
Abstract
Background
Walking impairment is a hallmark of multiple sclerosis (MS). It affects > 90% of individuals over time, reducing independence and negatively impacting health-related quality of life, productivity, and daily activities. Walking impairment is consistently reported as one of the most distressing impairments by individuals with MS. Prolonged-release (PR)-fampridine previously has been shown to improve objectively measured walking speed in walking-impaired adults with MS. The impact of PR-fampridine from the perspective of the individual with MS warrants full and detailed examination.
Objective
The objective of this study was to evaluate whether PR-fampridine has a clinically meaningful effect on self-reported walking ability in walking-impaired participants with MS.
Methods
ENHANCE was a phase III, randomized, double-blind, placebo-controlled study of PR-fampridine 10 mg twice daily in walking-impaired individuals age 18–70 years with either relapsing or progressive forms of MS and an Expanded Disability Status Scale (EDSS) score of 4.0–7.0 at screening. Participants were stratified by EDSS score (≤ 6.0 or 6.5–7.0) at randomization to ensure a balanced level of disability in the treatment groups. The primary endpoint was the proportion of participants with a mean improvement in the 12-item Multiple Sclerosis Walking Scale (MSWS-12) score exceeding the predefined threshold for clinically meaningful improvement (≥ 8 points) over 24 weeks. Secondary endpoints included the proportion with ≥ 15% improvement in Timed Up and Go (TUG) speed, and mean changes in Multiple Sclerosis Impact Scale physical impact subscale (MSIS-29 PHYS), Berg Balance Scale (BBS), and ABILHAND scores over 24 weeks.
Results
In total, 636 participants with MS were randomized (PR-fampridine, n = 317; placebo, n = 319; modified intention-to-treat sample: PR-fampridine, n = 315; placebo, n = 318). At baseline in the PR-fampridine and placebo groups, 46% and 51% had a progressive form of MS, median [range] EDSS scores were 6.0 [4.0–7.0] and 5.5 [4.0–7.0], mean [range] MSWS-12 scores were 63.6 [0–100] and 65.4 [0–100], and mean [range] TUG speed was 0.38 [0.0–1.0] and 0.38 [0.0–1.2] feet/s, respectively. A significantly higher percentage of PR-fampridine-treated participants (136/315 [43.2%]) had clinically meaningful improvement in MSWS-12 score over 24 weeks versus placebo (107/318 [33.6%]; odds ratio 1.61 [95% confidence interval 1.15–2.26]; p = 0.006). For PR-fampridine versus placebo, significantly more participants had a ≥ 15% improvement in TUG speed, and there was significantly greater mean improvement in MSIS-29 PHYS score (p < 0.05); numerical improvements that were not statistically significant were observed in BBS/ABILHAND. Adverse events that were more common in the PR-fampridine group than placebo group (difference ≥ 3%) by Medical Dictionary for Regulatory Activities (MedDRA®) Preferred Term were urinary tract infection and insomnia. There were no seizures reported.
Conclusions
PR-fampridine treatment resulted in sustained, clinically meaningful improvements over 24 weeks in self-reported walking and functional ability in walking-disabled participants with MS.
ClinicalTrials.gov Identifier
NCT02219932.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Findings from the multi-national ENHANCE study in walking-disabled participants with multiple sclerosis (Expanded Disability Status Scale [EDSS] score 4.0−7.0) showed that participants treated with prolonged-release (PR)-fampridine 10 mg twice daily were more likely than those treated with placebo to achieve clinically meaningful improvements in self-reported walking ability over 24 weeks. |
PR-fampridine was also associated with benefits in objectively measured mobility and self-reported physical functioning. |
Additional research is required to better understand the pathophysiologic differences in individuals who do and do not respond to PR-fampridine and to evaluate the impact of PR-fampridine on manual function, cognition, and fatigue in individuals with an EDSS score > 7.0. |
1 Introduction
Impaired walking is a hallmark of multiple sclerosis (MS); 93% of individuals with MS are walking impaired within 10 years of diagnosis [1]. Impaired walking and mobility have profoundly deleterious effects on independence, health-related quality of life, daytime functioning, and productivity [1, 2]. Maintaining mobility is a high priority for individuals with MS, irrespective of disease duration and disability level [2, 3].
Prolonged-release (PR)-fampridine (dalfampridine extended-release tablets in the USA), a PR formulation of 4-aminopyridine, is a twice-daily oral treatment indicated to improve walking in individuals with MS [4]. PR-fampridine is thought to improve conduction in demyelinated pathways by blocking voltage-dependent potassium channels [5]. Two pivotal phase III studies of PR-fampridine showed that walking speed, as measured by the Timed 25-Foot Walk (T25FW), improved in PR-fampridine responders (defined as participants with faster walking speed for at least three of four visits during the on-treatment period vs. the maximum speed from five off-drug visits) over 14 weeks [6] and 9 weeks [7] of treatment. These trials included individuals with clinically definite MS of any disease course who had objectively measured deficits in walking speed (i.e., T25FW time of 8–45 s) [6, 7]. Although these pivotal data were the foundation for the approval of PR-fampridine in the USA and European Union [4, 8], they were limited in terms of demonstrating the duration of effect and offered the opportunity to further build on the clinical meaningfulness of PR-fampridine. Subsequent studies, including the 12-month ENABLE and 6-month MOBILE studies [9, 10], showed that PR-fampridine also had beneficial effects on a broad range of other clinical and self-reported outcome assessments, including walking, balance, and aspects of life quality over longer treatment periods [9,10,11,12,13].
ENABLE was a single-arm, open-label study that showed that PR-fampridine was associated with statistically significant long-term improvements in self-perceived physical functioning and psychological health over time [9]. The exploratory randomized, double-blind, placebo-controlled MOBILE study was designed to further assess the effects of PR-fampridine beyond the 14-week period evaluated in the longest pivotal study, evaluate self-reported walking ability, and identify a clinically meaningful change threshold in the 12-item Multiple Sclerosis Walking Scale (MSWS-12) [10]. At entry to the study, MOBILE participants had a clinical diagnosis of MS of any course for at least 3 months’ duration with EDSS scores of 4.0–7.0 [10]. The T25FW test was not used as a screening measure in MOBILE, because this was considered to have been covered by previous studies and has implications for study design. Thus, MOBILE participants could have had any walking speed [10]. Results from MOBILE demonstrated that treatment with PR-fampridine resulted in early improvements in mobility and balance compared with placebo that were sustained over the 6-month treatment period [10]. Additionally, data from the MOBILE study were used to estimate the threshold for a patient-level clinically meaningful improvement in MSWS-12 score (i.e., ≥ 8-point mean score reduction) [14]. This threshold of improvement was then used as the primary endpoint for ENHANCE.
The novelty of ENHANCE over previous clinical studies of PR-fampridine is that it was designed to assess the effect of pharmacotherapy on the proportion of participants achieving a criterion for clinically meaningful change in walking using a self-reported outcome measure. The MSWS-12 assesses aspects of walking not captured by objective assessments [15]. While previous studies have shown benefits of PR-fampridine on self-reported outcomes, including walking ability [9, 10], ENHANCE was the first study that included formal statistical hypothesis testing in the setting of a rigorous study design (i.e., randomized, double-blind, placebo-controlled) to evaluate clinically meaningful improvement on a self-reported outcome. Furthermore, another objective of ENHANCE was to evaluate some of the broader effects of PR-fampridine that have been reported by patients in MS clinics using an expanded range of clinical outcome assessments over a longer treatment period. The main objective of the ENHANCE study was to determine whether PR-fampridine 10 mg twice daily has a clinically meaningful effect on self-reported walking ability when compared with placebo over 6 months of treatment.
2 Methods
2.1 Study Design
ENHANCE was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase III study of PR-fampridine versus placebo in participants with MS who had walking impairment. The study consisted of a 2-week screening period, a 24-week double-blind treatment period, and a 2-week post-dosing follow-up visit and was carried out at 92 centers in 11 countries (Fig. S1 and Table S1 in the Electronic Supplementary Material [ESM]). Independent ethics committees or institutional review boards approved the study protocol and all amendments. The trial was registered with ClinicalTrials.gov (NCT02219932). The first participant was treated on 29 September 2014 and the last participant’s last visit was 11 February 2016.
Participants were randomized (1:1) to PR-fampridine 10 mg twice daily or matched placebo for 24 weeks, and were stratified by Expanded Disability Status Scale (EDSS) score (≤ 6.0 or 6.5–7.0) according to a predefined randomization list to ensure a balanced level of disability. There was no placebo run-in phase. The protocol was amended on 3 December 2014 to add stratification by prior aminopyridine use (yes/no) because of concerns regarding potential bias. Enrollment caps were added based on stratification factors: enrollment of participants with prior aminopyridine use was limited to ~ 10% of the overall study population; enrollment of participants with an EDSS score > 6.0 was limited to ~ 35% of the overall study population. All participants, investigators, site personnel, and funder personnel were masked to treatment assignment.
Requests for the data supporting this study should be submitted to the Biogen Clinical Data Request Portal (www.biogenclinicaldatarequest.com).
2.2 Participants
Participant eligibility was assessed by a treating neurologist during a 14-day screening period. Key inclusion criteria were age 18–70 years, diagnosis of MS (any subtype), EDSS score of 4.0–7.0, and investigator-assessed walking impairment. Key exclusion criteria were recent exacerbation of MS (within 60 days of screening visit), recent initiation/change in the dosing of approved immunomodulatory therapies, and any history of seizure, epilepsy, or other convulsive disorder. The Methods section of the ESM reports full inclusion and exclusion criteria. Concomitant use of approved disease-modifying therapies and medications for fatigue or spasticity were allowed if the drug and dose remained stable throughout the study; physiotherapy and rehabilitation therapy were also allowed.
2.3 Assessments and Endpoints
Measurements of walking ability, physical impact of MS, balance, and manual ability were collected prospectively with widely used self-reported questionnaires (MSWS-12 [16], Multiple Sclerosis Impact Scale physical impact subscale [MSIS-29 PHYS] [17], and 56-item ABILHAND for manual ability [18]), performance measures (Timed Up and Go [TUG] speed [19]), and clinician-reported outcomes (Berg Balance Scale [BBS] [20, 21]). Participants completed questionnaires up to ten times and had at least ten clinic visits during the 26-week study. The MSWS-12, TUG, and MSIS-29 PHYS were evaluated at screening, day 1, and weeks 2, 4, 8, 12, 16, 20, and 24 during the 24-week double-blind treatment period. The BBS was collected at screening, day 1, and weeks 2, 12, and 24. The ABILHAND was collected at day 1 and weeks 2, 8, and 20. The MSWS-12 and TUG were also evaluated at the 2-week post-dose follow-up (Table 1). Translated questionnaires were provided by licensees where available (MSIS-29 PHYS and MSWS-12: Plymouth University, Plymouth, UK; TUG: American College of Rheumatology, Atlanta, GA, USA; ABILHAND: Catholic University of Louvain, Louvain-la-Neuve, Belgium).
The primary endpoint was the proportion of participants with a mean improvement in MSWS-12 score of ≥ 8 points [14] from baseline over 24 weeks, where improvement was defined as a decrease in score [14]. Clinically meaningful improvement in MSWS-12 score was previously estimated as an ≥ 8-point mean score reduction at the level of the individual based on triangulation of values obtained from both anchor- and distribution-based analyses using data from the MOBILE study [14]. Mean improvement in MSWS-12 was determined by calculating the mean change (i.e., mean on-treatment score over weeks 2–24 minus mean baseline score). MSWS-12 scores were transformed to a 0–100 scale.
Secondary endpoints were assessed over 24 weeks and rank ordered into two groups as a hierarchical testing approach. Rank group 1 secondary endpoints were the proportion of participants with a mean improvement in TUG speed of ≥ 15% from baseline, and mean change from baseline in MSIS-29 PHYS score (range 0–100). The threshold for a clinically important change in TUG speed was determined using data from the MOBILE study [10] and methods similar to those used in determining the threshold for a clinically important change for the MSWS-12 [14]. Both anchor- and distribution-based methods were used to determine what percentage improvement was clinically meaningful on the TUG speed at the level of the individual. Improvement on the Patient Global Impression of Change (PGIC) and a ≥ 8-point improvement on the MSWS-12 were used as anchors. The median percentage change in TUG speed in MOBILE study participants who had had one or more visits where they reported a score of ‘slightly improved’ on the PGIC was 16.83% (n = 81). The median percentage of change in TUG speed in MOBILE study participants who had a ≥ 8-point improvement in the MSWS-12 was 17.53% (n = 54). The distribution-based estimate, which uses estimates of measurement error based on within and between participant variability, was calculated using the standard error of measurement (SEM) = standard deviation (SD) × \(\sqrt{(1-{\text{Reliability}})}\) and was 10.0. The average of the three estimates was 15% and was used as the threshold for determining a clinically meaningful improvement in TUG speed in ENHANCE. Rank group 2 endpoints were the mean changes in BBS score (range 0–56) and ABILHAND score (range 0–100).
TUG speed, a performance mobility measure, was included, as it complements the self-reported MSWS-12, correlates with the T25FW (r = 0.85) [22], and detects changes in moderately impaired individuals with some precision [22, 23]. An exploratory analysis of TUG time was performed. The self-reported 20-item MSIS-29 PHYS assessed the physical impact of MS, the clinician-reported 14-item BBS measured static and dynamic balance, and the self-reported 56-item ABILHAND evaluated manual ability. Table 1 provides an overview of the questionnaires and estimated clinically important changes.
Subgroup analyses of MSWS-12 scores included assessment of PR-fampridine efficacy versus placebo in participants with lower (baseline EDSS score ≤ 6.0) or greater (EDSS score 6.5, 7.0) disability. Post hoc analyses evaluated ABILHAND data in participants with normal (≥ 80) [24] or abnormal (< 80) scores at baseline; results were compared between treatment groups.
Post hoc analyses of all outcomes were also conducted for those who responded to PR-fampridine as measured by MSWS-12 score (vs. PR-fampridine non-responders and placebo) to determine if these participants responded in other measurement domains. A PR-fampridine MSWS-12 responder was defined as an participant with a ≥ 8-point mean improvement in MSWS-12 score from baseline over 24 weeks (see the Methods section in the ESM for additional details).
Safety was evaluated via physical examination, electrocardiograms, vital signs, clinical laboratory tests, and adverse event (AE) reporting. A treatment-emergent AE was defined as any AE with an onset date on or after the first dose of study treatment, or any pre-existing condition that worsened in severity after the first dose of study treatment. A serious AE was any untoward medical occurrence that resulted in death/risk of death, hospitalization or prolonged hospitalization, persistent or significant disability/incapacity, or in a congenital anomaly/birth defect. AEs were spontaneously reported by participants and recorded using a specific AE collection form within the case report form. Serious AEs had to be reported to the sponsor within 24 h of the study staff becoming aware of the event. All AEs were recorded using Medical Dictionary for Regulatory Activities (MedDRA®; version 18.1) terms. Confirmatory urinary tract infection cultures were evaluated wherever possible to rule out infection or confirm bacterial infection. Compliance with dosing of study drug was calculated based on the number of days study drug was taken (number of tablets dispensed minus the number returned divided by 2) divided by the number of days of exposure multiplied by 100.
2.4 Statistical Analysis
Efficacy analyses were based on the modified intention-to-treat (mITT) sample, which comprised randomized participants who received at least one dose of study drug and had at least one post-baseline efficacy assessment. The planned sample size of 590 randomized participants was to provide ≥ 90% power at a two-sided 5% significance level and detect a minimum of 14.5% absolute improvement in the on-treatment response rate (i.e., ≥ 8-point mean improvement in MSWS-12 score over 24 weeks) for the PR-fampridine versus placebo groups, with an assumed 15% dropout rate. Data from one site that enrolled ten participants were deemed unreliable due to serious Good Clinical Practice non-compliance and were excluded from the analyses before unblinding. Pre-specified sensitivity analyses were conducted to compare the results with and without the data from these ten participants, which revealed no appreciable differences across all endpoints.
The pre-specified hypothesis test of the primary endpoint, PR-fampridine treatment effect (proportion of participants with a clinically significant ≥ 8-point mean improvement in MSWS-12 score), was based on a logistic regression model with treatment group as the classification variable and baseline MSWS-12 score, baseline TUG speed, age, screening EDSS score, and prior aminopyridine use as covariates (to increase the precision of the analysis and provide an unbiased estimate of treatment effect). A multiple imputation method [25] (50 times) was used to impute missing individual post-baseline MSWS-12 scores before deriving the primary endpoint. The electronic device used to administer the MSWS-12 did not allow component questions to go unanswered, so imputation was not required for individual MSWS-12 item scores. Therefore, imputation was only performed on assessments that were completely missing for the timepoint. The reasons for missing MSWS-12 data were: participant did not attend a study visit, participant was lost to follow-up or prematurely discontinued from the study, or participant attended the visit but responded ‘cannot walk at all’ on the MSWS-12, or the MSWS-12 was not performed or missing because of technical difficulties at the site. Baseline was defined as the mean score over screening and day 1.
A hierarchical testing approach was used to protect the overall type I error rate for the four secondary endpoints. Secondary endpoints were pre-specified as rank group 1 or rank group 2, as described in Sect. 2.3. Within each rank group, the statistical test was performed using the Hochberg procedure. Statistical tests for rank group 2 could only have been conducted if the tests for rank group 1 reached an overall p value threshold of 0.05 with the Hochberg adjustment.
The proportion of participants with ≥ 15% mean improvement in TUG speed was analyzed similarly to the primary endpoint. The changes from baseline over 24 weeks in MSIS-29 PHYS, BBS, and ABILHAND scores were analyzed using a mixed-effects model for repeated measures, with treatment group as the classification variable. Covariates in the model were baseline values for each measure, visit-by-treatment interaction, screening EDSS score, and prior aminopyridine use. Missing values were imputed using the multiple imputation method (50 times).
Subgroup analyses were performed using a similar model for each level of subgroup. Analyses of PR-fampridine MSWS-12 responders (vs. PR-fampridine MSWS-12 non-responders and placebo) used a similar model for each endpoint, except the responder group (PR-fampridine MSWS-12 responders, PR-fampridine MSWS-12 non-responders, and placebo) was specified in the model as a classification variable instead of a treatment group. A separate analysis was conducted on each endpoint for PR-fampridine MSWS-12 responders versus placebo MSWS-12 responders and PR-fampridine MSWS-12 non-responders versus placebo MSWS-12 non-responders (see Methods section in the ESM). Raw mean (SD) scores and floor and ceiling effects were calculated, along with Cohen’s effect size (evaluated using both mean change from baseline divided by pre-treatment SD and mean change from baseline divided by pooled SD) and standardized response mean (mean change from baseline divided by SD change from baseline). These scores were presented by treatment group and for PR-fampridine MSWS-12 responders and PR-fampridine MSWS-12 non-responders.
Safety analyses were based on the safety sample (i.e., all participants randomized and exposed to study drug), excluding participants from one site for the reasons described earlier. Any AE with a missing onset date and a resolution date after the first dose of study treatment was considered treatment emergent.
Statistical software (SAS® 9.4; SAS Institute Inc.; Cary, NC, USA) generated all summaries and statistical analyses.
3 Results
3.1 Participant Characteristics
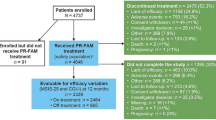
In total, 758 participants were screened and 636 randomized (Fig. 1). One participant randomized to PR-fampridine did not receive treatment. Of the 635 participants (safety sample), 633 completed at least one on-treatment efficacy assessment and were included in the mITT analyses (PR-fampridine, n = 315; placebo, n = 318). Most participants completed 24 weeks of treatment (PR-fampridine, 271/317 [85%]; placebo, 258/319 [81%]).

Participant disposition. AE adverse event, BID twice daily, PR prolonged-release. a1 patient randomized to PR-fampridine withdrew from the study before dosing (reason: consent withdrawn). bPatients who completed the 24-week double-blind treatment period and 2-week off-treatment follow-up visit calculated as follows: 317 − 51 = 266. A total of 271 participants completed the 24-week treatment period (Weeks 0–24). cPatients who completed the 24-week double-blind treatment period and 2-week off-treatment follow-up visit calculated as follows: 319 − 65 = 254. A total of 258 participants completed the 24-week treatment period (Weeks 0–24)
Baseline characteristics were similar between groups (Table 2); the most commonly used immunomodulatory medications were glatiramer acetate, fingolimod, interferon-β-1a, and natalizumab. At baseline, the treatment groups were balanced with respect to EDSS scores, distance walked, and MS-related symptoms that potentially affect walking ability (Table 2). Mean baseline EDSS scores implied disability severe enough to preclude full daily activities. Baseline MSWS-12 scores and TUG speed indicated moderately poor mobility. Concomitant medication and non-drug therapy use during the study were similar in the PR-fampridine and placebo groups, including anti-spasmodics and physiotherapy (Table S2 in the ESM).
Based on the mITT sample (n = 633), the following levels of post-baseline data were missing and were imputed for efficacy outcomes: MSWS-12 score: 12% (PR-fampridine, 10%; placebo, 14%); TUG speed: 9% (PR-fampridine, 7%; placebo, 12%); MSIS-29 PHYS score: 9% (PR-fampridine, 7%; placebo, 11%); BBS score: 9% (PR-fampridine, 8%; placebo, 11%); and ABILHAND score: 9% (PR-fampridine, 8%; placebo, 11%).
3.2 Primary Efficacy Analyses
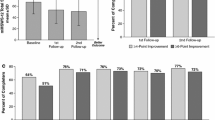
A significantly greater proportion of participants in the PR-fampridine group (43.2%) versus the placebo group (33.6%) had a clinically meaningful improvement in mean MSWS-12 score (odds ratio 1.61; 95% confidence interval [CI] 1.15–2.26; p = 0.006), which was the primary endpoint of the study (Table 3). Analysis of the primary endpoint using observed data without imputation provided similar findings. Figure 2 shows the cumulative proportion of MSWS-12 responders, which plots the proportion of responders for a range of responder threshold definitions. For every integer of MSWS-12 score point change from 0 to 10, the PR-fampridine group had a higher proportion of MSWS-12 responders. MSWS-12 score improvement ≥ 10 points was achieved by 38% of PR-fampridine and 27% of placebo-treated participants (p = 0.003). This implied a consistent treatment benefit with PR-fampridine regardless of responder definition. Figure 3 shows least squares mean (LSM) changes from baseline in MSWS-12 score per visit, showing improvements in MSWS-12 scores observed as early as 2 weeks after treatment initiation with benefits compared with placebo across the 24-week treatment period. Within 2 weeks of stopping PR-fampridine treatment, the effects of PR-fampridine on the MSWS-12 dissipated (Fig. 3). Participants in the PR-fampridine group had a LSM improvement in MSWS-12 score from baseline over the 24-week double-blind treatment period of 6.73 points versus an improvement of 2.59 points in the placebo group, a treatment difference that was statistically significant (LSM improvement vs. placebo 4.14 [95% CI − 6.22 to − 2.06]; p < 0.001; Table 3).

Estimated proportion of study participants who met each threshold of mean MSWS-12 score change over 24 weeks in the modified intention-to-treat sample. The MSWS-12 was transformed to a 0–100 scale; higher score = greater walking limitation. Negative change indicates improvement. Estimated percentages were based on binomial proportions. Multiple imputation was used for missing post-baseline data. Nominal p-values for PR-fampridine vs. placebo are from a logistic regression model adjusted for covariates (see Sect. 2). MSWS-12 12-item Multiple Sclerosis Walking Scale, PR prolonged-release

LSM changes in MSWS-12 score over 24 weeks in the modified intention-to-treat sample. Negative change indicates improvement. Multiple imputation was used for missing post-baseline data except for during follow-up where observed data were used. Error bars indicate SE. DB double-blind, LSM least squares mean, MSWS-12 12-item Multiple Sclerosis Walking Scale, PR prolonged-release, SE standard error
3.3 Secondary and Other Clinical Efficacy Analyses
A significantly higher proportion of participants in the PR-fampridine group (43.4%) versus the placebo group (34.7%) had a clinically meaningful improvement in TUG speed (≥ 15% mean increase from baseline [odds ratio 1.46; 95% CI 1.04–2.07; p = 0.03]). TUG speed improved from baseline over 24 weeks by a LSM of 0.05 feet/s in the PR-fampridine group compared with 0.03 feet/s in the placebo group (LSM treatment difference 0.02 [95% CI 0.01–0.03]; p < 0.001; Table 3). The PR-fampridine group also demonstrated significantly greater LSM improvements from baseline in MSIS-29 PHYS score (8.00 points) versus placebo (4.68 points) over 24 weeks (LSM improvement vs. placebo 3.31 [95% CI − 5.13 to − 1.50]; p < 0.001; Table 3). These results demonstrated significant improvements in both objective and subjective measures of functioning.
The rank 2 secondary endpoints of change in BBS and ABILHAND scores from baseline over 24 weeks were not statistically significant, although there were numerically greater improvements in both assessments in the PR-fampridine group than in the placebo group (Table 3). BBS scores improved from baseline over 24 weeks by an LSM of 1.75 points in the PR-fampridine group versus 1.34 in the placebo group, although the LSM treatment difference was not statistically significant (0.41 [95% CI − 0.13 to 0.95]; p = 0.141; Table 3). Similarly, ABILHAND scores improved from baseline by a LSM of 1.49 points in the PR-fampridine group and by 0.75 points in the placebo group over 24 weeks, which resulted in a non-significant LSM treatment difference of 0.74 (95% CI − 0.38 to 1.86; p = 0.197; Table 3).
Figure 4 shows the pre-specified analyses of percentage change in TUG speed per visit. The LSM percentage change from baseline over 24 weeks in TUG speed was greater with PR-fampridine than placebo (15.9% vs. 11.8%; LSM difference 4.17 [95% CI 0.43–7.91]; p = 0.029). LSM TUG time decreased from baseline over 24 weeks by − 3.30 s in the PR-fampridine group and by − 1.94 s in the placebo group (LSM treatment difference − 1.36 [95% CI − 2.85 to 0.12]; p = 0.073). Similar to what was observed on the MSWS-12, improvements in TUG speed subsided when treatment with PR-fampridine was stopped (Fig. 4).

LSM percentage change in TUG speed over 24 weeks in the modified intention-to-treat sample. TUG speed is given in feet/s. Positive change indicates improvement. Multiple imputation was used for missing post-baseline data except for during follow-up where observed data were used. Error bars indicate SE. DB double-blind, LSM least squares mean, PR prolonged-release, SE standard error, TUG Timed Up and Go
Table S3 in the ESM shows the distributional statistics of the PR-fampridine and placebo groups for all the reported efficacy outcomes in the mITT groups at baseline and on treatment, and change from baseline scores. Two effect size calculations (standardized change scores) are also reported. These calculations quantify the magnitude of the treatment effect contextualized by variance at baseline (Cohen’s effect size), or the variance of change (standardized response mean). Standardizing the change scores enables a meaningful comparison across different clinical outcome assessments with varying measurement processes. Both effect sizes were interpreted using Cohen’s widely used criteria [26], which are thresholds for small (> 0.20), moderate (> 0.50), and large (> 0.80) clinical change. Effect sizes were consistently greater for the PR-fampridine group versus placebo, with differences implying a clinically small to moderate change.
3.4 Efficacy Subgroup Analyses
Table 4 shows a pre-specified subgroup analysis of ABILHAND, including the LSM change stratified by disability level (EDSS score ≤ 6.0 vs. 6.5 and 7.0). While the differences between the PR-fampridine and placebo groups were not statistically significant, there were small numerical improvements that were larger in the more disabled participants. Further post hoc subgroup analyses examined improvements in manual ability, stratified by baseline ABILHAND score: ‘normal’ was ≥ 80; ‘abnormal’ was < 80. As predicted clinically, greater numerical improvements were observed in participants with abnormal manual ability. Formal significance testing was not undertaken for subgroup analyses.
3.5 Multiple Sclerosis Walking Scale (MSWS-12) Responder Efficacy Analyses
Table 5 compares outcomes of the post hoc PR-fampridine MSWS-12 responders with PR-fampridine non-responders and the placebo group. Here, an MSWS-12 responder was defined as an individual from the PR-fampridine group with an improvement (decrease) in MSWS-12 score of ≥ 8 points from baseline. PR-fampridine MSWS-12 responders had greater benefits than PR-fampridine MSWS-12 non-responders and the placebo group across all the efficacy outcome measures. The size of the numerical differences between the PR-fampridine MSWS-12 responder, MSWS-12 non-responder, and placebo groups varied (Table 5). Because the MSWS-12 responder analyses were conducted in non-randomized groups, significance testing was not undertaken.
Table S3 in the ESM shows the effect sizes for PR-fampridine MSWS-12 responders and non-responders. PR-fampridine MSWS-12 responders had clinically large improvements in MSWS-12 and MSIS-29 PHYS scores, and clinically small to moderate improvements in BBS and ABILHAND scores. Improvements observed in PR-fampridine MSWS-12 responders exceeded those of PR-fampridine MSWS-12 non-responders and placebo, except for TUG time. These findings indicate a clinical effect across a range of mobility and non-mobility parameters.
Table S4 in the ESM shows additional post hoc analyses of both the PR-fampridine and placebo groups based on MSWS-12 response. These results demonstrated benefits for PR-fampridine MSWS-12 responders across the main mobility outcome measures. PR-fampridine MSWS-12 responders showed higher numerical improvements across all the reported efficacy endpoints compared with other treatment groups. Significance testing was not performed on these MSWS-12 responder analyses.
3.6 Safety Results
Table 6 shows that treatment-emergent AEs, serious treatment-emergent AEs, and AEs leading to treatment discontinuation were similar in the PR-fampridine and placebo groups. AEs that were more common in PR-fampridine-treated participants (difference vs. placebo ≥ 3%) by MedDRA® Preferred Term were urinary tract infection (13% vs. 9%) and insomnia (4% vs. < 1%). Urine culture was performed in 53 participants in the PR-fampridine group and 33 in the placebo group; culture-positive urinary tract infections were reported in similar proportions of participants in each group (PR-fampridine: 8 [15%]; placebo: 4 [12%]). Among participants who reported an AE within the category of urinary tract infections (PR-fampridine, n = 56; placebo, n = 37; see Table 6), 28 participants in the PR-fampridine group and 15 participants in the placebo group had a urine culture performed and eight and four were positive, respectively.
No seizures or convulsions were reported. There were four deaths. One death occurred in each treatment group during the 2-week study follow-up period and was a result of coronary artery stenosis in the PR-fampridine group and acute myocardial infarction in the placebo group. These deaths were considered unrelated to study treatment by the investigators. One death also occurred in each treatment group ≥ 20 days after the last dose of study treatment. The death in the PR-fampridine group was a result of lung cancer with liver and brain metastasis and the death in the placebo group was a result of ovarian endometrioid carcinoma; both were considered unrelated to study treatment by the investigators.
Mean (SD) compliance with dosing of study drug was 98.7% (3.9%) in the PR-fampridine group and 98.4% (4.6%) in the placebo group for the ITT population (n = 633). Mean compliance rates were the same as the above for each treatment group in the safety population (n = 635).
4 Discussion
ENHANCE was one of the first studies to use a previously defined criterion of clinically meaningful change on a self-reported outcome as its primary endpoint. Results from ENHANCE show that PR-fampridine treatment resulted in a higher proportion of participants achieving clinically meaningful improvements in self-reported walking ability, clinician-measured mobility, and self-reported physical impact of MS over 24 weeks compared with placebo. These effects of PR-fampridine were statistically significant when compared with placebo. Placebo-treated participants demonstrated some benefits on efficacy measures, but the magnitude of improvement was consistently greater with PR-fampridine.
Results from ENHANCE not only demonstrate that PR-fampridine has clinically meaningful effects on self-reported walking, balance, and physical functioning in comparison to placebo, but also that a self-reported outcome measure can be used as a sensitive primary endpoint in a controlled clinical trial evaluating treatment effects. Similar to previous studies [10, 13, 27, 28], benefits from PR-fampridine over placebo were apparent as early as 2 weeks after treatment initiation and were sustained over 24 weeks of treatment in ENHANCE. The fast action of PR-fampridine supports the use of the MSWS-12, which is based on the experience of the previous 2 weeks, in this study. Of the multiple studies that have assessed the efficacy of PR-fampridine, ENHANCE was the largest, most geographically diverse trial to date, and at least 10 weeks longer than the pivotal phase III trials [6, 7]. Together, these studies provide a consistent body of evidence demonstrating that PR-fampridine is a beneficial treatment for a proportion of individuals with disabling MS [10, 13, 27, 28].
ENHANCE explored the effect of PR-fampridine on manual ability. Improvements in the ABILHAND were small and the treatment difference between PR-fampridine and placebo was not statistically significant. ABILHAND subgroup analyses showed greater numerical benefits for PR-fampridine than placebo in participants with greater clinician-rated disability (EDSS) and worse self-reported hand function, but formal significant testing was not performed for subgroup analyses. Because deterioration of manual dexterity occurs with increasing disability [29], this could explain why more impaired participants would have a greater potential to show improvements in ABILHAND scores. Pre-treatment ABILHAND score distributions were skewed towards fewer functional limitations (the scale’s ‘ceiling’). Therefore, the sample had less potential for measurable change in manual ability, a limitation of the ABILHAND that has been shown previously in individuals with MS [30, 31]. This suboptimal sample-to-scale targeting in less disabled participants [30, 31] may have contributed to the lack of significant treatment effect in ENHANCE.
Changes in dynamic and static balance (BBS score) in the PR-fampridine and placebo groups were similar over 24 weeks and the treatment difference was not statistically significant, consistent with previous findings [10, 12]. Similar to the ABILHAND, pre-treatment BBS score distributions were skewed towards better balance (the BBS’s ceiling), limiting its ability to measure change [32]. Participants experiencing improved balance with PR-fampridine would have subsequently moved further towards the scale’s ceiling, where the ability to detect change is weakened and the ability to convert ‘true’ change in dynamic and static balance to a change in BBS score may have been reduced [30,31,32]. Additional post hoc exploration of the BBS is warranted to determine if the results were a true reflection of the impact of PR-fampridine on balance, or an erroneous effect due to limitations of the BBS in this population of walking-disabled participants.
There was a significant difference between PR-fampridine and placebo in the analysis of mean change from baseline in TUG speed over 24 weeks, but the treatment group difference was not significant in the analysis of TUG time (Table 3). The best explanation for this discrepancy is that these were exploratory endpoints and the study was not powered to discern a treatment effect.
Results from effect size and standard response mean analyses to determine sample-to-scale targeting for the BBS and ABILHAND scores deserve additional consideration. Pre-treatment mean scores of both clinical outcome assessments were above the scale midpoint. Both distributions were skewed towards better functioning and away from the best point of measurement of the scale. Moreover, PR-fampridine seeks to improve function and move the mean score further to the right, towards the extreme of the scale range where scales are weaker at detecting change. We believe these distributional properties of the data contribute to the smaller change scores and effect sizes demonstrated for the BBS and ABILHAND, and therefore these may have been suboptimal instruments for examining the impact of PR-fampridine in this sample of walking-disabled participants.
Given the impact of balance in the context of walking, the BBS results may have been anomalous, as the effects of PR-fampridine on the BBS were small. While the limited targeting of the BBS to the ENHANCE population may have affected the findings, we believe that internal measurement problems also contributed. A Rasch measurement theory analysis of BBS data from the PR-fampridine MOBILE study has shown important limitations [33].
The efficacy benefits of PR-fampridine were accompanied by a favorable safety profile, again consistent with other studies [28]. The incidence of positive urine cultures was slightly higher for PR-fampridine than placebo. In contrast, previous findings reported the incidence of laboratory-confirmed urinary tract infections as 2.8% (PR-fampridine) and 4.2% (placebo) [12, 34]. Because PR-fampridine is excreted in urine, bladder irritation may be confounded with bladder infection in some participants.
The subgroup analyses of PR-fampridine MSWS-12 responders showed these participants also had benefits in TUG speed, MSIS-29 PHYS score, and ABILHAND score. Again, these findings allude to MSWS-12 walking responders gaining improvements in other aspects of functioning (non-self-reported functional parameters): walking speed, physical limitations in dynamic and static balance, and manual ability. However, it must be noted that formal statistical testing of the treatment difference was not undertaken in subgroup analyses. The wide range of benefits associated with PR-fampridine is consistent with its proposed mode of action as a blocker of voltage-dependent potassium channels in demyelinated nerve fibers [35]. Results also support the clinical meaningfulness of the MSWS-12 responder definition used in ENHANCE: ≥ 8-point mean improvement [14].
The PR-fampridine MSWS-12 responder effect sizes demonstrated large improvements in MSWS-12 and MSIS-29 PHYS, clinically small to moderate improvements in BBS score, and small improvements in TUG time and ABILHAND score. Importantly, there is no item overlap between the MSWS-12 and MSIS-29 PHYS, indicating that these scales measure different concepts, despite a strong observed correlation (r = 0.72). Although PR-fampridine MSWS-12 responders improved by ≥ 8 points, the group mean change of > 20 points from baseline, and the associated effect sizes (Cohen’s effect size, pre-treatment SD − 1.01, pooled SD − 1.94; standardized response mean − 1.68), represent striking numerical and clinical improvements. It should be noted that the thresholds used to identify clinically meaningful improvement on the MSWS-12 and TUG speed were based on changes at the level of the individual participant and were not meant to evaluate clinically meaningful change or treatment differences at the group level. The thresholds and criteria for a clinically important change in an assessment identified at the individual level may not be applicable at the group level [36].
The pathophysiological explanation for why only some individuals with MS respond to PR-fampridine remains unclear. Therefore, it is not yet possible to predict responders a priori, and clinicians need a clinically practical and meaningful responder definition [37]. There is no consensus, and different studies have used different criteria. More labor-intensive definitions are less clinically feasible. For example, the consistent T25FW responder definition of the pivotal phase III studies (faster walking speed for at least three of four treatment visits than the maximum speed of five off-treatment visits) [6, 7] cannot be incorporated easily into clinical practice, despite its scientific advantages.
One possible limitation of this study was that it did not include a conventional objective walking test, such as the T25FW. The T25FW, or another longer objective walking test, was not included in an effort to limit participant assessment burden and to obtain good-quality data on the expanded range of outcome measures in line with patients’ reports. The effect of PR-fampridine on T25FW speed had already been demonstrated [6, 7]. While one of the benefits of PR-fampridine is a rapid onset of effects, the converse also is true: when treatment stops, the effects of PR-fampridine are lost. This means that patients must be watchful when they discontinue PR-fampridine, as their functioning can worsen soon after discontinuation. In addition, this concept provides a mechanism for evaluating ongoing drug benefit in those people who have progressive disease and are therefore, by definition, likely to worsen over time and in whom it may be difficult to determine whether PR-fampridine is still working. A carefully controlled trial of discontinued treatment—or short drug holiday—can help to determine if PR-fampridine is still beneficial.
ENHANCE was designed with a self-reported primary outcome and thus was planned as a large study to overcome the high spontaneous variability associated with subjective measures. The high placebo response observed in this study may be a natural consequence of this variability. Self-reported measures provide unique information on how individuals feel and function, but there are trade-offs, including problems of stability and interpretability. The broad response categories are open to individual interpretation, based on internal frames of reference that may be influenced by circumstance and mood; and participation in clinical trials is known to be associated with a great deal of expectation [38], with the potential to have greater influence on subjective than objective measures. Learning effects [39, 40] or fatigue [22] may partly explain why mean scores do not return to baseline in the off-treatment period. This study highlights the importance of double-blind, randomized, placebo-controlled clinical trials when self-reported outcomes are the primary endpoints.
5 Conclusions
Results from ENHANCE demonstrate that PR-fampridine was associated with a greater likelihood of walking-impaired participants with MS experiencing clinically meaningful improvements in self-reported walking ability over 24 weeks versus placebo. The benefits of PR-fampridine also included improvements in objectively measured mobility and self-reported physical functioning. PR-fampridine has demonstrated clinically meaningful improvements across a range of study types and designs, and in clinical outcome measures that include the MSWS-12, TUG, MSIS-29 PHYS and psychological impact subscales, T25FW, and 36-Item Short-Form Health Survey physical component summary [6, 7, 9, 10]. Overall, findings from ENHANCE confirm that a self-reported outcome can be used effectively as a primary endpoint in a pharmacotherapy study and provide further evidence demonstrating the favorable risk–benefit profile of PR-fampridine.
References
van Asch P. Impact of mobility impairment in multiple sclerosis 2—patients’ perspectives. Eur Neurol Rev. 2011;6(2):115–20.
Sutliff MH. Contribution of impaired mobility to patient burden in multiple sclerosis. Curr Med Res Opin. 2010;26(1):109–19.
Heesen C, Bohm J, Reich C, Kasper J, Goebel M, Gold SM. Patient perception of bodily functions in multiple sclerosis: gait and visual function are the most valuable. Mult Scler. 2008;14(7):988–91.
European Medicines Agency. Fampyra 10 mg prolonged-release tablets [summary of product characteristics]. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002097/WC500109956.pdf. Accessed 22 Aug 2018.
Judge SI, Bever CT Jr. Potassium channel blockers in multiple sclerosis: neuronal Kv channels and effects of symptomatic treatment. Pharmacol Ther. 2006;111(1):224–59.
Goodman AD, Brown TR, Krupp LB, Schapiro RT, Schwid SR, Cohen R, et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;373(9665):732–8.
Goodman AD, Brown TR, Edwards KR, Krupp LB, Schapiro RT, Cohen R, et al. A phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann Neurol. 2010;68(4):494–502.
Acorda Therapeutics Inc. AMPYRA® (dalfampridine) extended-release tablets, for oral use [prescribing information]. https://ampyra.com/prescribing-information.pdf?v=2. Accessed 22 Aug 2018.
Macdonell R, Nagels G, Laplaud DA, Pozzilli C, de Jong B, Martins da Silva A, et al. Improved patient-reported health impact of multiple sclerosis the ENABLE study of PR-fampridine. Mult Scler. 2016;22(7):944–54.
Hupperts R, Lycke J, Short C, Gasperini C, McNeill M, Medori R, et al. Prolonged-release fampridine and walking and balance in MS: randomised controlled MOBILE trial. Mult Scler. 2016;22(2):212–21.
Gasperini C, Hupperts R, Lycke J, Short C, McNeill M, Zhong J, et al. Prolonged-release fampridine treatment improved subject-reported impact of multiple sclerosis: item-level analysis of the MSIS-29. J Neurol Sci. 2016;15(370):123–31.
Zörner B, Filli L, Reuter K, Kapitza S, Lorincz L, Sutter T, et al. Prolonged-release fampridine in multiple sclerosis: improved ambulation effected by changes in walking pattern. Mult Scler. 2016;22(11):1463–75.
Filli L, Zörner B, Kapitza S, Reuter K, Lörincz L, Weller D, et al. Monitoring long-term efficacy of fampridine in gait-impaired patients with multiple sclerosis. Neurology. 2017;88(9):832–41.
Mehta L, McNeill M, Hobart J, Wyrwich KW, Poon J-L, Auguste P, et al. Identifying an important change estimate for the Multiple Sclerosis Walking Scale-12 (MSWS-12v1) for interpreting clinical trial results. Mult Scler J Exp Transl Clin. 2015;1:2055217315596993.
Kieseier BC, Pozzilli C. Assessing walking disability in multiple sclerosis. Mult Scler. 2012;18(7):914–24.
Hobart JC, Riazi A, Lamping DL, Fitzpatrick R, Thompson AJ. Measuring the impact of MS on walking ability: the 12-Item MS Walking Scale (MSWS-12). Neurology. 2003;60(1):31–6.
Costelloe L, O’Rourke K, Kearney H, McGuigan C, Gribbin L, Duggan M, et al. The patient knows best: significant change in the physical component of the Multiple Sclerosis Impact Scale (MSIS-29 physical). J Neurol Neurosurg Psychiatry. 2007;78(8):841–4.
Penta M, Thonnard JL, Tesio L. ABILHAND: a Rasch-built measure of manual ability. Arch Phys Med Rehabil. 1998;79(9):1038–42.
Podsiadlo D, Richardson S. The timed “Up & Go”: a test of basic functional mobility for frail elderly persons. J Am Geriatr Soc. 1991;39(2):142–8.
Berg KO, Wood-Dauphinee SL, Williams JI, Maki B. Measuring balance in the elderly: validation of an instrument. Can J Public Health. 1992;83(Suppl 2):S7–11.
Cattaneo D, Regola A, Meotti M. Validity of six balance disorders scales in persons with multiple sclerosis. Disabil Rehabil. 2006;28(12):789–95.
Cavanaugh JT, Gappmaier VO, Dibble LE, Gappmaier E. Ambulatory activity in individuals with multiple sclerosis. J Neurol Phys Ther. 2011;35(1):26–33.
Learmonth YC, Paul L, McFadyen AK, Mattison P, Miller L. Reliability and clinical significance of mobility and balance assessments in multiple sclerosis. Int J Rehabil Res. 2012;35(1):69–74.
Simone A, Rota V, Tesio L, Perucca L. Generic ABILHAND questionnaire can measure manual ability across a variety of motor impairments. Int J Rehabil Res. 2011;34(2):131–40.
Little RA, Rubin DB. Estimation of imputation uncertainty. In: Statistical analysis with missing data. 2nd ed. New York: Wiley-Interscience; 2002. p. 75–96.
Cohen J. Statistical power analysis for the behavioral sciences. New York: Academic Press; 1969.
Allart E, Benoit A, Blanchard-Dauphin A, Tiffreau V, Thevenon A, Zephir H, et al. Sustained-released fampridine in multiple sclerosis: effects on gait parameters, arm function, fatigue, and quality of life. J Neurol. 2015;262(8):1936–45.
Goodman AD, Bethoux F, Brown TR, Schapiro RT, Cohen R, Marinucci LN, et al. Long-term safety and efficacy of dalfampridine for walking impairment in patients with multiple sclerosis: results of open-label extensions of two phase 3 clinical trials. Mult Scler. 2015;21(10):1322–31.
Bertoni R, Lamers I, Chen CC, Feys P, Cattaneo D. Unilateral and bilateral upper limb dysfunction at body functions, activity and participation levels in people with multiple sclerosis. Mult Scler. 2015;21(12):1566–74.
Barrett LE, Cano SJ, Zajicek JP, Hobart JC. Can the ABILHAND handle manual ability in MS? Mult Scler. 2013;19(6):806–15.
Barrett L, Cano S, Zajicek J, Hobart J. Lending a hand: can DASH items help ABILHAND improve manual ability measurement in multiple sclerosis? Mult Scler. 2015;21(5):612–21.
Hobart J, Cano S. Improving the evaluation of therapeutic interventions in multiple sclerosis the role of new psychometric methods. Health Technol Assess. 2009;13(12):1–177.
Hobart J. Clinical outcomes assessment in MS trials: on balance, the Berg Balance Scale is a precarious measure. In: 34th Congress of the European Committee for Treatment and Research in Multiple Sclerosis; 10–12 Oct 2018; Berlin.
Kantor D, Chancellor MB, Snell CW, Henney HR 3rd, Rabinowicz AL. Assessment of confirmed urinary tract infection in patients treated with dalfampridine for multiple sclerosis. Postgrad Med. 2015;127(2):218–22.
Dunn J, Blight A. Dalfampridine: a brief review of its mechanism of action and efficacy as a treatment to improve walking in patients with multiple sclerosis. Curr Med Res Opin. 2011;27(7):1415–23.
Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9(2):105–21.
Rodriguez-Leal FA, Haase R, Thomas K, Eisele JC, Proschmann U, Schultheiss T, et al. Fampridine response in MS patients with gait impairment in a real-world setting: need for new response criteria? Mult Scler. 2018;24(10):1337–46.
Institute of Medicine (US) Forum on Drug Discovery, Development, and Translation. Transforming clinical research in the United States: challenges and opportunities: workshop summary. Forum on Drug Discovery, Development, and Translation; 2010. https://www.ncbi.nlm.nih.gov/books/NBK50889/. Accessed 19 Jul 2017.
Oken BS. Placebo effects: clinical aspects and neurobiology. Brain. 2008;131(Pt 11):2812–23.
Lyders Johansen K, Derby Stistrup R, Skibdal Schjøtt C, Madsen J, Vinther A. Absolute and relative reliability of the Timed ‘Up & Go’ test and ‘30second Chair-Stand’ test in hospitalised patients with stroke. PLoS One. 2016;11(10):e0165663.
Hobart J, Lamping D, Fitzpatrick R, Riazi A, Thompson A. The Multiple Sclerosis Impact Scale (MSIS-29): a new patient-based outcome measure. Brain. 2001;124(Pt 5):962–73.
Phillips GA, Wyrwich KW, Guo S, Medori R, Altincatal A, Wagner L, et al. Responder definition of the Multiple Sclerosis Impact Scale physical impact subscale for patients with physical worsening. Mult Scler. 2014;20(13):1753–60.
Cattaneo D, Jonsdottir J, Repetti S. Reliability of four scales on balance disorders in persons with multiple sclerosis. Disabil Rehabil. 2007;29(24):1920–5.
Conradsson M, Lundin-Olsson L, Lindelöf N, Littbrand H, Malmqvist L, Gustafson Y, et al. Berg balance scale: intrarater test-retest reliability among older people dependent in activities of daily living and living in residential care facilities. Phys Ther. 2007;87(9):1155–63.
Muir SW, Berg K, Chesworth B, Speechley M. Use of the Berg Balance Scale for predicting multiple falls in community-dwelling elderly people: a prospective study. Phys Ther. 2008;88(4):449–59.
Newstead AH, Hinman MR, Tomberlin JA. Reliability of the Berg Balance Scale and balance master limits of stability tests for individuals with brain injury. J Neurol Phys Ther. 2005;29(1):18–23.
Batcho CS, Durez P, Thonnard JL. Responsiveness of the ABILHAND questionnaire in measuring changes in rheumatoid arthritis patients. Arthritis Care Res (Hoboken). 2011;63(1):135–41.
Wang TN, Lin KC, Wu CY, Chung CY, Pei YC, Teng YK. Validity, responsiveness, and clinically important difference of the ABILHAND questionnaire in patients with stroke. Arch Phys Med Rehabil. 2011;92(7):1086–91.
Penta M, Tesio L, Arnould C, Zancan A, Thonnard JL. The ABILHAND questionnaire as a measure of manual ability in chronic stroke patients: Rasch-based validation and relationship to upper limb impairment. Stroke. 2001;32(7):1627–34.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Funding
This study was funded by Biogen. Biogen provided funding for medical writing support in the development of this paper; Malcolm Darkes and Juliet Bell from Excel Scientific Solutions wrote the first draft of the manuscript based on input from authors, and Kristen DeYoung of Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the paper to the authors. The authors had full editorial control of the paper, and provided their final approval of all content. The open access fee was paid by Biogen.
Conflict of interest
Jeremy Hobart has received consulting/advisor fees, grants, honoraria, support for clinical service delivery, or research support from Acorda, Biogen, Brickell Biotech, GBT, Merck Serono, MS Society of Great Britain and Northern Ireland, Novartis, Roche, Tigercat, and Vantia; his hospital received a grant from Biogen to assist in the development of clinical service for people with MS and the university received a grant to conduct data analysis, and he received honoraria for attending PR-fampridine advisory boards and payment from Biogen for lectures on MS and PR-fampridine. Tjalf Ziemssen has received grants and personal fees from Bayer, Biogen, Genzyme, Novartis, and Teva; and personal fees from Almirall, GlaxoSmithKline, Merck, and Roche. Peter Feys has received advisory board fees from Biogen; speaker fees from Excemed and Paradigms; and fees for providing educational materials from Neurocompass. Michael Linnebank has received grants to fund a distinct study on PR-fampridine from Biogen, and support for travel and fees paid to his department for attendance at a PR-fampridine steering board. Andrew D. Goodman has received personal fees from AbbVie, Acorda, Adamas, Atara, Avanir, Bayer, Biogen, Celgene, EMD Serono, Novartis, Roche, Sanofi-Genzyme, Sun, and Teva. His institution (University of Rochester) has received research support for conducting clinical trials from Acorda, Biogen, EMD Serono, Novartis, Ono, Roche, Sanofi-Genzyme, and Teva. Rachel Farrell has received advisory fees from Biogen; consulting and principal investigator fees from Canbex; and speaker fees from Allergan PLC, GW Pharma, Merck, and Teva. Raymond Hupperts has received grants and honoraria for advisory boards from Biogen, Merck, and Sanofi-Genzyme. Andrew R. Blight was an employee of and holds stock/stock options in Acorda. Veronica Englishby, Manjit McNeill, Ih Chang, and Jacob Elkins are employees of and hold stock/stock options in Biogen. Gabriel Lima was an employee of Biogen at the time the study was conducted and the manuscript drafted; holds stock/stock options in Biogen; and is a current employee of Amgen.
Ethical approval
The study was conducted according to the US Code of Federal Regulations Parts 50, 54, 56, and 312 Subpart D; the International Conference on Harmonisation guideline on Good Clinical Practice (E6); the European Union Clinical Trial Directive 2001/20/EC; the principles of the Declaration of Helsinki; and, where applicable, the European Directives 2001/20 and 2005/28 in relation to conduct of clinical trials and investigational medicinal products. The trial was registered with ClinicalTrials.gov (NCT02219932).
Informed consent
Written informed consent was obtained from each individual before study participation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hobart, J., Ziemssen, T., Feys, P. et al. Assessment of Clinically Meaningful Improvements in Self-Reported Walking Ability in Participants with Multiple Sclerosis: Results from the Randomized, Double-Blind, Phase III ENHANCE Trial of Prolonged-Release Fampridine. CNS Drugs 33, 61–79 (2019). https://doi.org/10.1007/s40263-018-0586-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-018-0586-5