
Abstract
Background
Psychostimulants remain first-line treatment options for the management of attention-deficit/hyperactivity disorder (ADHD). A multilayer extended-release bead methylphenidate capsule (provisional name Aptensio XR™, MPH-MLR) with unique release properties is being investigated for the treatment of ADHD.
Objective
The aim of this study was to assess the efficacy (primary) and safety and tolerability (secondary) of MPH-MLR compared with placebo in children and adolescents aged 6–18 years with ADHD.
Methods
This study was a parallel, double-blind, multicenter, placebo-controlled, forced-dose, phase III study in which patients were randomized to placebo or MPH-MLR 10, 15, 20, or 40 mg given once daily. There were four study phases: (1) 4-week screening/baseline; (2) 1-week, double-blind treatment (DBP); (3) 11-week, open-label, dose-optimization period; and (4) 30-day follow-up call. During the open-label dose-optimization period all patients started with MPH-MLR 10 mg, unless the investigator deemed it necessary to begin at a higher dose, and were titrated to an optimized dose (10, 15, 20, 30, 40, 50, 60 mg; all given once daily) based on response and adverse events (AEs). The primary endpoint was the change from baseline to end of DBP in ADHD Rating Scale, 4th Edition (ADHD-RS-IV) total score. Secondary endpoints included changes in ADHD-RS-IV subscales and Clinical Global Impression–Improvement Scale (CGI-I) at the end of the DBP. The primary analysis was an analysis of covariance including terms for treatment, site, and baseline ADHD-RS-IV total score.
Results
A total of 221 patients completed the DBP. The primary endpoint had a statistically significant difference among treatments (p = 0.0046) and sites (p = 0.0018), and baseline covariate made a significant contribution (p < 0.0001). As the MPH-MLR dose increased, the ADHD-RS-IV total score improved; the 20 and 40 mg doses were statistically different (p = 0.0145 and p = 0.0011, respectively) from placebo. Females responded differently than did males (p = 0.0238); there was a significant difference among treatments for males but not for females, partly because only one-third of subjects were female and partly because some females who received placebo had considerable improvement during the DBP. Similarly, the ADHD-RS-IV subscales and CGI-I scores at the end of the DBP also showed more improvement as the dose of MPH-MLR increased. During the open-label phase, ADHD-RS-IV total scores improved (mean change from baseline −22.5) and correlated as the dose of MPH-MLR increased; CGI-I scores also improved. No unexpected AEs were noted.
Conclusions
Dose-related improvements in ADHD-RS-IV scores that exceeded those of placebo were observed in patients treated with MPH-MLR. No new safety signals were noted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Methylphenidate (MPH) multilayer bead extended-release (ER) capsules (MPH-MLR; Aptensio XR™) administered once daily demonstrated significant dose-related improvements in Attention-Deficit/Hyperactivity Disorder Rating Scale, 4th Edition (ADHD-RS-IV) scores compared with placebo in children and adolescents with ADHD. |
The safety profile of MPH-MLR is consistent with other ER MPH formulations. |
The results of this phase III study indicate that MPH-MLR, with a novel release profile, offers a valuable option for the treatment of ADHD in children and adolescents. |
1 Introduction
Nearly 6 million children and adolescents aged 3–17 years in the USA have been diagnosed with attention-deficit/hyperactivity disorder (ADHD) [1]. The practice guideline from the American Academy of Pediatrics recommends medication, including psychostimulants, for the management of ADHD in this population [2]. Methylphenidate (MPH) is a psychostimulant medication that is used extensively for the management of ADHD in children and adolescents. Many formulations of MPH are available to address the unique needs of this population.
Available extended-release (ER) formulations of MPH provide different portions of the total dose as the immediate-release (IR) component (20, 22, 30, and 50 %), as well as different durations of action, allowing the prescriber to tailor therapy to individual needs and responses [3–5]. Inter- and intra-patient variabilities have been described with these agents and contribute to the need for MPH dose titration [6]. Response to treatment for ADHD is individualized [7]; thus, new formulations of MPH continue to be investigated.
A novel ER formulation that incorporates multilayer beads comprising 37 % of the labeled dose as IR MPH into ER capsules (MPH-MLR; Aptensio XR™)Footnote 1 has been developed. Once-daily MPH-MLR is administered orally as intact capsules or the capsule can be opened and sprinkled on food, such as applesauce. Pharmacokinetic studies have demonstrated that following once-daily MPH-MLR administration, a biphasic release profile of MPH is observed [8, 9]. The initial maximum (peak) concentration (C max) occurs approximately 2 h post-dose. A moderate decline in MPH concentration occurs until approximately 5 h after dose administration and then a second C max occurs at about hour 7. Overall, MPH-MLR produced a better fluctuation index (less variability) than IR MPH (Ritalin®, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA) administered three times daily [8, 9]. Intra-subject coefficients of variation were slightly higher (44–136 %) for MPH-MLR during the first 2 h post-dose in the absorption phase compared with the IR product (20–112 %) on day 1 of a 4-day steady-state pharmacokinetic study, but were quite similar after reaching C max and demonstrably tighter through day 4 for MPH-MLR (28–56 %) compared with the IR reference comparator (26–108 %) [8, 9]. Clinical benefit of MPH-MLR, measured in children aged 6–12 years in a laboratory classroom setting, was evident from at least hour 1 (first assessment point) through to the hour 12 final study assessment point [10].
To better understand the potential application of MPH-MLR in the outpatient setting, the pharmacokinetic profile of MPH-MLR must be linked with clinical outcomes. The objective of this study was to assess the efficacy of MPH-MLR compared with placebo in the clinic setting as measured by the clinician-administered ADHD Rating Scale, 4th Edition (ADHD-RS-IV) in children and adolescents aged 6–18 years with ADHD. Evaluation of the safety and tolerability of MPH-MLR during the 1-week double-blind study period was a secondary objective.
2 Methods
2.1 Study Conduct
This study was conducted at 16 sites in the USA from December 2010 to November 2011 (ClinicalTrials.gov identifier NCT01239030 [11]). The study protocol, amendments, and informed consent form were reviewed and approved by an Institutional Review Board for each study site. The study was conducted in compliance with Good Clinical Practice (GCP) guidelines of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, the United States Code of Federal Regulations that relate to clinical trial conduct, and the principles of the Declaration of Helsinki. All patients and/or their guardians provided informed consent prior to screening assessments.
2.2 Study Patients
Children and adolescents (male and female) aged 6–18 years at time of consent with an ADHD diagnosis of all subtypes (except Not Otherwise Specified) as defined in the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM-IV-TRTM) [12] were included if they met defined inclusion and exclusion criteria. ADHD diagnosis was supported by the Schedule for Affective Disorders and Schizophrenia for School-Age Children—Present and Lifetime version (K-SADS-PL) [13]. Recorded baseline ADHD-RS-IV total or subscale scores had to be ≥90th percentile relative to the general population of children by age and sex at screening or baseline. Patients had to require pharmacological treatment for ADHD.
Exclusion criteria included an Estimated Full Scale intellectual level <80 using the four-subtest form of the Wechsler Abbreviated Scale of Intelligence™ (WASI™) [14], and a current primary psychiatric diagnosis of severe anxiety disorder, conduct disorder, psychotic disorder, pervasive developmental disorder, eating disorder, obsessive-compulsive disorder, major depressive disorder, bipolar disorder, substance use disorder, chronic tic disorder, or a personal or family history of Tourette’s syndrome as defined by the DSM-IV-TR criteria and supported by the K-SADS-PL. Patients with a chronic medical illness (seizure, cardiac disorders, untreated thyroid disease, glaucoma), using monoamine oxidase inhibitors or psychotropic medication within 14 days of screening or another experimental drug or device within 30 days of screening, who had a clinically significant electrocardiogram (ECG) or clinical laboratory abnormality at screening and/or baseline, or who were pregnant or lactating were also excluded from the study.
2.3 Study Treatments
All study treatments (MPH-MLR 10, 15, 20, 30, 40, 50, 60 mg, placebo) were given orally once daily in the morning, no later than 10 a.m. and were packaged in bottles of ten capsules for a 1-week dispensing interval and bottles of 30 for 4- and 8-week dispensing intervals. Lot numbers used during the double-blind phase were A07983-002L01 (10 mg), A07983-002L02 (15 mg), A07983-002L03 (20 mg), A07983-002L04 (40 mg), and A07983-001L02 (placebo). During the open-label phase the following lot numbers were used: 10 mg, A07983-003L01 (10 Ct), A07983-005L01 (30 Ct); 15 mg, A07983-003L03 (10 Ct), A07983-003L03 (30 Ct); 20 mg, A07983-003L06 (10 Ct), A07983-005L05 (30 Ct); 30 mg, A07983-003L08 (10 Ct), A07983-005L07 (30 Ct); 40 mg, A07983-003L10 (10 Ct), A07983-005L09 (30 Ct); 50 mg, A07983-006L01 (30 Ct); and 60 mg, A07983-006L0 (30 Ct).
2.4 Study Design
The study included four distinct phases: screening, double-blind, open-label, and safety follow-up.
The screening phase (up to day −28, visit 1) comprised the initial study visit. During this visit, informed consent and medical and psychiatric histories were obtained, vital signs, baseline physical examination and ECG were performed, and serum chemistry and hematology measurements were collected. The K-SADS-PL, WASI™, and the baseline Columbia Suicide Severity Rating Scale (C-SSRS) [15] were assessed. For patients receiving ADHD medications at study entry, a washout period of 48 h (minimum) was initiated prior to beginning the double-blind phase.
The double-blind, forced-dose phase began on day 0 (visit 2), which included baseline assessments (Table 1), recording of body weight and vital signs, and 12-lead ECG. Patients received their randomized, fixed dose of MPH-MLR or placebo for the 1-week, double-blind phase. Dosing began at home in the morning on day 1. During this phase, patients were randomized (1:1:1:1:1) to receive MPH-MLR 10, 15, 20, or 40 mg or placebo following a computer-generated randomization schedule with patients assigned the next random number arranged in an ABCDE block design with each letter representing one of the five treatment groups. There was no site stratification in randomization. Patients weighing ≤25 kg were not assigned to receive the 40 mg dose. This phase concluded on day 7 or later (+3 days, visit 3). At this time, post-double-blind assessments were done (Table 1) and patients who completed the double-blind phase had the option of continuing on to the open-label phase.
Patients choosing to continue to the open-label (dose-optimization) phase had treatment initiated with a once-daily MPH-MLR 10 mg dose unless the investigator deemed it necessary to begin at a higher dose based on previous treatment experience. Patients returned to the clinic at weekly intervals (range 3–7 days) through the first 3 weeks (weeks 2, 3, 4; visits 4, 5, and 6) of the open-label period. Additional mandatory visits occurred at week 8 (visit 7) and the end of week 12 (visit 8, end of open-label period). Unscheduled visits were permitted for additional dose titration as needed. The investigator titrated (up or down) the dose of MPH-MLR based on ADHD-RS-IV and Clinical Global Impression (CGI)–Improvement Scale (CGI-I) scores, tolerability, and clinical judgment until the optimal dose was achieved. MPH-MLR capsules of 10, 15, 20, 30, 40, 50, and 60 mg were available in the open-label phase. The Since-Last version of the C-SSRS was used at all visits during this phase.
A follow-up telephone call was made 30 days (±7 days) following the patient’s last dose of study medication. During the call, patients and/or their guardians were asked about new or unresolved adverse events (AEs) since the last study visit and new medications started since the last study visit.
AEs were collected at each visit. Safety assessments included vital signs, physical examination, ECG, clinical laboratory evaluations, C-SSRS, and AEs.
2.5 Assessments
To interview the parent or guardian, clinicians at each site were instructed to use the ADHD-RS-IV, an 18-item scale that rates symptoms of ADHD as outlined in the DSM-IV-TR [16]. Each symptom is rated on a scale of 0–3: 0 = never or rarely; 1 = sometimes; 2 = often; and 3 = very often. A total score is calculated from a sum of individual scores with total score ranging from 0 (no impairment) to 54 (maximal impairment). Scoring can also be broken into subcategories of inattention and hyperactivity-impulsivity. During the double-blind and open-label phases, clinician raters were instructed to do their best to perform ratings on the same children and adolescents.
The CGI is a clinician-administered tool used to assess the symptoms of ADHD [17]. The tool provides a scoring of initial severity on the CGI–Severity Scale (CGI-S) from 1 to 7; subsequent improvements over time during treatment are rated using the CGI-I. Scoring for the CGI-S is as follows: 1 = normal, not ill at all; 2 = borderline ill; 3 = mildly ill; 4 = moderately ill; 5 = markedly ill; 6 = severely ill; and 7 = among the most extremely ill patients. At subsequent study visits, clinicians used the CGI-I 7-point scale to rate the patients’ total improvement based on comparison with their baseline assessment from 1 = very much improved to 7 = very much worse. In this study, each clinical rater established target symptoms on which to anchor the CGI using baseline clinical problems specific to each individual child enrolled in the study.
2.6 Endpoints
The primary efficacy endpoint measure was change from baseline to end of the double-blind period in the clinician-rated ADHD-RS-IV total score. Secondary endpoints included change from baseline to end of the double-blind period in ADHD-RS-IV subscales of Inattention and Hyperactivity and CGI-I at the end of the double-blind period. Exploratory endpoints included the ADHD-RS-IV total score and subscale scores during weeks 3 through 12 (during the open-label period), assessment of CGI-I at the same timepoints, and an evaluation of the final open-label MPH-MLR dose vs. body weight.
2.7 Statistical Analysis
The safety/intent-to-treat (ITT) population included all patients who took at least one dose of study medication. The efficacy population included all patients who completed the double-blind phase.
The primary efficacy analysis was an analysis of covariance (ANCOVA) of the decrease in ADHD-RS-IV total score during the double-blind phase using the efficacy population. The model had class terms for treatment (five levels), site (sites with less than ten patients were combined into a pseudo site), and a covariate term for baseline ADHD-RS-IV total score. The same model was applied to the ITT population as a sensitivity analysis. Subjects not completing the double-blind phase had the decrease in total score set to zero for the sensitivity analysis.
The key secondary efficacy analysis was a follow-up to the primary analysis using the efficacy population, and each MPH-MLR dose level was compared to placebo using Dunnett’s multiple comparison to control procedure with the family-wise type I error (two-sided) rate set at 0.05. A sensitivity analysis was conducted using the ITT population. Secondary analyses analyzed the ADHD-RS-IV subscales and CGI-I during the double-blind phase using the efficacy population. The ANCOVA model had terms for treatment and site, and the covariate was either the baseline score for the ADHD-RS-IV subscale or the baseline CGI-S score. Comparison of treatments at baseline was accomplished using an analysis of variance (ANOVA) with terms for treatment and site.
The ANCOVA for the subgroup analysis of ADHD-RS-IV included the main effects of treatment, site, age, sex, race, and the interactions of site by treatment, age by treatment, sex by treatment, and race by treatment, as well as the covariate baseline score.
Open-label phase data were summarized descriptively. A linear regression of the final open-label dose on body weight was performed.
It was estimated that a total sample size of 225 would have 80 % power to detect a mean difference in ADHD-RS-IV score between treatment and placebo of 8, based on a standard deviation (SD) of 14.
3 Results
3.1 Patients
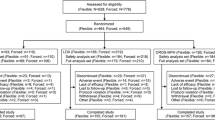
Of the 280 children who were screened, 230 entered the double-blind phase and were administered one of the four strengths of MPH-MLR or placebo (10 mg, n = 49; 15 mg, n = 44; 20 mg, n = 45; 40 mg, n = 45; placebo, n = 47). Most patients were male (67 %) and were predominantly white (69 %) (Table 2). The average age was 10.8 years: 60 children were 6–8 years, 76 were 9–11 years, 63 were 12–14 years, and 31 were 15–18 years. The most common ADHD diagnosis subtype was combined (61 %), with 33 % diagnosed as predominantly inattentive. The most common psychiatric co-morbidities were oppositional defiant disorder (n = 22) and enuresis (n = 14). 221 children completed the 1-week double-blind phase and were included in the efficacy population (Fig. 1) and 200 completed the open-label phase.

Patient disposition
3.2 Efficacy
At baseline (visit 2), there was no significant difference among treatments for either ADHD-RS-IV total score (ANOVA, p = 0.1284) or CGI-S scores (ANOVA, p = 0.1122) (Table 3). There was a significant difference among sites for ADHD-RS-IV total score (ANOVA, p = 0.0197) and CGI-S (ANOVA, p < 0.0001). CGI-S scores revealed a population with substantial disease as >90 % of patients scored as moderately or markedly ill.
The primary efficacy analysis showed that the mean decrease in ADHD-RS-IV total score from baseline to the end of the double-blind phase differed among treatments (Fig. 2a). There were statistically significant differences (ANCOVA) among treatments (p = 0.0046), significant differences among sites (p = 0.0018), and the baseline covariate made a significant contribution to the model (p < 0.0001). The sensitivity analysis produced similar results, reporting a statistically significant difference among treatments (p = 0.0078).

Mean decrease in ADHD-RS-IV total score (a); hyperactivity subscale score (b); and inattention subscale score (c) from baseline (day 0) to the end of the double-blind phase (day 7; efficacy population; n = 221). ADHD-RS-IV Attention-Deficit/Hyperactivity Disorder Rating Scale, 4th Edition, MPH-MLR methylphenidate multilayer extended-release capsules. *p = 0.0145 vs. placebo; **p = 0.0011 vs. placebo; † p = 0.0840 vs. placebo; †† p = 0.0061 vs. placebo; ‡ p = 0.0118 vs. placebo; ‡‡ p = 0.0026 vs. placebo
Key secondary endpoint analysis demonstrated decreases in ADHD-RS-IV total score that were significantly different from placebo for the 20 mg (p = 0.0145) and 40 mg (p = 0.0011) MPH-MLR doses, while the 10 mg (p = 0.2083) and 15 mg (p = 0.0769) doses were not significantly different from placebo.
The mean decrease during the double-blind phase for hyperactivity/impulsivity and inattention subscale scores demonstrated between-treatment differences that were similar to the primary endpoint (Fig. 2b, c, respectively). ANCOVA testing found statistically significant differences among treatments for hyperactivity/impulsivity (p = 0.0240) and inattention (p = 0.0080). The follow-up pairwise comparison of treatments showed that the MPH-MLR 40 mg dose was significantly different from placebo for both subscales (p = 0.0061 hyperactivity/impulsivity, p = 0.0026 inattention), and the MPH-MLR 20 mg dose was significantly different from placebo for the inattention subscale (p = 0.0118).
Subset analyses that examined the decrease in ADHD-RS-IV total score over the double-blind period revealed no difference among treatment groups for all sites, all age groups, and all races. Females responded differently than males (p = 0.0238); there was a significant difference among treatments for males but not for females, partly because only one-third of subjects were females and partly because some females who received placebo had considerable improvement during the double-blind phase. Three of 17 female patients in the placebo group had markedly high values for the ADHD-RS-IV total score at baseline and very low scores for the ADHD-RS-IV total score at the end of the double-blind period.
CGI-I scores at the end of the double-blind phase also showed more improvement as the dose of MPH-MLR increased (Fig. 3). From the ANCOVA, there was a significant difference among treatments (p = 0.0121) and among sites (p = 0.0004), but the covariate baseline CGI-S did not have a significant contribution (p = 0.1029). Pairwise difference from placebo was significant for both the 20 mg (p = 0.0311) and 40 mg (p = 0.0072) doses but not for the 10 mg (p = 0.7391) or the 15 mg (p = 0.5518) doses.

Mean CGI-I scores at end of the double-blind phase (day 7: efficacy population; n = 221). CGI-I Clinical Global Impression-Improvement Scale, MPH-MLR methylphenidate multilayer extended-release capsules, SD standard deviation. *p = 0.0311 vs. placebo; **p = 0.0072 vs. placebo
Throughout the open-label period, ADHD-RS-IV total scores decreased (Fig. 4). At the end of the open-label period, mean (SD) ADHD-RS-IV total scores were 13.5 (8.65), a decrease of 22.5 from baseline.

Arithmetic mean ADHD-RS-IV total scores (lower numbers indicate improvement) from 1 week following the beginning of open-label dose optimization (visit 4) to the end of the open-label phase (visit 8) (efficacy population; n = 221). ADHD-RS-IV Attention-Deficit/Hyperactivity Disorder Rating Scale, 4th Edition
CGI-I scores improved during the open-label period (Fig. 5). The ADHD-RS-IV total score and the mean MPH-MLR dose during the open-label period are plotted in Fig. 6. The mean ADHD-RS-IV total score decreased and the MPH-MLR doses increased as the open-label phase proceeded.

Arithmetic mean CGI-I scores from 1 week following the beginning of open-label dose optimization (visit 4) to the end of the open-label phase (visit 8) (efficacy population; n = 221). CGI-I Clinical Global Impression-Improvement Scale

Mean ADHD-RS-IV total score and mean MPH-MLR dose from 1 week following the beginning of open-label dose optimization (visit 4) to the end of the open-label phase (visit 8) (patients completing open-label phase, n = 200). ADHD-RS-IV Attention-Deficit/Hyperactivity Disorder Rating Scale, 4th Edition, MPH-MLR methylphenidate multilayer extended-release capsules
3.3 Final Open-Label Dose
The most common final open-label dose was 30 mg (27.7 %) followed by 40 mg (25.2 %), 50 mg (17.8 %), 20 mg (16.8 %), 60 mg (8.9 %), 15 mg (2.0 %), and 10 mg (1.5 %). No relationship between the final open-label phase dose and body weight was observed (linear regression, slope p = 0.1039).
3.4 Safety
The most common AEs during the double-blind phase included headache, insomnia, and upper abdominal pain (Table 4). Most events were mild or moderate in severity. Three severe AEs were reported: insomnia by two patients receiving MPH-MLR 40 mg and crying in one patient receiving MPH-MLR 10 mg. Similarly, the most common AEs during the open-label phase were decreased appetite (19.0 %), headache (17.6 %), insomnia (11.8 %), upper abdominal pain (10.9 %), upper respiratory tract infection (6.3 %), irritability (5.4 %), and fatigue (5.0 %); most were mild or moderate in severity. Four patients experienced a severe AE during the open-label phase: viral gastroenteritis (n = 1) and viral infection (n = 1), both of which were unrelated to study treatment, and aggression (n = 1) and mood swings (n = 1), which were related to study treatment.
One serious AE was reported during the double-blind phase. One patient receiving MPH-MLR 15 mg was hospitalized for adjustment disorder with mixed disturbance of emotion and conduct not considered to be related to the study drug. Additionally, one serious AE was reported during the open-label phase. A patient receiving MPH-MLR 30 mg was diagnosed with an injury-related migraine headache not considered to be related to study drug. Both patients with serious AEs withdrew from the study at the time of the event. Additional safety assessments did not reveal any unexpected results beyond the known AE profile for this class of medication.
4 Discussion
Results of this randomized, parallel, double-blind, placebo-controlled study demonstrated that MPH-MLR produced symptom improvement evidenced by ADHD-RS-IV total scores when given to children and adolescents aged 6–18 years with ADHD. Improvements in ADHD-RS-IV total scores correlated with increasing doses of MPH-MLR. Similarly, CGI-I scores indicated bigger improvements as the MPH-MLR dose increased. The two highest MPH-MLR doses yielded significant improvement on the ADHD-RS-IV total score and CGI-I compared with placebo. Although the two lower MPH-MLR doses did not show improvement that was significantly different from placebo, the five treatments descriptively indicated more improvement with higher MPH-MLR doses. The study was not powered to demonstrate a significant difference from placebo for every MPH-MLR treatment group. The finding that the two lowest-dose groups were not significantly different from placebo during the double-blind phase may reflect that only 3.5 % of patients had dose levels of 10 or 15 mg at the end of the open-label (dose-optimization) phase.
MPH-MLR composition (37 % immediate release) in a 10 or 15 mg tablet would result in 3.7 or 5.55 mg of MPH being immediately available, respectively. Treatment improvement in all patients with ADHD would not be expected at these doses. Rather, lower doses are important for dose optimization in the naturalistic clinic setting to permit slow titration to efficacy; this is particularly important for treatment-naïve and younger patients. The randomization forced-dose schedule used in this study did not account for age or previous medication use.
It is difficult to directly compare ER formulations of MPH across independent clinical research studies as study designs and outcome measures vary. However, evaluations of other ER MPH formulations using the ADHD-RS-IV have reported improvements similar to those observed in this study [18, 19]. In our study of MPH-MLR, when each patient’s dose was optimized, the mean ADHD-RS-IV total score was 13.5 for a decrease of 22.5 from baseline. Similarly, at the end of the open-label period of a randomized controlled study using MPH ER oral suspension (MEROS) in children and adolescents aged 6–12 years, those patients receiving MEROS had improved from a mean (SD) of 39.3 (7.6) at baseline to 12.6 (6.3) [19]. In a phase III study of MPH transdermal system (MTS), ADHD-RS-IV was assessed in children aged 6–12 years with ADHD [18]. Significant improvements in ADHD-RS-IV total scores were reported for children randomized to MTS as well as to the active comparator [osmotic-release oral system (OROS)] compared with placebo over the 7-week study period. ADHD-RS-IV scores improved from 43.0, 43.9, and 41.9 to 18.18, 12.8, and 32.1 for MTS, OROS, and placebo, respectively.
We found improvements in ADHD-RS-IV and CGI-I appeared to be dose-related, although the 10 and 15 mg doses did not achieve statistical significance during the 1-week double-blind phase. Further statistical analysis did not identify a relationship between the optimal dose at the end of the open-label period and body weight. However, a separate analysis using a population pharmacokinetic model including data from this study showed a correlation between body weight [ranging from 20 to 80 pounds (9–36 kg)] and change from baseline in ADHD-RS-IV score [20]. Although additional studies are needed to investigate this relationship, this information may be useful to clinicians who are initiating therapy with MPH-MLR.
One treatment goal of pharmacotherapy for ADHD is to provide children and adolescents with the ability to function more effectively. Since school is one of the key settings for this age group, functionality in children and adolescents with ADHD has often been evaluated in the laboratory school setting [21]. MPH-MLR was evaluated in this setting in a randomized, double-blind, controlled, crossover study that included 20 children aged 6–12 years [10]. Once-daily administration of MPH-MLR resulted in significant decrease in Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) scores (p = 0.0001) compared with placebo, suggesting that symptoms of ADHD in the studied children improved with treatment. The current study conducted in a less-structured clinic setting adds to these previously published data.
When administered once daily, MPH-MLR was safe and well-tolerated during the fixed-dose, double-blind phase. Overall, there were no new or unexpected safety signals identified in the study and reported AEs are consistent with the known AE profile of MPH. While there are many psychostimulants available on the market, the unique release pattern of MPH-MLR offers clinicians another option for their patients with ADHD.
Strengths of this study included the large number of patients from multiple sites and use of validated efficacy measures to evaluate response to treatment. However, study participation criteria, particularly the exclusion of patients with a co-morbid psychiatric condition requiring medication treatment, although similar to other ADHD clinical trials, limit the generalizability of the findings to the general population of children and adolescents with ADHD. Also, fixed dosing used during the double-blind phase might have resulted in study patients who were over- or under-treated with MPH-MLR. The double-blind period was short and, overall, the study duration was only 12 weeks, thus limiting a clear understanding of long-term efficacy. The brief washout period (2 days), while equaling or exceeding at least five half-lives for ADHD medications patients may have been using prior to study entry, may have resulted in inflated baseline scores for some patients as it is possible that there were residual pharmacodynamic changes at receptor sites.
5 Conclusions
In this study, the use of MPH-MLR in children and adolescents aged 6–18 years with ADHD led to improvements in ADHD-RS-IV scores that were larger than those observed in placebo-treated patients during a 1-week double-blind evaluation in a dose-related pattern. Improvements continued over a subsequent 11-week open-label study period. No new safety signals were observed. Results indicate that MPH-MLR is a viable treatment option, with a release profile that differs from currently marketed psychostimulants, significantly improving symptoms of ADHD, with an AE profile comparable to other MPH products.
Notes
Rhodes Pharmaceuticals L.P. has received conditional acceptance from the US FDA to use the name Aptensio XR™ for this ER MPH product.
References
Centers for Disease Control and Prevention. Attention deficit hyperactivity disorder (ADHD). http://www.cdc.gov/nchs/fastats/adhd.htm. Accessed 17 June 2014.
Wolraich M, Brown L, Brown RT, DuPaul G, Earls M, Feldman HM, et al. ADHD: clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2011;128(5):1007–22. doi:10.1542/peds.2011-2654.
Markowitz JS, Straughn AB, Patrick KS. Advances in the pharmacotherapy of attention-deficit-hyperactivity disorder: focus on methylphenidate formulations. Pharmacotherapy. 2003;23(10):1281–99.
Quillivant XR™ (methylphenidate hydrochloride) product information. New York: NextWave Pharmaceuticals (2013). http://www.quillivantxr.com. Accessed 24 July 2013.
Wigal SB, Wigal TL, Kollins SH. Advances in methylphenidate drug delivery systems for ADHD therapy. Adv ADHD. 2006;1:4–7.
Ermer JC, Adeyi BA, Pucci ML. Pharmacokinetic variability of long-acting stimulants in the treatment of children and adults with attention-deficit hyperactivity disorder. CNS Drugs. 2010;24(12):1009–25. doi:10.2165/11539410-000000000-00000.
National Institute of Mental Health. Attention deficit hyperactivity disorder (ADHD) (2012). http://www.nimh.nih.gov/health/publications/attention-deficit-hyperactivity-disorder/adhd_booklet_CL508_144426.pdf. Accessed 17 June 2014.
Adjei A, Teuscher NS, Kupper RJ, Chang W-W, Greenhill L, Newcorn J, et al. Single-dose pharmacokinetics of methylphenidate extended-release administered as intact capsule or sprinkles versus methylphenidate immediate-release tablets (Ritalin®) in healthy adult volunteers. J Child Adolesc Psychopharmacol. 2014;24(10):570–8.
Adjei A, Kupper RJ, Teuscher NS, Wigal S, Sallee FR, Childress A, et al. Steady-state bioavailability of methylphenidate extended-release (MPH-MLR) capsule versus methylphenidate immediate-release tablets (Ritalin®) in healthy adult volunteers. Clin Drug Invest. 2014;34(11):795–805.
Wigal SB, Greenhill LL, Nordbrock E, Connor DF, Kollins SH, Adjei A, et al. A randomized placebo-controlled double-blind study evaluating the time course of response to methylphenidate hydrochloride extended-release capsules in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2014;24(10):562–9.
Rhodes Pharmaceuticals, L.P. Efficacy and safety of methylphenidate HCl ER capsules in children and adolescents with ADHD [ClinicalTrials.gov identifier NCT01239030]. US National Institutes of Health, ClinicalTrials.gov. http://www.clinicaltrials.gov. Accessed 22 March 2015.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders, fourth edition, text revision (DSM-IV-TR). Washington, DC: American Psychiatric Association; 2000.
Kaufman J, Birmaher B, Brent D, Rao U, Flynn C, Moreci P, et al. Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL): initial reliability and validity data. J Am Acad Child Adolesc Psychiatry. 1997;36(7):980–8. doi:10.1097/00004583-199707000-00021.
Axelrod BN. Validity of the Wechsler abbreviated scale of intelligence and other very short forms of estimating intellectual functioning. Assessment. 2002;9(1):17–23.
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–77. doi:10.1176/appi.ajp.2011.10111704.
DuPaul GJ, Power TJ, Anastopoulos AD, Reid R. ADHD Rating Scale-IV: checklist, norms, and clinical interpretation. New York: Guilford Press; 1998.
Busner J, Targum SD. The Clinical Global Impressions Scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4(7):28–37.
Findling RL, Bukstein OG, Melmed RD, Lopez FA, Sallee FR, Arnold LE, et al. A randomized, double-blind, placebo-controlled, parallel-group study of methylphenidate transdermal system in pediatric patients with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2008;69(1):149–59.
Robb AS, Findling RL, Childress AC, Berry SA, Belden HW, Wigal SB. Efficacy, safety, and tolerability of a novel methylphenidate extended-release oral suspension (MEROS) in ADHD. J Atten Disord. 2014. doi:10.1177/1087054714533191.
Teuscher NS, Adjei A, Findling RL, Greenhill L, Kupper RJ, Wigal SB. Population pharmacokinetics of methylphenidate hydrochloride extended-release multiple layer beads (MPH-MLR) in pediatric subjects with attention-deficit/hyperactivity disorder. Drug Des Devel Ther (in press).
Wigal SB, Wigal TL. The laboratory school protocol: its origin, use, and new applications. J Atten Disord. 2006;10(1):92–111. doi:10.1177/1087054705286049.
Acknowledgments
This research was funded by Rhodes Pharmaceuticals L.P. The authors would like to acknowledge the contributions of all of the investigators, patients, and parents/guardians of patients. Medical writing assistance was provided by Linda Wagner, PharmD, from Excel Scientific Solutions and funded by Rhodes Pharmaceuticals L.P.
Conflict of interest
Dr. Wigal has been an advisory board and speakers bureau member/consultant for Eli Lilly, Ironshore, Neos, NextWave, Noven, NuTec, Pfizer, Purdue, Rhodes Pharmaceuticals L.P., Shionogi, Shire, and Tris and has received grant and research support from Eli Lilly, Forest, Ironshore, the National Institutes of Health, NextWave, Noven, NuTec, Purdue, Rhodes Pharmaceuticals L.P., Shire, Sunovion, and Tris. Dr. Nordbrock is a consultant for Rhodes Pharmaceuticals L.P. Dr. Adjei and Dr. Kupper are employees of Rhodes Pharmaceuticals L.P. Dr. Childress has received research support from Shire, Novartis, NextWave, Lilly, Forest Research Institute, Johnson & Johnson, Sepracor, Otsuka, Sunovion, Pfizer, Shionogi, Noven, Ironshore, Rhodes, Theravance, Neurovance, Neos, Arbor, Tris, and Purdue. She has received consulting fees or honoraria from Ironshore, Pfizer, Shionogi, Rhodes, Shire, Novartis, and Neos; support for travel from Ironshore, Pfizer, Shire, Novartis, and NextWave; writing assistance on projects from Shire, Novartis, NextWave, Pfizer, Ironshore, and Arbor; and payment for lectures from Shire, Novartis, and Pfizer. Dr. Greenhill has received research support from the National Institute on Drug Abuse/National Institutes of Health and Shire and is on the advisory board for BioBehavioral Diagnostics.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial Registration: ClinicalTrials.gov identifier NCT01239030.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Wigal, S.B., Nordbrock, E., Adjei, A.L. et al. Efficacy of Methylphenidate Hydrochloride Extended-Release Capsules (Aptensio XR™) in Children and Adolescents with Attention-Deficit/Hyperactivity Disorder: A Phase III, Randomized, Double-Blind Study. CNS Drugs 29, 331–340 (2015). https://doi.org/10.1007/s40263-015-0241-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-015-0241-3