–51.
Jacobsen C, Hagemeier J, Myhr K-M, Nyland H, Lode K, Bergsland N, et al. Brain atrophy and disability progression in multiple sclerosis patients: a 10-year follow-up study. J Neurol Neurosurg Psychiatry. 2014;85(10):1109–15.
Tekok-Kilic A, Benedict RH, Weinstock-Guttman B, Dwyer MG, Carone D, Srinivasaraghavan B, et al. Independent contributions of cortical gray matter atrophy and ventricle enlargement for predicting neuropsychological impairment in multiple sclerosis. Neuroimage. 2007;36(4):1294–300.
Houtchens MK, Benedict RHB, Killiany R, Sharma J, Jaisani Z, Singh B, et al. Thalamic atrophy and cognition in multiple sclerosis. Neurology. 2007;69(12):1213–23.
Fisher E, Nakamura K, Lee JC, You X, Sperling B, Rudick RA. Effect of intramuscular interferon beta-1a on gray matter atrophy in relapsing−remitting multiple sclerosis: a retrospective analysis. Mult Scler. 2016;22(5):668–76.
Patti F, Nicoletti A, Messina S, Bruno E, Fermo SL, Quattrocchi G, et al. Prevalence and incidence of cognitive impairment in multiple sclerosis: a population-based survey in Catania. Sicily J Neurol. 2015;262(4):923–30.
Sumowski JF, Benedict R, Enzinger C, Filippi M, Geurts JJ, Hamalainen P, et al. Cognition in multiple sclerosis: state of the field and priorities for the future. Neurology. 2018;90(6):278–88.
Fischer JS, Priore RL, Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, et al. Neuropsychological effects of interferon beta-1a in relapsing multiple sclerosis. Ann Neurol. 2000;48(6):885–92.
Kinkel RP, Laforet G, You X. Disease-related determinants of quality of life 10 years after clinically isolated syndrome. Int J MS Care. 2014;17(1):26–34.
Amato MP, Ponziani G, Siracusa G, Sorbi S. Cognitive dysfunction in early-onset multiple sclerosis: a reappraisal after 10 years. Arch Neurol. 2001;58(10):1602–6.
Benedict RH, Cohan S, Lynch SG, Riester K, Wang P, Castro-Borrero W, et al. Improved cognitive outcomes in patients with relapsing-remitting multiple sclerosis treated with daclizumab beta: results from the DECIDE study. Mult Scler. 2018;24(6):795–804.
Benedict RHB, Arnold DL, Rose J, Cohan S, Lynch SG, Kappos L, et al. Change in cognitive processing speed is associated with cortical grey matter and thalamic volume loss in patients with relapsing-remitting multiple sclerosis. ECTRIMS; September 11-13, 2019; Stockholm, Sweden.
Landmeyer NC, Bürkner P-C, Wiendl H, Ruck T, Hartung H-P, Holling H, et al. Disease-modifying treatments and cognition in relapsing-remitting multiple sclerosis. Neurology. 2020;94(22):e2373.
Kalincik T, Butzkueven H. Observational data: understanding the real MS world. Mult Scler. 2016;22(13):1642–8.
Cohan S, Smoot K, Kresa-Reahl K, Garland R, Yeh W-S, Wu N, et al. Outcomes of stable multiple sclerosis patients staying on initial interferon beta therapy versus switching to another interferon beta therapy: a US claims database study. Adv Ther. 2018;35(11):1894–904.
Viswanathan M, Golin CE, Jones CD, Ashok M, Blalock SJ, Wines RC, et al. Interventions to improve adherence to self-administered medications for chronic diseases in the United States: a systematic review. Ann Intern Med. 2012;157(11):785–95.
Poulos C, Kinter E, Yang J-C, Bridges JFP, Posner J, Reder AT. Patient preferences for injectable treatments for multiple sclerosis in the United States: a discrete-choice experiment. Patient. 2016;9(2):171–80.
Fox RJ, Salter AR, Tyry T, Sun J, You X, Laforet G, et al. Treatment discontinuation and disease progression with injectable disease-modifying therapies: findings from the North American Research Committee on Multiple Sclerosis database. Int J MS Care. 2013;15(4):194–201.
Katsarava Z, Ehlken B, Limmroth V, Taipale K, Patel SN, Niemczyk G, et al. Adherence and cost in multiple sclerosis patients treated with IM IFN beta-1a: impact of the CARE patient management program. BMC Neurol. 2015;15:170.
Rudick RA, Lee JC, Simon J, Ransohoff RM, Fisher E. Defining interferon beta response status in multiple sclerosis patients. Ann Neurol. 2004;56(4):548–55.
Bustamante MF, Morcillo-Suárez C, Malhotra S, Rio J, Leyva L, Fernández O, et al. Pharmacogenomic study in patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2015;2(5):e154.
Rudick RA, Simonian NA, Alam JA, Campion M, Scaramucci JO, Jones W, et al. Incidence and significance of neutralizing antibodies to interferon beta-1a in multiple sclerosis. Multiple Sclerosis Collaborative Research Group (MSCRG). Neurology. 1998;50(5):1266–72.
Deisenhammer F. Neutralizing antibodies to interferon-β and other immunological treatments for multiple sclerosis. CNS Drugs. 2009;23(5):379–96.
Pachner AR, Warth JD, Pace A, Goelz S. Effect of neutralizing antibodies on biomarker responses to interferon beta: the INSIGHT study. Neurology. 2009;73(18):1493–500.
Hartung HP, Munschauer FI, Schellekens H. Significance of neutralizing antibodies to interferon beta during treatment of multiple sclerosis: expert opinions based on the proceedings of an international consensus conference. Eur J Neurol. 2005;12(8):588–601.
Einarson TR, Bereza BG, Machado M. Comparative effectiveness of interferons in relapsing-remitting multiple sclerosis: a meta-analysis of real-world studies. Curr Med Res Opin. 2017;33(3):579–93.
White JT, Newsome SD, Kieseier BC, Bermel RA, Cui Y, Seddighzadeh A, et al. Incidence, characterization, and clinical impact analysis of peginterferon beta1a immunogenicity in patients with multiple sclerosis in the ADVANCE trial. Ther Adv Neurol Disord. 2016;9(4):239–49.
Reder AT, Oger JF, Kappos L, O’Connor P, Rametta M. Short-term and long-term safety and tolerability of interferon b-1b in multiple sclerosis. Mult Scler Relat Disord. 2014;3(3):294–302.
Herndon RM, Rudick RA, Munschauer FE, Mass MK, Salazar AM, Coats ME, et al. Eight-year immunogenicity and safety of interferon beta-1a-Avonex®treatment in patients with multiple sclerosis. Mult Scler. 2005;11(4):409–19.
Minagar A, Murray TJ, for the PROOF Study Investigators. Efficacy and tolerability of intramuscular interferon beta-1a compared with subcutaneous interferon beta-1a in relapsing MS: results from PROOF. Curr Med Res Opin. 2008;24(4):1049–55.
Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nature Commun. 2014;5(1):3864.
Killip MJ, Fodor E, Randall RE. Influenza virus activation of the interferon system. Virus Res. 2015;209:11–22.
O’Rourke KET, Hutchinson M. Stopping beta-interferon therapy in multiple sclerosis: an analysis of stopping patterns. Mult Scler. 2005;11(1):46–50.
Zhao Y, Chen K, Ramia N, Sahu S, Kumar A, Naylor ML, et al. Bioequivalence of intramuscular and subcutaneous peginterferon beta-1a: results of a phase I, open-label crossover study in healthy volunteers. Ther Adv Neurol Disord. 2021;14:1756286420975227.
Filipi ML, Beavin J, Brillante RT, Costello K, Hartley GC, Hartley K, et al. Nurses’ perspective on approaches to limit flu-like symptoms during interferon therapy for multiple sclerosis. Int J MS Care. 2014;16(1):55–60.
McEwan L, Brown J, Poirier J, Spring J, Brouillette J, Carr K, et al. Best practices in skin care for the multiple sclerosis patient receiving injectable therapies. Int J MS Care. 2010;12(4):177–89.
Niemczyk G, Begus-Nahrmann Y, Washeim N, Grundler M-T, Naylor ML, Mäurer M. Real-world characterisation of peginterferon beta-1a associated flu-like symptoms and injection site reactions and impact of mitigation strategies coached in an individualized patient support program in Germany. ECTRIMS; October 10–12, 2018; Berlin, Germany.
Halper J, Centonze D, Newsome SD, Huang D, Robertson C, You X, et al. Management strategies for flu-like symptoms and injection-site reactions associated with peginterferon beta-1a: obtaining recommendations using the Delphi technique. Int J MS Care. 2016;18(4):211–8.
Hendin B, Huang D, Wray S, Naismith RT, Rosenblatt S, Zambrano J, et al. Subcutaneous peginterferon beta-1a injection-site reaction experience and mitigation: Delphi analysis of the ALLOW study. Neurodegener Dis Manag. 2017;7(1):39–47.
Houtchens MK, Edwards NC, Schneider G, Stern K, Phillips AL. Pregnancy rates and outcomes in women with and without MS in the United States. Neurology. 2018;91(17):e1559–69.
Hellwig K, Geissbuehler Y, Sabidó M, Popescu C, Adamo A, Klinger J, et al. Pregnancy outcomes in interferon-beta-exposed patients with multiple sclerosis: results from the European Interferon-beta Pregnancy Registry. J Neurol. 2020;267(6):1715–23.
Vattulainen P, Burkill S, Geissbuehler Y, Meritxell Sabidó M, Catrinel Popescu C, Suzart-Woischnik K, et al. Prevalence of infant outcomes at birth after exposure to interferon beta prior to or during pregnancy: a register-based cohort study in Finland and Sweden among women with MS. ECTRIMS; September 11–13, 2019; Stockholm, Sweden.
Hakkarainen KM, Juuti R, Burkill S, Geissbühler Y, Sabidó M, Popescu C, et al. Pregnancy outcomes after exposure to interferon beta: a register-based cohort study among women with MS in Finland and Sweden. Ther Adv Neurol Disord. 2020;13:1756286420951072.
Burkill S, Vattulainen P, Geissbuehler Y, Sabido Espin M, Popescu C, Suzart-Woischnik K, et al. The association between exposure to interferon-beta during pregnancy and birth measurements in offspring of women with multiple sclerosis. PLoS ONE. 2019;14(12):e0227120.
Hale TW, Siddiqui AA, Baker TE. Transfer of interferon β-1a into human breastmilk. Breastfeed Med. 2011;7(2):123–5.
Houtchens M, Manieri MC, Mahlanza T, Ciplea A, Ramia N, Zhao Y, et al. Analysis of peginterferon beta-1a in the breast milk of lactating patients with multiple sclerosis. American Academy of Neurology Annual Meeting [online]; 2020.
Avonex [summary of product characteristics]. Badhoevedorp, The Netherlands: Biogen Netherlands BV; 2019.
Rebif [summary of product characteristics]. Amsterdam, The Netherlands: Merck Europe BV; 2020.
Extavia [summary of product characteristics]. Dublin, Ireland: Novartis Europharm Ltd.; 2019.
Nowinski CJ, Miller DM, Cella D. Evolution of patient-reported outcomes and their role in multiple sclerosis clinical trials. Neurotherapeutics. 2017;14(4):934–44.
Miller DM, Weinstock-Guttman B, Bourdette D, You X, Foulds P, Rudick RA. Change in quality of life in patients with relapsing–remitting multiple sclerosis over 2 years in relation to other clinical parameters: results from a trial of intramuscular interferon β-1a. Mult Scler. 2011;17(6):734–42.
Jongen PJ, Sindic C, Carton H, Zwanikken C, Lemmens W, Borm G, et al. Improvement of health-related quality of life in relapsing remitting multiple sclerosis patients after 2 years of treatment with intramuscular interferon-beta-1a. J Neurol. 2010;257(4):584–9.
Fernández O, Duran E, Ayuso T, Hernández L, Bonaventura I, Forner M, et al. Treatment satisfaction with injectable disease-modifying therapies in patients with relapsing-remitting multiple sclerosis (the STICK study). PLoS ONE. 2017;12(10):e0185766.
Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am. 2009;29(2):247–64.
Janssen HLA, Brouwer JT, van der Mast RC, Schalm SW. Suicide associated with alfa-interferon therapy for chronic viral hepatitis. J Hepatol. 1994;21(2):241–3.
Renault PF, Hoofnagle JH, Park Y, Mullen KD, Peters M, Jones DB, et al. Psychiatric complications of long-term interferon alfa therapy. Arch Intern Med. 1987;147(9):1577–80.
Udina M, Castellví P, Moreno-España J, Navinés R, Valdés M, Forns X, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry. 2012;73(8):1128–38.
Kirzinger SS, Jones J, Siegwald A, Crush AB. Relationship between disease-modifying therapy and depression in multiple sclerosis. Int J MS Care. 2013;15(3):107–12.
Kim S, Foley FW, Picone MA, Halper J, Zemon V. Depression levels and interferon treatment in people with multiple sclerosis. Int J MS Care. 2012;14(1):10–6.
Alba Palé L, León Caballero J, Samsó Buxareu B, Salgado Serrano P, Pérez SV. Systematic review of depression in patients with multiple sclerosis and its relationship to interferonβ treatment. Mult Scler Relat Disord. 2017;17:138–43.
Hermann BP, Vickrey B, Hays RD, Cramer J, Devinsky O, Meador K, et al. A comparison of health-related quality of life in patients with epilepsy, diabetes and multiple sclerosis. Epilepsy Res. 1996;25(2):113–8.
Rudick RA, Miller D, Clough JD, Gragg LA, Farmer RG. Quality of life in multiple sclerosis. Comparison with inflammatory bowel disease and rheumatoid arthritis. Arch Neurol. 1992;49(12):1237–42.
Patten SB, Beck CA, Williams JV, Barbui C, Metz LM. Major depression in multiple sclerosis: a population-based perspective. Neurology. 2003;61(11):1524–7.
Lee CH, Giuliani F. The role of inflammation in depression and fatigue. Front Immunol. 2019;10:1696.
Leonard BE. The concept of depression as a dysfunction of the immune system. Curr Immunol Rev. 2010;6(3):205–12.
Hart S, Fonareva I, Merluzzi N, Mohr DC. Treatment for depression and its relationship to improvement in quality of life and psychological well-being in multiple sclerosis patients. Qual Life Res. 2005;14(3):695–703.
Mohr DC, Boudewyn AC, Goodkin DE, Bostrom A, Epstein L. Comparative outcomes for individual cognitive-behavior therapy, supportive-expressive group psychotherapy, and sertraline for the treatment of depression in multiple sclerosis. J Consult Clin Psychol. 2001;69(6):942–9.
Mohr DC, Goodkin DE, Likosky W, Gatto N, Baumann KA, Rudick RA. Treatment of depression improves adherence to interferon beta-1b therapy for multiple sclerosis. Arch Neurol. 1997;54(5):531–3.
Nathoo N, Mackie A. Treating depression in multiple sclerosis with antidepressants: a brief review of clinical trials and exploration of clinical symptoms to guide treatment decisions. Mult Scler Relat Disord. 2017;18:177–80.
Vidal-Jordana A. New advances in disease-modifying therapies for relapsing and progressive forms of multiple sclerosis. Neurol Clin. 2018;36(1):173–83.
Noyes K, Weinstock-Guttman B. Impact of diagnosis and early treatment on the course of multiple sclerosis. Am J Manag Care. 2013;19(17 Suppl):s321–31.
Ontaneda D, Tallantyre E, Kalincik T, Planchon SM, Evangelou N. Early highly effective versus escalation treatment approaches in relapsing multiple sclerosis. Lancet Neurol. 2019;18:973–80.
Ruggieri S, Pontecorvo S, Tortorella C, Gasperini C. Induction treatment strategy in multiple sclerosis: a review of past experiences and future perspectives. Mult Scler Demyelinating Disord. 2018;3(1):5.
Merkel B, Butzkueven H, Traboulsee AL, Havrdova E, Kalincik T. Timing of high-efficacy therapy in relapsing-remitting multiple sclerosis: a systematic review. Autoimmun Rev. 2017;16(6):658–65.
Ontaneda D, Tallantyre EC, Raza PC, Planchon SM, Nakamura K, Miller D, et al. Determining the effectiveness of early intensive versus escalation approaches for the treatment of relapsing-remitting multiple sclerosis: the DELIVER-MS study protocol. Contemp Clin Trials. 2020;95:106009.
Fox RJ, Cree BA, De Seze J, Gold R, Hartung HP, Jeffery D, et al. MS disease activity in RESTORE: a randomized 24-week natalizumab treatment interruption study. Neurology. 2014;82(17):1491–8.
Hong J, Tejada-Simon MV, Rivera VM, Zang YC, Zhang JZ. Anti-viral properties of interferon beta treatment in patients with multiple sclerosis. Mult Scler. 2002;8(3):237–42.
Sallard E, Lescure F-X, Yazdanpanah Y, Mentre F, Peiffer-Smadja N. Type 1 interferons as a potential treatment against COVID-19. Antiviral Res. 2020;178:104791.
Hung IF-N, Lung K-C, Tso EY-K, Liu R, Chung TW-H, Chu M-Y, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020;395(10238):1695–704.
Sormani MP, De Rossi N, Schiavetti I, Carmisciano L, Cordioli C, Moiola L, et al. Disease modifying therapies and coronavirus disease 2019 severity in multiple sclerosis. Ann Neurol. 2021;89(4):780–9.
Baker D, Amor S, Kang AS, Schmierer K, Giovannoni G. The underpinning biology relating to multiple sclerosis disease modifying treatments during the COVID-19 pandemic. Mult Scler Relat Disord. 2020;43:102174.
Berger JR, Brandstadter R, Bar-Or A. COVID-19 and MS disease-modifying therapies. Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e761.
Hartung H-P, Aktas O. COVID-19 and management of neuroimmunological disorders. Nat Rev Neurol. 2020;16(7):347–8.
Magyari M, Sorensen PS. The changing course of multiple sclerosis: rising incidence, change in geographic distribution, disease course, and prognosis. Curr Opin Neurol. 2019;32(3):320–6.
Marrie RA, Yu N, Blanchard J, Leung S, Elliott L. The rising prevalence and changing age distribution of multiple sclerosis in Manitoba. Neurology. 2010;74(6):465.
Vaughn CB, Jakimovski D, Kavak KS, Ramanathan M, Benedict RHB, Zivadinov R, et al. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat Rev Neurol. 2019;15(6):329–42.
Ostolaza Ibanez A, Corroza Lavineta J, Ayuso Blanco T. Multiple sclerosis and aging: comorbidity and treatment challenges. Mult Scler Relat Disord. 2021;50:102815.
Hua LH, Hersh CM, Tian F, Mowry EM, Fitzgerald KC. Clinical characteristics of a large multi-center cohort of people with multiple sclerosis over age 60. Mult Scler Relat Disord. 2021;47:102637.
Grebenciucova E, Berger JR. Immunosenescence: the role of aging in the predisposition to neuro-infectious complications arising from the treatment of multiple sclerosis. Curr Neurol Neurosci Rep. 2017;17(8):61.
Costantini E, D’Angelo C, Reale M. The role of immunosenescence in neurodegenerative diseases. Mediators Inflamm. 2018;2018:6039171.
Rodriguez IJ, Lalinde Ruiz N, Llano León M, Martínez Enríquez L, Montilla Velásquez MDP, Ortiz Aguirre JP, et al. Immunosenescence study of T cells: a systematic review. Front Immunol. 2021;11:604591.
Schweitzer F, Laurent S, Fink GR, Barnett MH, Reddel S, Hartung H-P, et al. Age and the risks of high-efficacy disease modifying drugs in multiple sclerosis. Curr Opin Neurol. 2019;32(3):305–12.
Vandenbark AA, Huan J, Agotsch M, La Tocha D, Goelz S, Offner H, et al. Interferon-beta-1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis. J Neuroimmunol. 2009;215(1–2):125–8.
Gajofatto A, Bacchetti P, Grimes B, High A, Waubant E. Switching first-line disease-modifying therapy after failure: impact on the course of relapsing–remitting multiple sclerosis. Mult Scler. 2009;15(1):50–8.
Rudick RA, Lee JC, Cutter GR, Miller DM, Bourdette D, Weinstock-Guttman B, et al. Disability progression in a clinical trial of relapsing-remitting multiple sclerosis: eight-year follow-up. Arch Neurol. 2010;67(11):1329–35.
Simon JH, Jacobs LD, Campion M, Wende K, Simonian N, Cookfair DL, et al. Magnetic resonance studies of intramuscular interferon beta-1a for relapsing multiple sclerosis. Ann Neurol. 1998;43(1):79–87.
Piehl F. A changing treatment landscape for multiple sclerosis: challenges and opportunities. J Intern Med. 2014;275(4):364–81.
Krause CD, Pestka S. Evolution of the Class 2 cytokines and receptors, and discovery of new friends and relatives. Pharmacol Ther. 2005;106(3):299–346.
Acknowledgements
Medical writing support for the development of this manuscript, under the direction of the authors, was provided by John Watson, PhD, of Ashfield MedComms, an Ashfield Health company, and funded by Biogen. Joshua Safran of Ashfield MedComms copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the manuscript to the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Biogen provided funding for medical writing in the development of this manuscript and for payment of open-access fees.
Conflict of interest
Stanley L. Cohan has received research support from AbbVie, Biogen, Mallinckrodt, MedDay, Novartis, Roche Genentech, and Sanofi Genzyme and has received compensation for serving as a consultant, advisory board member, or paid speaker for AbbVie, Bristol Myers Squibb, Biogen, EMD Serono, Novartis, Roche Genentech, and Sanofi Genzyme. Barry A. Hendin is a paid consultant and/or speaker for Acorda, Biogen, EMD Serono, Genzyme, Mallinckrodt, Novartis, and Teva. Anthony T. Reder has received personal compensation for consulting, serving on a scientific advisory board, clinical trials, or other activities from AbbVie, Bayer, Biogen, Genentech, Mallinckrodt, EMD Serono, Novartis, and TG Therapeutics and has received personal compensation in an editorial capacity from MedLink. Kyle Smoot has received research support from Biogen, IMS Health, and MedDay and has received speaking and consulting fees from Biogen, EMD Serono, Genentech, Genzyme, Novartis, and Teva. Robin Avila and Jason P. Mendoza are employees of and may own stock and/or stock options in Biogen. Bianca Weinstock-Guttman has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities from AbbVie, Biogen, Bristol Myers-Squibb, EMD Serono, Genentech, Mallinckrodt, and Novartis and has received research support from Biogen, EMD Serono, Genentech, and Novartis.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors' contributions
All authors participated in conceptualization of the review and interpretation of the literature and in critical review and revision of the manuscript. All authors read and approved the final version of the manuscript. All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this manuscript and take responsibility for the integrity of the work as a whole.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cohan, S.L., Hendin, B.A., Reder, A.T. et al. Interferons and Multiple Sclerosis: Lessons from 25 Years of Clinical and Real-World Experience with Intramuscular Interferon Beta-1a (Avonex). CNS Drugs 35, 743–767 (2021). https://doi.org/10.1007/s40263-021-00822-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00822-z