
Abstract
Background
Prolonged-release fampridine (PR-FAM) 10-mg tablet twice daily is the only approved pharmacological treatment for improvement of walking ability in adults with multiple sclerosis (MS). LIBERATE assessed the safety/effectiveness of PR-FAM in the real-world.
Objectives
The aim of this study was to collect additional safety data, including the incidence rate of seizures and other adverse events (AEs) of interest, from patients with MS taking PR-FAM in routine clinical practice (including patients aged ≥ 65 years and those with pre-existing cardiovascular risk factors). Other objectives included change over time in patient-reported evaluation of physical and psychological impact of MS while taking PR-FAM, and change over time in physician-reported assessment of walking ability in MS patients taking PR-FAM.
Methods
Patients with MS newly prescribed PR-FAM were recruited (201 sites, 13 countries). Demographic/safety data were collected at enrolment through 12 months. Physician-rated Clinical Global Impression of Improvement (CGI-I) scores for walking ability, and Multiple Sclerosis Impact Scale-29 (MSIS-29) were assessed.
Results
Safety analysis included 4646 patients with 3534.8 patient-years of exposure; median (range) age, 52.6 (21–85) years, 87.3% < 65 years, and 65.7% women. Treatment-emergent AEs (TEAEs) were reported in 2448 (52.7%) patients, and serious TEAEs were reported in 279 (6.0%) patients, of whom 37 (< 1%) experienced treatment-emergent serious AEs (TESAEs) considered related to PR-FAM. AEs of special interest (AESI) occurred in 1799 (38.7%) patients, and serious AESI in 128 (2.8%) patients. Seventeen (< 1%) patients experienced actual events of seizure. Overall, 1158 (24.9%) patients discontinued treatment due to lack of efficacy. At 12 months, a greater proportion of patients on-treatment had improvement from baseline in CGI-I for walking ability versus those who discontinued (61% vs. 11%; p < 0.001). MSIS-29 physical impact score improved significantly for patients on-treatment for 12 months versus those who discontinued (mean change, baseline to 12 months: − 9.99 vs. − 0.34 points; p < 0.001). Results were similar for MSIS-29 psychological impact.
Conclusion
No new safety concerns were identified in this real-world study, suggesting that routine risk-minimization measures are effective. CGI-I and MSIS-29 scores after 12 months treatment with PR-FAM treatment show clinical benefits consistent with those previously reported.
Trial Registration
ClinicalTrials.gov: NCT01480063.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Results from LIBERATE demonstrate that long-term (12 months) treatment with prolonged-release fampridine (PR-FAM) in a large number of patients in routine clinical practice was well tolerated. |
The nature and types of adverse events reported in the study were consistent with the known safety profile of PR-FAM, and, overall, no new safety concerns were identified. |
The LIBERATE results add to previous findings on the benefits of PR-FAM on quality of life and walking ability by demonstrating that PR-FAM therapy improves physician-reported walking ability and patient-reported well-being in real life. |
1 Introduction
Impaired walking ability is common in multiple sclerosis (MS) and negatively impacts patients’ lives. Within 10 years of diagnosis, up to 93% of patients with MS have walking impairment and consequent loss of mobility, which have a profound negative impact on day-to-day functioning, independence, and quality of life [1, 2]. Maintenance of walking ability is a high priority for individuals with MS, regardless of the duration of their disease or level of disability [3, 4].
Prolonged-release fampridine (PR-FAM) tablets (AMPYRA®; known as dalfampridine extended release in the United States (US), and as fampridine sustained or modified release [FAMPYRA®] in the European Union (EU) and other countries) are indicated for the improvement of walking in adult patients with MS with walking disability (measured by the Expanded Disability Status Scale [EDSS] 4.0–7.0). PR-FAM is approved in over 50 countries, and as of 31 December 2020, approximately 396,747 patients had been treated with PR-FAM worldwide, representing 561,888 patient-years of exposure (data on file). Although several drugs are available for treating various symptoms of MS, such as fatigue and pain, no other drugs have been approved for the treatment of walking disability in patients with MS.
Two pivotal phase III studies of PR-FAM showed that walking speed, measured by the Timed 25-Foot Walk, was improved in PR-FAM responders over 9 and 14 weeks of treatment [5, 6]. These pivotal data led to the approval of PR-FAM in the US and EU, however these trials were limited with respect to demonstrating the duration of effect and clinical meaningfulness of PR-FAM treatment. Subsequently, the 12-month ENABLE, 6-month MOBILE, and 6-month ENHANCE studies showed that PR-FAM also had beneficial effects on other clinical and patient-reported outcome assessments, including walking and functional ability, balance, and aspects of quality of life over longer treatment periods [7,8,9]. ENABLE, a single-arm, open-label study, showed that PR-FAM was associated with significant long-term improvements in patient-reported physical functioning and psychological health over time [9]. The MOBILE study was an exploratory, double-blind, placebo-controlled study to further assess the effects of PR-FAM beyond 14 weeks (the longest follow-up in the pivotal studies), evaluate patient-reported walking ability, and identify a clinically meaningful change threshold in the 12-item Multiple Sclerosis Walking Scale [8]. ENHANCE was a multinational, phase III, randomised, double-blind, placebo-controlled study of PR-FAM in walking-disabled participants with MS (EDSS score 4.0−7.0) [7]. Findings from ENHANCE showed that participants treated with PR-FAM were more likely to achieve clinically meaningful improvements in self-reported walking ability over 6 months than those treated with placebo (responder rate 43.2% for PR-FAM, vs. 33.6% for placebo); benefits in mobility and self-reported physical functioning were also reported. The explanation for why only some individuals with MS respond to PR-FAM remains unclear, and, as yet, it is not possible to predict who will be a responder. It is acknowledged that a clinically practical and meaningful responder definition is needed [10].
LIBERATE is a long-term, multicenter, post-authorization study of PR-FAM in patients with MS. Due to the inherent constraints of clinical studies, at the time of conditional approval in the EU in 2011, the PR-FAM safety database established during the development phase had its limitations; for example, the number of patients ≥ 65 years of age was limited, and patients with pre-existing cardiovascular risk factors were excluded from the studies. This observational study was conducted in a postmarketing routine practice setting to provide additional safety and effectiveness data on those subpopulations not fully studied during the development phase. Specifically, premarketing clinical studies were not powered to evaluate the incidence of rare adverse events (AEs), including seizure. LIBERATE was powered to detect a doubling in the incidence of seizures in the MS patient population to further assess whether PR-FAM treatment was associated with an increased seizure rate. Other objectives included change over time in patient-reported evaluation of physical and psychological impact of MS while taking PR-FAM, and change over time in physician-reported assessment of walking ability in MS patients taking PR-FAM.
2 Methods
2.1 Standard Protocol Approvals, Registrations, and Patient Consents
The trial was registered with ClinicalTrials.gov (NCT01480063). Biogen and the investigators (see supplementary information) complied with the principles set forth by the Declaration of Helsinki; the instructions, regulations, and agreements in the study protocol; and the applicable International Conference on Harmonisation and Good Clinical Practice guidelines in conducting Study 218MS401 in a manner consistent with local regulations. Independent Ethics Committee approval was obtained by the investigators for the study protocol, any applicable protocol amendments, and the informed consent form employed.
2.2 Patients and Study Design
LIBERATE was a prospective, non-interventional, multicenter, observational, post-authorization study. Patients with MS newly prescribed PR-FAM (10-mg tablets, twice daily) were recruited at 201 sites in 13 countries (Argentina, Canada, Czech Republic, France, Germany, Ireland, Israel, Lebanon, The Netherlands, Norway, Portugal, Spain, and United Arab Emirates) from 16 April 2012 through 5 February 2019 (last patient, last visit). The main phase of the study was regarded as the study period from the initiation of patient enrolment in 2012 through February 2019. There was also an extension phase of 12 months from the end of the conditional reimbursement period in The Netherlands on 1 April 2018 through 1 April 2019. The overall study was the aggregate period comprising the main and extension phases. Patients were observed for approximately 1 year. Data were collected at baseline and at any time point between baseline and month 12 according to local routine clinical practice, either by telephone contact (except for the assessment of Clinical Global Impression of Improvement [CGI-I]) or during routine local clinical practice site visits. Safety data were collected from the first dose of PR-FAM until study completion or early discontinuation.
Patients were enrolled between 16 April 2012 and 31 December 2017. Eligible patients were those who were newly prescribed PR-FAM according to market authorization, but who had not yet started PR-FAM treatment, and who were able to provide written informed consent.
Patients who discontinued PR-FAM, regardless of the reasons and time of discontinuation, were encouraged to remain in the study for the full 1-year observation period, unless the patients withdrew consent to participate in the study. For this reason, patients who discontinued PR-FAM could have completed the study. A patient was considered lost to follow-up after three failed documented attempts to contact, with one attempt occurring via certified letter.
2.3 Objectives
The primary objective was to collect additional safety data, including the incidence rate of seizure and other specific AEs of special interest (AESI), from patients with MS taking PR-FAM in routine clinical practice (including patients aged ≥ 65 years and those with pre-existing cardiovascular risk factors). All recorded AE verbatim terms were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 18.1. Serious AEs, all AEs leading to PR-FAM dose change or discontinuation, and all AEs experienced by the patient at the time of PR-FAM overdose were recorded, and relatedness to PR-FAM treatment was assessed by the reporting investigator. AESI in this study were those related to seizure, serious hypersensitivity reactions, urinary tract infection (UTI) and urinary symptoms, depression and suicide attempt, cardiovascular disorders, severe infections other than UTI, anxiety, central nervous system stimulation, and clinically significant hematologic abnormalities. Seizure-related events were reported based on the Biogen search strategy using the Standard Medical Dictionary for Regulatory Activities Query (MedDRA SMQ) for Convulsions. The SMQ comprises a group of event terms, some of which are not specifically seizure, seizure disorder, or diagnoses of seizure (e.g., muscle twitching and trismus). Treatment-emergent AEs (TEAEs) were defined as any AE with an onset date on or after the first dose of PR-FAM and within 28 days after the last dose or any pre-existing condition that has worsened in severity after the first dose of PR-FAM. For overall AEs in this study, the period is relative from the first dose of treatment and within 28 days after the last dose in the extension study.
Secondary objectives included characterization of utilization patterns of PR-FAM in routine clinical practice and the assessment of the effectiveness of risk-minimization measures in the PR-FAM Risk Management Plan. In LIBERATE, data were collected on patients with renal impairment and on patients taking 4-aminopyridine (4-AP) or organic cation transporter-2 (OCT2) inhibitors concomitantly with PR-FAM to assess adherence to the product label, the effectiveness of risk-minimization measures, and the incidence of seizure events in these subpopulations of patients. The pattern of PR-FAM use (dose and duration) was summarised descriptively, inclusive of disease subtype, dose change, or treatment discontinuation. In addition, dosing deviations from the local PR-FAM label, product information, or prescription information (including incidences of overdoses) were summarised. For measurement of risk minimisation, the number and percentage of patients with a neurological assessment of walking within 14 days after the start of dosing were evaluated to assess whether appropriate follow-up of effectiveness was performed. Adherence to the product label was assessed by review of available data collected by the Investigators (i.e., reason for PR-FAM prescription, dosing information, medical history, creatinine clearance [CrCl] at baseline and change from baseline, and concomitant medications). Walking ability was assessed using the physician-rated CGI-I scale of walking ability [11]. A patient’s overall walking ability was assessed using the CGI-I 1 question scale [11] (‘how much has the patient’s overall walking ability improved or worsened relative to a baseline status at the beginning of PR-FAM treatment?’); the physician rated improvement or worsening of the patients’ overall walking ability relative to baseline; responses ranged from 1 (very much improved) to 7 (very much worse) over 12 months. In order to make the CGI-I measure as objective as possible, this assessment could only be performed in-person by a neurologist (as opposed to by telephone consultation, for example). Furthermore, the baseline assessment had to be performed prior to the first dose of PR-FAM to ensure that the comparison was relative to pretreatment walking ability.
The impact of MS on the patients and their quality of life was assessed using the 29-item Multiple Sclerosis Impact Scale (MSIS-29), which provides scores for both physical and psychological impact [12]. The validated, self-administered MSIS-29 questionnaire contains a 20-item physical impact and a 9-item psychological impact subscale. The physical scores provided data on the impact of PR-FAM on symptoms associated with walking, such as stiffness, spasms, and difficulties moving or with balance. The psychological score provides data on the impact of PR-FAM on symptoms such as fatigue, sleep disturbance, and feeling depressed [13, 14]. For each visit, the MSIS-29 score was calculated by summing 20 items and transforming the score to a scale with a range of 0 (no impact of MS) to 100 (extreme impact of MS). Negative changes on the MSIS-29 physical and psychological impact subscales indicate improvement in physical and psychological health, respectively.
2.4 Statistical Analyses
The sample size for LIBERATE was calculated based on the known rates of AESI, including seizures and cardiovascular events, in patients with MS. A sample of 3308 was estimated to achieve 80% power to detect a doubling of seizure rates (assuming a background incidence of 0.003 per patient-year [15] at a 0.05 level of significance). With a background incidence of cardiovascular outcomes such as 1.0% of arrhythmias and conduction disorders in patients ≥ 65 years of age [16] and 0.1% of atrial fibrillation/flutter [17], the power to detect a doubling of events after 12 months’ follow-up was estimated to be 99% and 46%, respectively. Therefore, sample size was determined to be approximately 5000 patients followed for 1 year after treatment initiation, allowing for 3308 patient-years of exposure, and assuming a dropout rate of 34%. All patients enrolled in the study who received at least one dose of PR-FAM were included in the safety population for which all analyses were presented, including safety and effectiveness analyses. CGI-I data were analysed by treatment status (on-treatment and off-treatment) over visit windows for patients who were still in the study at 12 months. The p-value and 95% confidence interval (CI) values were presented using the Chi-square test to test the difference in the proportion of patients with an improvement between the on- and off-treatment groups. MSIS-29 data were analysed using the same approach. In addition, the mean change of MSIS-29 from baseline at each visit window was analysed by status groups (on-treatment and off-treatment) using analysis of covariance with treatment effect and adjusted by baseline MSIS-29 values. An improvement in the patient’s overall walking ability was indicated by a negative change from baseline.
3 Results
3.1 Patients
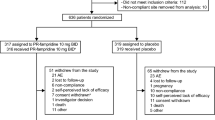
A total of 4737 patients were enrolled in LIBERATE, and 4646 patients received at least one dose of PR-FAM and were included in the safety population (Fig. 1). Overall, 3251 (70%) patients completed the study, 2475 (53%) discontinued treatment, and 1395 (30%) did not complete the study (Fig. 1). The most common reasons for treatment discontinuation were lack of efficacy (n = 1158 [24.9%]) and AEs (n = 753 [16.2%]). Of 1158 patients who discontinued due to lack of effectiveness, 1140 were from the main phase of the study and 18 were from the extension phase; of the 1158 patients, 163 and 995 patients discontinued treatment ≤ 14 days and > 14 days, respectively, from the start of treatment.

Patient disposition. Data cut-off date: 21 June 2019. Percentages are calculated using the total number of patients (N) as the denominator. aSafety population: patients who enrolled in the study and received ≥ 1 dose of PR-FAM. CGI-I Clinical Global Impression of Improvement, MSIS-29 Multiple Sclerosis Impact Scale-29, PR-FAM prolonged-release fampridine
Patient baseline characteristics are described in Table 1. Overall, the mean (standard deviation) patient age was 52.5 (10.5) years, median (minimum, maximum) EDSS at enrolment was 5.5 (1, 9), and the majority (65.7% [n = 3051]) of patients were female; 4057 patients (87.3%) were < 65 years of age and 589 patients (12.7%) were ≥ 65 years of age. Race was not reported for the 4439 patients enrolled in EU countries due to confidentiality regulations; of the 207 patients enrolled in non-European countries, the majority were White (n = 198 [95.7%]).
Of the 4646 patients in the safety population, 4058 (87.3%) patients were taking at least one concomitant medication. Symptomatic medications taken by ≥ 10.0% of patients included baclofen (18.9%). Other MS disease-modifying therapies that were used concomitantly by ≥ 2% of patients included interferon (13.8%), fingolimod (10.7%), natalizumab (7.0%), glatiramer acetate (5.0%), and fumaric acid/dimethyl fumarate (4.3%). Low rates of concomitant physiotherapy (14%) were observed in this study.
3.2 Safety
Overall, TEAEs were reported in 2448 (52.7%) patients (Table 2). The incidence rate for TEAEs was 141.3 per 100 patient-years based on 3534.8 patient-years of exposure. The most commonly reported TEAEs occurring in ≥ 2.0% of patients are presented in Table 2; the majority of these events are known adverse reactions included in the EU Summary of Product Characteristics (SmPC) for PR-FAM, or were due to the patient’s underlying MS disease.
TEAEs considered related to PR-FAM treatment were reported in 1626 (35.0%) patients (Table 3). At least one serious TEAE (TESAE) was reported in 279 (6.0%) patients (Table 4); the most common TESAEs, reported in > 2 patients, were MS relapse (n = 61 [1.3%]) and fall (n = 25 [< 1%]). In total, 37 (< 1%) patients experienced TESAEs that were considered related to PR-FAM treatment. Related TESAEs in > 2 patients were epilepsy (n = 7 [0.2%]); UTI (n = 5 [0.1%]); seizure (n = 4 [< 0.1%]); urosepsis (n = 3 [< 0.1%]); and Uhthoff’s phenomenon, generalised tonic-clonic seizure, sepsis, and cystitis (n = 2 patients each [< 0.1%]).
Of the 4646 patients in the safety population, 751 (16.2%) reported 1255 TEAEs leading to discontinuation of PR-FAM, with an overall incidence rate of 35.5 per 100 patient-years (based on 3534.8 patient-years of exposure). The most commonly reported TEAEs (reported by ≥ 1% of patients) included dizziness (n = 136 [2.9%]), insomnia (n = 98 [2.1%]), headache (n = 77 [1.7%]), nausea (n = 73 [1.6%]), vertigo (n = 63 [1.4%]), and paraesthesia (n = 51 [1.1%]). In total, 296 (6.4%) patients experienced a TEAE leading to withdrawal from the study, and 46 (1.0%) patients experienced a TEAE leading to a change in PR-FAM dose. Four patients (< 0.1%) experienced a TEAE leading to death. The events with fatal outcome reported in these four patients were duodenitis, erosive duodenitis, hemorrhagic erosive gastritis, nausea, vomiting, pneumonia, and sepsis (all in 1 patient); chordae tendinae rupture (1 patient); suicide (1 patient); and death (1 patient; cause of death unknown).
Of the 4646 patients in the safety population, 32 (0.7%) patients reported seizure-related events, based on the Biogen search strategy using the MedDRA SMQ for Convulsions. The SMQ comprises a group of event terms, some of which are not specifically seizure, seizure disorder, or diagnoses of seizure (e.g., muscle twitching and trismus). Of these 32 patients, only 17 (< 1%) experienced actual events of seizure, including epilepsy (n = 8 [0.2%]), seizure (n = 5 [0.1%]), generalised tonic-clonic seizure (n = 2 [< 0.1%]), and partial seizure and absence seizure (n = 1 each [< 0.1%]) (Table 4). The nature and types of other AESIs reported, including serious hypersensitivity reactions, and urinary symptoms, cardiovascular disorders, depression and suicide attempt, severe infections other than UTI, anxiety, events suggestive of central nervous system stimulation, and clinically significant hematologic abnormalities, were consistent with the known safety profile of PR-FAM (Table 4).
Analysis of the events for patients taking PR-FAM concomitantly with either OCT2 inhibitors (n = 896 [19.3%]) or OCT2 substrates (n = 313 [6.7%]) showed that the nature and types of events reported were in line with the overall study population. The incidence of seizure-related events for patients taking OCT2 inhibitors (0.7 per 100 patient-years) or substrates (0.4 per 100 patient-years) were comparable with the incidence rate for seizure-related events reported in the overall study population (0.5 per 100 patient-years). Therefore, although there is a risk of seizure due to increased plasma levels in patients taking PR-FAM concomitantly with OCT2 inhibitors, and a theoretical risk of increased seizure due to increased plasma levels in patients taking PR-FAM concomitantly with OCT2 substrates, the data did not demonstrate a higher incidence of seizure in these patients.
Of the 2152 patients who took PR-FAM concomitantly with medications that have the potential to lower the seizure threshold, only 18 (< 1%) patients experienced seizure-related events. Of these 18 patients, 11 patients experienced actual events of seizure. Despite the increased risk of seizure in patients taking PR-FAM concomitantly with medications that have the potential to lower seizure threshold, the number of reported seizure events was low.
When stratified by age, the incidence of TEAEs was comparable between patients ≥ 65 and < 65 years of age (52.7% vs. 52.6%, respectively) (Table 5); the most common TEAEs were also comparable between patients ≥ 65 and < 65 years of age. In this study, no new safety concerns have been identified with respect to the use of PR-FAM in patients ≥ 65 years of age.
Of the 4646 patients included in the safety population, baseline data on renal function were available for 2212 patients (47.6%), of whom 1672 patients (75.6%) had normal renal function (CrCl ≥ 80 mL/min), 533 patients (24.1%) had mild renal impairment (CrCl < 80 mL/min to ≥ 50 mL/min), 5 patients (0.2%) had moderate renal impairment (CrCl < 50 mL/min to ≥ 30 mL/min), and 1 patient (< 0.1%) had severe impairment (CrCl < 30 mL/min). Among the 2212 patients, post-baseline data on renal function were available for 448 (20.3%) patients, of whom 125 showed a change in renal function status: 124 patients (27.7%) showed a change from normal renal function to mild impairment and 1 patient (0.2%) showed a change from normal renal function to severe impairment.
Analysis of the safety data showed that the incidence of TEAEs, including seizure and generalised tonic-clonic seizure, were comparable between patients with normal renal function (n = 963 [57.6%]; 2084 TEAEs; incidence rate: 164.9 per 100 patient-years), patients with mild renal impairment (n = 302 [56.7%]; 672 TEAEs; incidence rate: 157.2 per 100 person-years), and patients with moderate renal impairment (n = 3 [60.0%]; 6 TEAEs; incidence rate: 185.1 per 100 patient-years). Overall, no new safety concerns were identified in patients with renal impairment.
When stratified by age and renal function, the most commonly reported TEAEs for patients, and the incidence rates of reported TEAEs, were similar irrespective of patient renal function status and age. The nature of events reported, based on the system organ class (SOC) and preferred terms (PT) stratification, is consistent with those reported in the overall safety population.
Overall, 1936 patients with 12 months of exposure to PR-FAM were included in the study. Of these, 977 patients (50.5%) reported TEAEs and the data did not show an increase in the incidence of TEAEs with increased exposure. The nature and types of events reported were consistent with the events reported for the overall safety population and no new safety concerns were identified.
3.3 Drug Utilization and Effectiveness of Risk-Minimization Measures
The potential for seizure is the primary safety risk of treatment with PR-FAM. The risk appears to be dose-dependent and increases at dosages of above 10 mg twice daily. The possibility of a dose-dependent risk of seizure is also relevant in patients with moderate or severe chronic kidney disease and in patients taking other medications containing 4-AP or OCT2 inhibitors concomitantly with PR-FAM.
Due to the observational nature of this study, patients with renal impairment were enrolled and administered PR-FAM despite the approved labelling for PR-FAM contraindicating its use in patients with mild, moderate, or severe renal impairment. Although TEAEs (including seizure and generalised tonic-clonic seizure) in patients with mild renal impairment were similar to those in patients with normal renal function, PR-FAM should be used with caution, and monitoring of renal function should be considered in patients with mild renal impairment. Similarly, again due to the observational study design, concomitant use of other medications containing 4-AP and OCT2 inhibitors with PR-FAM is contraindicated but patients taking these concomitant medications were included in the study. In patients taking other formulations of 4-AP with PR-FAM (n = 23), the incidence of TEAEs was similar to that observed in the overall population. For patients coadministered ≥ 1 OCT2 inhibitor and PR-FAM (n = 896), the incidence of TEAEs overall was greater than that among the overall study population.
3.4 Physician-Assessed Clinical Global Impression of Improvement of Walking Ability
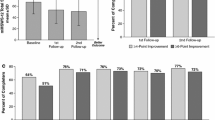
In total, 3329 patients were included in the 12-month analysis of CGI-I, of whom 2464 were on-treatment and 865 patients had discontinued PR-FAM but remained in the study. Using the CGI-I measurement, at 12 months a total of 730 (61%) patients on-treatment demonstrated improvement in walking ability relative to baseline, compared with 45 (11%) patients off-treatment (p < 0.001) [Fig. 2]. Analysis of CGI-I scores between European and non-European countries showed similar proportions of patients who reported improvement at 12 months (47.4% vs. 50.5%).

Patients on-treatment with PR-FAM showed improved walking ability compared with patients off-treatment. Data cut-off date: 21 June 2019. P value is presented to test the difference in proportion of patients with an improvement between the on-treatment and off-treatment groups using the Chi-square test. Nominal p value is presented. Percentages are calculated using the total number of patients with non-missing values as the denominator. PR-FAM prolonged-release fampridine
3.5 Patient’s Assessment of the Physical and Psychological Impact of Multiple Sclerosis: Multiple Sclerosis Impact Scale-29
A total of 3329 patients had 12-month MSIS-29 data, of whom 2464 were still on-treatment and 865 had discontinued PR-FAM but remained in the study. Patients receiving PR-FAM therapy at 12 months showed a significant improvement in the MSIS-29 PHYS and MSIS-29 PSYCH scales at 12 months compared with patients who had discontinued treatment. The mean change from baseline to month 12 in the MSIS-29 PHYS score was − 9.99 compared with − 0.34 for patients who had discontinued treatment (p < 0.001) [Fig. 3a], while the mean change from baseline to month 12 in the MSIS-29 PSYCH score was − 7.61 compared with − 1.38 for patients who had discontinued treatment (p < 0.001) (Fig. 3b).

MSIS-29 PHYS and MSIS-29 PSYCH scores improved in patients treated with PR-FAM compared with patients off-treatment: a MSIS-29 PHYS; b MSIS-29 PSYCH. Data cut-off date: 21 June 2019. P-values are presented to test the difference in change from baseline between on-treatment and off-treatment groups using an analysis of covariance model with baseline value as covariate and a fixed term for treatment status at 12 months. MSIS-29 Multiple Sclerosis Impact Scale, PHYS physical impact subscale, PSYCH psychological impact subscale, PR-FAM prolonged-release fampridine
4 Discussion
Results from LIBERATE demonstrate that long-term treatment with PR-FAM in a large number of patients in routine clinical practice was well tolerated. The nature and types of AEs reported in the study were consistent with the known safety profile of PR-FAM [18]. At the prescribed dose, there was no increased risk of seizure over long-term follow-up. Although some particular AESI such as seizures and UTIs should be monitored, they are well-documented in the PR-FAM prescribing information and can be adequately managed by routine risk-minimization activities. Drug utilization results from LIBERATE showed that the majority (≥ 95%) of investigators prescribed PR-FAM according to the product label and adhered to the contraindications and recommendations. Taken together with the rare reporting of seizure (comparable with the background incidence of seizure in patients with MS), the results demonstrate that routine risk-minimization measures through labelling are effective. Overall, no new safety concerns were identified. The LIBERATE results also add to previous findings on the benefits of PR-FAM on quality of life and walking ability by demonstrating that PR-FAM therapy improves physician-reported walking ability and patient-reported well-being in real life.
Although randomised clinical trials are considered the gold standard in evaluating the efficacy of new therapies, their generalisability to patients in the real world is often unclear. As a post-authorization safety study, LIBERATE further characterized the safety and effectiveness of PR-FAM in real-world clinical settings. Specifically, premarketing clinical studies were not powered to evaluate the incidence of rare AEs, including seizure. This observational study was powered to detect a doubling in the incidence of seizures in the patient population with MS to further assess whether PR-FAM treatment was associated with an increase in seizure rate. In our study, < 1% of patients in the safety population experienced TEAEs of seizure, with an incidence rate of 0.5 per 100 patient-years. Only one patient experienced seizure events potentially related to an overdose of PR-FAM treatment, which supports the finding that the incidence of seizure is uncommon in routine clinical practice and is consistent with the information in the approved label.
UTI is known to be one of the most common AEs associated with PR-FAM treatment. In this study, the incidence of UTIs (8.7%) is similar to the rate of UTIs observed in placebo-controlled studies reported in the EU SmPC (12.0%) [19].
Pivotal clinical studies during the development of PR-FAM did not include sufficient numbers of patients ≥ 65 years of age to determine whether they responded differently to younger patients. Overall, in LIBERATE, the incidence and types of events observed in patients ≥ 65 years of age were largely comparable with those < 65 years of age. The current EU SmPC does not contain any warnings about use in patients ≥ 65 years of age, and, overall, the data suggest that the current labelling is appropriate because no increased risk inherent with advanced age itself was evident.
Our results also suggest that there were no new safety risks in patients with mild renal impairment. Because PR-FAM is primarily excreted unchanged through the kidneys, patients with renal impairment have higher plasma concentrations, which may be associated with adverse neurologic effects. For this reason, determination of renal function before treatment and its regular monitoring during treatment is recommended in all patients.
PR-FAM therapy was also found to be associated with improvements in patient-reported well-being and physician-reported walking ability. These results support those observed in the ENABLE, MOBILE, and ENHANCE studies in which PR-FAM therapy demonstrated significant and clinically meaningful improvements measured by a broad range of MS-specific and patient-reported endpoints, over 6 and 12 months in patients who received PR-FAM [7,8,9].
As an open-label, observational study conducted in a routine clinical practice setting, there are typical methodologic limitations associated with confounding factors. The main limitation is the lack of randomisation. Although this contributes to the high external validity of the data, it reduces the internal validity of the data. In addition, without a controlled clinical study environment, there may be less control over the quality of the data collection. However, the study was conducted in multiple centers and countries and in a large cohort. The authors acknowledge that there is a shortage of formal effectiveness data in this study; a limitation of a scoring system such as the Physician-Assessed CGI-I of walking ability is that it may not have relied on objective documented data from prior visits to inform the assessment. Furthermore, analysis of effectiveness data by treatment status (on-treatment, or off-treatment but still followed in the study) was subject to potential bias by non-responders in the off-treatment group. An additional limitation was that the effects of concomitant physiotherapy were not considered in conjunction with effectiveness analyses. A specific analysis on the timing of AEs is not included in this manuscript but is considered of interest and will be further explored.
5 Conclusions
LIBERATE was primarily conducted to collect additional safety data and to characterise utilization patterns of PR-FAM in routine clinical practice, including whether the descriptions of potential risks properly reflect the incidence of adverse reactions. The data collected show that the incidence and intensity of potential adverse effects of treatment with PR-FAM are consistent with those described in the product label. The drug utilisation results from LIBERATE showed that the majority of Investigators (≥ 95%) prescribed PR-FAM according to the product label and adhered to the contraindications and recommendations, and the safety results showed that reports of seizure with PR-FAM use are rare and the incidence of seizure is comparable with the background incidence of seizure in patients with MS. Taken together, the results demonstrate that routine risk-minimisation measures through labelling are effective. No unexpected safety concerns were seen in patients treated with PR-FAM for 12 months in routine clinical practice. Furthermore, these results demonstrate that longer-term treatment with PR-FAM improves physician-reported walking ability and patient-reported well-being in patients with MS, which is consistent with clinical benefits reported previously in patients with MS.
References
Asch P. Impact of mobility impairment in multiple sclerosis 2—patient perspectives. Eur Neurol Rev. 2011;6(2):115–20. https://doi.org/10.17925/ENR.2011.06.02.115.
Panitch H, Applebee A. Treatment of walking impairment in multiple sclerosis: an unmet need for a disease-specific disability. Expert Opin Pharmacother. 2011;12(10):1511–21. https://doi.org/10.1517/14656566.2011.586338.
Heesen C, Bohm J, Reich C, Kasper J, Goebel M, Gold SM. Patient perception of bodily functions in multiple sclerosis: gait and visual function are the most valuable. Mult Scler. 2008;14(7):988–91. https://doi.org/10.1177/1352458508088916.
Sutliff MH. Contribution of impaired mobility to patient burden in multiple sclerosis. Curr Med Res Opin. 2010;26(1):109–19. https://doi.org/10.1185/03007990903433528.
Goodman AD, Brown TR, Edwards KR, Krupp LB, Schapiro RT, Cohen R, et al. A phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann Neurol. 2010;68(4):494–502. https://doi.org/10.1002/ana.22240.
Goodman AD, Brown TR, Krupp LB, Schapiro RT, Schwid SR, Cohen R, et al. Fampridine MS-F203 Investigators. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;373(9665):732–8. https://doi.org/10.1016/S0140-6736(09)60442-6.
Hobart J, Ziemssen T, Feys P, Linnebank M, Goodman AD, Farrell R, et al. Assessment of clinically meaningful improvements in self-reported walking ability in participants with multiple sclerosis: results from the randomized, double-blind, phase III ENHANCE trial of prolonged-release fampridine. CNS Drugs. 2019;33(1):61–79. https://doi.org/10.1007/s40263-018-0586-5.
Hupperts R, Lycke J, Short C, Gasperini C, McNeill M, Medori R, et al. Prolonged-release fampridine and walking and balance in MS: randomised controlled MOBILE trial. Mult Scler. 2016;22(2):212–21. https://doi.org/10.1177/1352458515581436.
Macdonell R, Nagels G, Laplaud DA, Pozzilli C, de Jong B, Martins da Silva A, et al. Improved patient-reported health impact of multiple sclerosis: the ENABLE study of PR-fampridine. Mult Scler. 2016;22(7):944–54. https://doi.org/10.1177/1352458515606809.
Rodriguez-Leal FA, Haase R, Thomas K, Eisele JC, Proschmann U, Schultheiss T, et al. Fampridine response in MS patients with gait impairment in a real-world setting: Need for new response criteria? Mult Scler. 2018;24(10):1337–46. https://doi.org/10.1177/1352458517720043.
Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: US Department of Heath, Education, and Welfare Public Health Service Alcohol, Drug Abuse, and Mental Health Administration; 1976.
Hobart J, Lamping D, Fitzpatrick R, Riazi A, Thompson A. The Multiple Sclerosis Impact Scale (MSIS-29): a new patient-based outcome measure. Brain. 2001;124(Pt 5):962–73. https://doi.org/10.1093/brain/124.5.962.
Allart E, Benoit A, Blanchard-Dauphin A, Tiffreau V, Thevenon A, Zephir H, et al. Sustained-released fampridine in multiple sclerosis: effects on gait parameters, arm function, fatigue, and quality of life. J Neurol. 2015;262(8):1936–45. https://doi.org/10.1007/s00415-015-7797-1.
Bakirtzis C, Konstantinopoulou E, Langdon DW, Grigoriadou E, Minti F, Nikolaidis I, et al. Long-term effects of prolonged-release fampridine in cognitive function, fatigue, mood and quality of life of MS patients: The IGNITE study. J Neurol Sci. 2018;395:106–12. https://doi.org/10.1016/j.jns.2018.10.004.
Eriksson M, Ben-Menachem E, Andersen O. Epileptic seizures, cranial neuralgias and paroxysmal symptoms in remitting and progressive multiple sclerosis. Mult Scler. 2002;8(6):495–9. https://doi.org/10.1191/1352458502ms812oa.
Fleming ST, Blake RL Jr. Patterns of comorbidity in elderly patients with multiple sclerosis. J Clin Epidemiol. 1994;47(10):1127–32. https://doi.org/10.1016/0895-4356(94)90099-x.
Christiansen CF, Christensen S, Farkas DK, Miret M, Sorensen HT, Pedersen L. Risk of arterial cardiovascular diseases in patients with multiple sclerosis: a population-based cohort study. Neuroepidemiology. 2010;35(4):267–74. https://doi.org/10.1159/000320245.
Goodman AD, Bethoux F, Brown TR, Schapiro RT, Cohen R, Marinucci LN, et al.; MS-F203, MS-F204, and Extension Study Investigators. Long-term safety and efficacy of dalfampridine for walking impairment in patients with multiple sclerosis: results of open-label extensions of two phase 3 clinical trials. Mult Scler. 2015;21(10):1322–31. https://doi.org/10.1177/1352458514563591.
European Medicines Agency. Fampyra 10 mg prolonged-release tablets (summary of product characteristics). https://www.ema.europa.eu/en/documents/product-information/fampyra-epar-product-information_en.pdf. Accessed 26 June 2020.
Acknowledgements
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript and take responsibility for the integrity of the work as a whole. The authors thank the patients who volunteered for this study and the many site staff members who helped to conduct the study. Biogen provided funding for medical writing support in the development of this paper; Katherine Ayling-Rouse, from Excel Scientific Solutions, wrote the first draft of the manuscript based on input from the authors, and Cindy Bush from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the paper to the authors. The authors had full editorial control of the paper and provided their final approval of all content.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Biogen, Cambridge, MA, USA. Open access fee funded by Biogen.
Conflict of interest
Giovanni Castelnovo has received research/educational grants from Allergan, Biogen, Merz, and Novartis, and speaking/consulting fees from Allergan, Biogen, Ipsen, Merck, Merz, Novartis, and Sanofi-Genzyme. Mark S. Freedman has received research/educational grants from EMD (Canada), Roche, and Sanofi-Genzyme (Canada); consulting fees from Actelion (Janssen/J&J), Alexion, Biogen, Celgene (BMS), EMD, Merck Serono, Novartis, Roche, Sanofi-Genzyme, and Teva Canada Innovation; advisory board/board of directors/similar group for Actelion (Janssen/J&J), Alexion, Atara Biotherapeutics, Bayer, Biogen, Celgene (BMS), Clene Nanomedicine, GRI Bio, Magenta Therapeutics, Merck Serono, MedDay, Novartis, Roche, Sanofi-Genzyme, and Teva Canada Innovation; and speaker bureaus for EMD Serono and Sanofi-Genzyme. Vladimiro Sinay has received reimbursement for developing educational presentations, educational/research grants, and consultation fees/travel stipends from Bayer, Biogen, Biosidus, Gador, Genzyme, Merck, Novartis, Raffo/Asofarma, and Roche. Tamara Castillo-Triviño has received speaking/consulting fees and/or travel funding from Bayer, Biogen, Merck, Novartis, Roche, Sanofi-Genzyme, and Teva. Joep Killestein has received speaking/consulting fees from Biogen, Merck, Novartis, Roche, Sanofi-Genzyme, and Teva. George Kong, Thijs Koster, Heather Williams and Arie R. Gafson are employees of, and hold stock/stock options in, Biogen. Oliver Gerlach and Arnfin Bergmann have no disclosures to declare.
Availability of data and material
Data supporting this article can be requested via the corresponding author.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. LIBERATE was registered at ClinicalTrials.gov (NCT01480063).
Consent
Informed consent was obtained from all individual participants included in this study.
Code availability (software application or custom code)
Not applicable.
Author contributions
All authors were involved in critically reviewing the manuscript for intellectual content, and have read and provided approval of the final submitted version.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Castelnovo, G., Gerlach, O., Freedman, M.S. et al. Safety, Patient-Reported Well-Being, and Physician-Reported Assessment of Walking Ability in Patients with Multiple Sclerosis for Prolonged-Release Fampridine Treatment in Routine Clinical Practice: Results of the LIBERATE Study. CNS Drugs 35, 1009–1022 (2021). https://doi.org/10.1007/s40263-021-00840-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00840-x