
Abstract
Background
Pentobarbital pharmacokinetics (PK) remain elusive and the therapeutic windows narrow. Administration is frequent in critically ill children with refractory status epilepticus (SE) and severe traumatic brain injury (sTBI).
Objectives
To investigate pentobarbital PK in SE and sTBI patients admitted to the paediatric intensive care unit (PICU) with population-based PK (PopPK) modelling and dosing simulations.
Methods
Develop a PopPK model with non-linear mixed-effects modelling (NONMEM®) with retrospective data (n = 36; median age 1.3 years; median weight 10 kg; 178 blood samples) treated with continuous intravenous pentobarbital. An independent dataset was used for external validation (n = 9). Dosing simulations with the validated model evaluated dosing regimens.
Results
A one-compartment PK model with allometrically scaled weight on clearance (CL; 0.75) and volume of distribution (Vd; 1) captured data well. Typical CL and Vd values were 3.59 L/70 kg/h and 142 L/70 kg, respectively. Elevated creatinine and C-reactive protein (CRP) levels significantly correlated to decreased CL, explaining 84% of inter-patient variability, and were incorporated in the final model. External validation using stratified visual predictive checks showed good results. Simulations demonstrated patients with elevated serum creatinine and CRP failed to achieve steady state yet progressed to toxic levels with current dosing regimens.
Conclusions
The one-compartment PK model of intravenous pentobarbital described data well whereby serum creatinine and CRP significantly correlated with pentobarbital CL. Dosing simulations formulated adjusted dosing advice in patients with elevated creatinine and/or CRP. Prospective PK studies with pharmacodynamic endpoints, are imperative to optimise pentobarbital dosing in terms of safety and clinical efficacy in critically ill children.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Pentobarbital demonstrates high PK variability, which is challenging in the clinical context given its narrow therapeutic window, which serves mainly to prevent toxicity than it does to achieve efficacy. |
The population-based pentobarbital PK model demonstrated that creatinine and C-reactive protein significantly influence pentobarbital clearance in critically ill children with SE and sTBI and could explain the majority of the variability. The significance of creatinine as a marker of renal function or overall severity of illness requires further prospective investigation. |
Dosing simulations with the population-based pentobarbital PK model suggest weight-based dosing in children with SE and sTBI should be lower in children with elevated serum creatinine and C-reactive protein values given their failure to achieve steady-state and progress to toxic levels instead. |
1 Introduction
Pentobarbital is an oxybarbiturate analogue of barbituric acid and a potent central nervous system (CNS) depressant mediating its action via γ-aminobutyric acid-sensitive chloride channels [1,2,3]. This short-acting barbiturate has a half-life ranging from 5 to 50 h [3, 4]. Similar to all barbiturates, pentobarbital is metabolised by the hepatic microsomal enzyme system to inactive metabolites [2, 3]. Elimination is by urinary excretion and less commonly in faeces [2, 3]. Intravenously administered pentobarbital quickly distributes into the CNS due to its relatively high lipophilicity (log P 2.16), allowing rapid onset of action [3, 5]. After prolonged infusions, pentobarbital may accumulate in adipose tissue due its lipophilicity, resulting in decreased drug elimination and protracted sedative effects [3, 5].
Pentobarbital has traditionally been used as a sedative-hypnotic and anticonvulsant agent as well as premedication in anaesthetic procedures [1]. Pentobarbital exhibits a dose-dependent effect producing all sedation levels, from drowsiness to deep coma [2, 3]. High-dose pentobarbital administration induces cardiorespiratory depressive effects. As such, pentobarbital intoxication is associated with severe morbidity and mortality [3].
Pentobarbital prescription has been restricted to specific therapeutic applications given its narrow therapeutic window and toxicity profile [1, 3]. In intensive care settings, pentobarbital remains a therapeutic option for the emergency control of refractory seizures in status epilepticus (SE) and refractory intracranial hypertension after severe traumatic brain injury (sTBI) [2, 3, 5,6,7,8,9,10,11]. The therapeutic endpoint of pentobarbital can vary depending on the underlying disease and its course. For example, the goal in refractory SE is burst suppression on electroencephalogram (EEG), whereas in sTBI improved intracranial pressure control (ICP) does not necessarily equate to burst suppression on EEG. High-dose pentobarbital can cause loss of brainstem reflexes and an isoelectric EEG pattern in combination with reduction in cerebral metabolic rate [3, 12]. The latter reduces oxygen demand and cerebral blood flow resulting in ICP decrease, albeit the exact mechanisms of ICP-reduction are unknown [3, 13].
Limited paediatric pharmacokinetic (PK) data are available to guide pentobarbital dosing in paediatric SE and sTBI. The clinical application of pentobarbital in this population is debatable, given safety issues including systemic hypotension, feeding intolerance and propylene glycol toxicity that may counterbalance its clinical benefit, or even lead to multiorgan failure and death [6, 14]. For SE, recommendations for inducing and maintaining adequate plasma pentobarbital levels have been published; however, these are based solely on adult data [6]. In paediatric sTBI, evidence for refractory ICP treatment with pentobarbital is limited, low-level and dated [15, 16].
Children pose unique pharmacological challenges because of significant physiological differences due to rapid growth and developmental changes in comparison to adults [17]. In addition, SE and sTBI paediatric patients are critically ill and have significant PK alterations secondary to illness-related issues, of which inflammation is an increasingly recognised factor [2, 18, 19]. These dynamic developmental and illness-related changes could significantly impact pentobarbital PK. Understanding pentobarbital PK is essential for rational drug dosing in this particularly vulnerable patient group. The aim of this study was to describe pentobarbital PK in critically ill children with SE and sTBI, and to explore how to optimise individual pentobarbital dosing in this population by means of dosing simulations.
2 Methods
2.1 Study Design and Population
This is a single-centre, retrospective study (January 2007–September 2021) at a 28-bed university-based paediatric intensive care unit (PICU) of the Erasmus MC, Sophia Children’s Hospital in Rotterdam, The Netherlands. Patients admitted were aged < 18 years. Inclusion criteria: refractory SE or sTBI defined by a Glasgow Coma Scale (GCS) of ≤ 8 with refractory intracranial hypertension requiring pentobarbital infusion. Per-protocol pentobarbital dosing at our centre is: (1) for SE patients: a 5-mg/kg loading dose of pentobarbital followed by a 3-mg/kg/h continuous infusion; (2) for TBI patients: a 1-mg/kg loading dose of pentobarbital followed by a 2-mg/kg/h continuous infusion. Exclusion criteria: pentobarbital infusion for another diagnosis, incomplete documentation of pentobarbital administration or no documented pentobarbital concentrations in blood serum. Pharmacokinetic model external validation was performed on an independent patient dataset (n = 9) treated with pentobarbital from October 2019 to February 2023 in our hospital. These children were selected with the same inclusion criteria and not included in the initial model building dataset. Study approval was granted by the institutional review board (IRB) with waiver of signed consent (MEC-2019-0072).
2.2 Data Collection
Demographic and clinical data were derived from hospital electronic medical records. Patients who received pentobarbital were identified from the pharmacy database. Demographic data included diagnosis, gender, age, weight, height and PICU length of stay. Missing values for height were resolved using the P50 value for gender-based height-for-age validated growth curves. Laboratory data consisted of documented renal function (serum creatinine and urea), liver assays (albumin, bilirubin, aspartate transaminase (ASAT), alanine transaminase (ALAT), gamma-glutamyl-transferase (γ-GT), alkaline phosphatase (ALP) C-reactive protein (CRP) and blood serum pentobarbital concentrations. Renal function was also evaluated by calculating glomerular filtration rate (GFR) using the Schwartz formula: eGFR = k × L/Scr, whereby eGFR is the estimated GFR in mL/min/1.73 m2, L is height in centimetres, Scr is serum creatinine in milligrams per decilitre and k is an empirical constant that is determined by comparing the L/Scr ratio against measured GFR [20]. A value of 0.365 is used as the k constant in our clinical chemistry department and therefore used for our calculations. Kidney Disease Improving Global Outcomes (KDIGO) criteria (a staging system for renal disease that incorporates creatinine change and diuresis over time; Supplementary Information, Table 1) [21]. Data collected on pentobarbital administration included number and amount of pentobarbital boluses as well as amount and duration of pentobarbital infusion.
2.3 Laboratory Analysis
Pentobarbital was analysed in 100 µL plasma. Ultraviolet detection was performed at 220 nm. Pentobarbital was measured using a high-performance liquid chromatography with diode-array detection (HPLC-DAD) method, which was validated according to Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines. The lower limit of quantification (LLOQ) was 0.5 mg/L and the upper limit of quantification (ULOQ) was 90 mg/L. Internal standard was secobarbital. All validation parameters were within the requirements (amongst all of e.g., precision/accuracy < 15%).
Per local protocol, pentobarbital samples of SE patients are collected 1, 6 and 24 h after initiation of pentobarbital continuous infusion and then once daily. In sTBI patients, pentobarbital samples are collected once daily due to lower dosing practice. In daily practice, the actual frequency of sampling tends to vary.
2.4 Population PK Modelling
Pharmacokinetic analysis was performed by non-linear mixed-effects modelling using NONMEM® Version 7.2.0 (ICON Development Solutions, Ellicott City, MD, USA) and PsN® Version 4.6.0. Pirana® software (version 2.9.5) was used as an interface between NONMEM®, R (version 4.2.1) and Xpose (version 4). Data were analysed using the first-order conditional estimation method with interaction (FOCE-I).
2.4.1 Base Model Development
One- and two-compartment models were tested to fit the pentobarbital plasma concentration data based on visual data inspection, objective function value (OFV) and literature review. Typical values for clearance (CL), volume of distribution (Vd) and inter-compartmental clearance (Q) were estimated. Inter-individual variability (IIV) for each PK parameter was evaluated using an exponential model and residual variability was described as an additive and proportional error. Pharmacokinetic parameters were allometrically scaled with fixed exponents (0.75 for CL and 1 for Vd) to account for variability due to bodyweight differences in a paediatric population [22]. Exponents for allometric scaling were also further estimated during covariate analysis. Shrinkage was calculated for all model parameters with estimated IIV and residual error. A shrinkage below 25% was considered acceptable [23]. Model selection was based on minimum OFVs, parameter precision, error estimates, shrinkages and visual inspection of the goodness-of-fit plots.
2.4.2 Covariate Model Development
Demographic and laboratory characteristics were evaluated as potential model covariates after base model selection. Covariates tested: diagnosis, age, gender, urea, creatinine, eGFR, albumin, bilirubin, ASAT, ALAT, γ-GT, ALP, CRP and KDIGO criteria [21]. The relationship between covariates and ETA distribution was first examined graphically, albeit all covariates were singly added to the model. Continuous covariates were described using an exponential model normalised to population median values or cut-off values when used. Categorical covariates were described using a proportional model [24]. The forward inclusion-backward elimination method was used [25]. Covariates that significantly improved the model with an OFV decrease ≥ 3.84 (p < 0.05 with 1 degree of freedom) were added to the full model. A backward elimination process was performed with a statistical significance of p < 0.001 (ΔOFV ≥ 10.83). Covariate effect on IIV of the PK parameter involved was also evaluated to assess covariate significance.
2.4.3 Model Validation
First, a bootstrap analysis was performed whereby 1000 bootstrap datasets were generated by randomly sampling from the original dataset with replacement [26]. The model was evaluated for its robustness and validity by comparing median values and their corresponding 95% confidence intervals (CI) of the bootstrap samples with the estimates from the original dataset. Second, the model was evaluated using the visual predictive check (VPC) by simulating 1000 datasets [27]. Furthermore, a normalised prediction distribution errors (NPDE) analysis was performed using 1000 simulations. Finally, an independent dataset comprising 9 children treated with pentobarbital for SE or sTBI was used for external validation using a VPC. The VPCs for internal and external validation were stratified for final model covariates.
2.5 Dosing Simulations
Simulations were performed based on covariate value variations included in the final PK model and stratified by body weight. The goal of the simulations was to reach and maintain a pentobarbital concentration of 25 mg/L, which reflects a pentobarbital concentration within the known safety margin. Simulations compared 2 different loading dose strategies and adapted continuous infusions rates accordingly in 0.5 m/kg/h steps. The time interval between loading doses (5 mg/kg) was 5 min and repeated a maximum of 6 times (within a 30-min time span). The rationale of allowing 5-min intervals between loading doses is to allow for observation of the haemodynamic impact of a loading dose before repeating in clinical practice. It must be emphasised that the efficacy of pentobarbital loading doses and continuous infusion rate in terms of alterations in EEG patterns or ICP control was not investigated in these dosing simulations. However, loading doses were incorporated in the simulations as these are common practice for the aforementioned reasons.
3 Results
3.1 Patient Characteristics
Forty-eight patients received intravenous pentobarbital for SE or sTBI at our PICU between January 2007 and February 2023. Two patients were excluded from PK analysis: one patient had an unrealistic documented pentobarbital dose compared to normal dosing reference and two patients had pentobarbital infusion whilst receiving extracorporeal membrane oxygenation (ECMO). No patient had hypothermia (targeted temperature management < 35 degrees Celsius) during pentobarbital infusion. Of the 45 included patients, 36 patients were used for the base model development group of which 22 and 14 received pentobarbital for SE and sTBI, respectively. The external validation group consisted of 9 patients: SE (n = 3) and sTBI (n = 6). Patient characteristics for each model group were evaluated (Table 1).
A total of 178 blood samples were used for the population PK model, and 60 blood samples for external validation. Seven of these samples were beyond the quantification limits (3 < LLOQ and 4 > ULOQ, respectively) but not excluded as this constituted 3% of all samples. Pentobarbital concentrations excluded from PK analysis were from patients receiving concurrent haemodialysis or, if it was unclear, which barbiturate was administered at the time of a documented pentobarbital concentration (e.g., pentobarbital vs phenobarbital). The median measured pentobarbital concentration was 27.5 mg/L (range 0.1–106 mg/L) in the model building group and 33.5 mg/L (range 2–79 mg/L) in the model validation group. Status epilepticus median infusion dose was 3 mg/kg/h (range 0.5–10 mg/kg/h) and the sTBI median infusion dose was 2 mg/kg/h (range 0.05–5 mg/kg/h). Six patients from the external validation group had sTBI, reflecting similarities in loading and infusion dose with the sTBI cohort of our PK model. Regarding adverse events, 3 patients (1 in base model group and 2 in external validation group; all sTBI patients) required continuous veno-venous hemofiltration (CVVH) due to high pentobarbital levels (> 40 mg/L) in combination with refractory shock.
3.2 Base Model
Data were equally well-described by a one- or two-compartment model. For the simplicity of modelling, a one-compartment model was chosen for further model refinement. Including an IIV resulted in a model improvement for CL only. We described the residual error with a combined error model first. The residual error was described as an additive error, as the proportional error was estimated close to zero. In total, 7 samples (3%) were beyond quantification limits (3 < LLOQ and 4 > ULOQ), for which extrapolated concentrations were used. Allometric scaling with fixed exponents (0.75 for CL and 1 for Vd) significantly improved the model (p < 0.001) compared to no allometric scaling. Parameter estimates of the base and final model with their respective standard errors are presented in Table 2.
3.3 Covariate Analysis
The base one-compartment model with allometric scaling was used as a reference for covariate analysis. Estimation of the exponent for allometric scaling of bodyweight was taken into account as covariate. After visual inspection of the covariates with the PK parameters, the univariate analysis resulted in the following significant covariates: diagnosis, age, creatinine, γ-GT, CRP, KDIGO criteria and body weight as presented in Table 3. As for eGFR, it must be noted 67% of height measurements were missing and were estimated based on the P50-value for age. For body weight, no significantly different exponent was estimated, the CI included the fixed value of 0.75. Regarding CRP, relative lower pentobarbital CL was seen in the higher range of CRP. To describe this effect, several cut-off values were evaluated, of which a cut-off value of > 70 mg/L resulted in the best correlation and highest decrease in OFV. Creatinine and CRP significantly correlated with CL (p < 0.001) after backward elimination and hence incorporated in the final model. The covariance step was performed successfully. Using this covariate model, no trends in covariates were found. Inter-individual variability on CL decreased by 84% in the final model with incorporation of creatinine and CRP when compared to the base model. The following equation described the final model for estimation of pentobarbital CL (L/h), whereby the effect of CRP was only included for patients with increased CRP > 70 mg/L:
If CRP ≤ 70 mg/L:
If CRP > 70 mg/L:
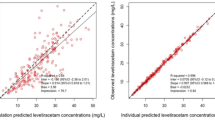
Goodness-of-fit plots of the final model showed good model performance (Supplementary information, Fig. 1), whereby population and individual predictions were evenly distributed around the unity line when plotted against the observations. Trends in interpatient variability of pentobarbital clearance, as seen in the base model, were no longer present after incorporation of the covariates (Supplementary information, Fig. 2). The control stream of the final model is added in the Supplementary information, Fig. 3.
3.4 Model Validation
Bootstrap results showed that the model-based parameter estimates were similar to median values and within the 95% CIs of the bootstrap analysis, indicative of final model stability (Table 2). The VPCs (Fig. 1) and NPDE (Supplementary Information, Fig. 4) demonstrate that observed pentobarbital concentrations (median and variability) fall within the corresponding simulations when stratified by diagnosis with creatinine or CRP, respectively. Some small deviations in variability were seen, which can be explained by the sample size. External validation using stratified VPCs showed good results (Supplementary information, Fig. 5).

Visual predictive check (VPC) of the final model. The VPC’s illustrate how the average trend (solid red line) and variability (two dashed red lines) of the pentobarbital observed concentrations fall within the model-based simulations average trend (red semi-transparent area) variability (blue semi-transparent areas) represented as a 95% confidence interval (CI). A VPC of the final model per diagnosis for creatinine B VPC of the final model per diagnosis for C-reactive protein
3.5 Dosing Simulations
Different dosing regimens were simulated using the final pentobarbital PK model developed in this study for a 6 kg SE patient and a 40 kg sTBI patient as these were the median weight per diagnosis for our study cohort. Initial creatinine and CRP values for these simulations were kept at a value of 26 µmol/L and ≤ 70 mg/L, respectively.
First of all, dosing simulations of continuous pentobarbital infusion (without a loading dose) led to the differing pentobarbital dosing advice based on body weight (Fig. 2). Clearance was lower in patients with a higher body weight. Therefore, lower pentobarbital continuous infusion doses were required to achieve concentrations around 25 mg/L. At the same time, steady state took longer to reach in higher weight categories. Overall, it took at least 48 h to reach steady state in all patients. This could be shortened using loading doses.

Pentobarbital dose adjusted for bodyweight. Dosing simulations were performed to reach a target pentobarbital concentration of 25 mg/L (simulations performed with creatinine values 26 µmol/L and CRP ≤ 70 mg/L)
The effect of loading doses on achieving pentobarbital steady state for each diagnosis and its respective median weight is visualised in Fig. 3 and Supplementary information Figure 6, which includes 90 percentile ranges. A single loading dose (5 mg/kg) did not contribute substantially to achieving steady state. Repeated loading doses with a minimum time interval of 5 min facilitated a more rapid achievement of steady state. The median weight difference between the SE and sTBI patient categories resulted in a different continuous pentobarbital infusion dose between diagnosis groups. This was to ensure the desired steady state concentration (25 mg/L) was not exceeded in the higher weight group (sTBI). For practical purposes at the bedside, the infusion rates during simulations were rounded off to values of 0.5 mg/kg/h and as such the target concentrations ultimately reached 27 mg/L instead of the targeted 25 mg/L. The effect of body weight, creatinine and CRP on pentobarbital concentration is depicted in Fig. 4. In this simulation a pentobarbital maintenance infusion of 3 mg/kg/h without loading dose was used to evaluate the effect on reaching a pentobarbital steady state concentration of 25 mg/L. The effect of elevated creatinine (range 15–180 µmol/L) and CRP (range ≤ 70 to 400 mg/L) on pentobarbital concentrations is more pronounced than weight-based dosing: instead of reaching a steady state, pentobarbital concentrations progress to toxic levels (Fig. 4).

Effect of pentobarbital loading dose simulations on achieving steady state per diagnosis. CRP C-reactive protein, SE status epilepticus, TBI traumatic brain injury

Effect of bodyweight, creatinine and C-reactive protein (CRP) on pentobarbital concentrations using a continuous infusion of 3 mg/kg/h. Simulations were performed for a median patient of 20 kg, creatinine 26 µmol/L and CRP ≤70 mg/L, of which A bodyweight, B creatinine or C CRP was adjusted each time to evaluate the effect on pentobarbital concentrations
3.5.1 Dose Adjustment Recommendations
The simulations yield the following suggestions for dose adjustment of the continuous infusion rate to keep pentobarbital concentrations within the safety margin of 20–40 mg/L: a doubling of creatinine results roughly in halving of pentobarbital clearance. Thus, halving of the pentobarbital continuous infusion rate dose seems necessary. As for CRP, it seemed that no dose adjustments were required for a CRP value ≤ 70 mg/L. Dose adjustments are recommended for a CRP > 70 mg/L as presented in Table 4. It must be noted these dose adjustments are based solely on safety targets with the goal to prevent toxicity and do not reflect clinical efficacy.
If CRP ≤ 70 mg/L:
If CRP > 70 mg/L:
4 Discussion
To our knowledge, this is the first pentobarbital population-based PK study in PICU patients with refractory SE or sTBI. A one-compartment PK model including allometric scaling and IIV on CL described data well. Our pentobarbital PK model demonstrated that serum creatinine and CRP (> 70 mg/L) significantly influence pentobarbital CL requiring pentobarbital dose adjustments based on simulations. Importantly, dosing simulations demonstrated that patients with elevated serum creatinine and CRP failed to reach a pentobarbital steady state and progressed to toxic concentrations without dose adjustments.
The following observations were made concerning pentobarbital pharmacokinetics based on our pentobarbital PK model. First of all, the identification of creatinine and CRP as significant covariates on pentobarbital CL has not been previously documented. Regarding CRP, there is an increasing body of evidence that inflammation significantly impacts cytochrome P450 (CYP) activity leading to alterations in drug clearance [18, 19, 28]. Our findings concerning elevated CRP and decreased pentobarbital CL mirror these results. In the context of pentobarbital CL, the liver has a pivotal role in metabolism. Pentobarbital is subject to low hepatic intrinsic CL and hence its systemic CL would not be primarily sensitive to fluctuations in hepatic blood, which usually occur in critical illness, but rather to alterations in intrinsic enzyme activity or plasma binding [13]. Pentobarbital is mainly metabolised by the hepatic microsomal enzyme system [2], therefore alterations in the activity of hepatic enzymes, such as CYP450, secondary to inflammation, may explain the altered pentobarbital CL of our population [18, 19]. Another indirect marker of a decrease in hepatic function could be reflected by protein synthesis, such as albumin levels. Hypothetically, this could influence pentobarbital protein binding, which can range from 35% to 70% [3]. However, hepatic markers were not significant covariates in our study. Thus, we hypothesise that pentobarbital protein binding alterations are not of clinical significance in the context of pentobarbital CL. As for serum creatinine, this has been reported as a significant covariate on CL in children receiving phenobarbital for seizure management [29]. Phenobarbital is a barbiturate often compared to pentobarbital when administered for refractory SE; thus, our findings could reflect these barbiturate PK similarities. The question remains whether creatinine as a significant covariate solely represents a marker for kidney dysfunction or severity of critical illness in general. To explore this hypothesis, we tested our pentobarbital PK model with other estimates of renal function by estimating eGFR using the Schwartz formula and categorizing patients with the KDIGO criteria. However, these proved to have limited value as a marker of renal function in this retrospective study given the need to estimate most anthropometric data (height), which is commonplace in the acute setting of an intensive care admission, in combination with missing diuresis data. Furthermore, the use of one k-coefficient for all paediatric ages, such as in our clinic, is debatable as it might overestimate eGFR in younger children as proposed by de Souza et al [30]. In addition, neonates and infants show steady GFR increase up to 18 months of age when full maturity is reached [31]. This is of interest, as the median age on our SE cohort was 0.3 years. Thus, eGFRs based on standard creatinine-based equations could be misleading in specific populations, not only based on age but also gender, dietary factors, catabolic and disease state [32]. Second, in terms of pentobarbital CL we identified no dose-dependent effect on pentobarbital CL and half-life within the known range of 5–50 h (24.68 h) as described in the literature [2, 15]. The CL values we determined (3.59 L/h/70 kg) were higher than values reported in a paediatric population PK study in patients who received pentobarbital after open heart surgery (2.96 L/h/70 kg) [2]. A possible reason for this observed CL variation was the younger age in the post-cardiac surgery cohort (median age 6.3 months, range 3.0 days to 4.4 y) which could relate to substantial variation in body weight and/or kidney maturation [2, 31]. Analogous to Zuppa et al, allometric scaling significantly improved our PK model. However, their study also suggested that an age effect on CL remained for subjects aged < 12 months after accounting for difference in body weight. We tested this hypothesis by using an age cut-off (< and ≥ 12 months) to assess kidney maturation and this did not result in further improvement of our pentobarbital PK model.
Third, pentobarbital is a highly lipophilic drug with a rapid blood brain barrier (BBB) penetration and CNS distribution after intravenous administration, ensuring its rapid onset of action [3, 5]. However, continuous infusions lead to rapid drug redistribution into peripheral tissues and result in pentobarbital storage in adipose tissue [3, 5]. Despite this knowledge, it was remarkable to find that a two-compartment model did not provide superior data fit in our study. A possible explanation could be the lack of high frequency sampling at the onset of pentobarbital dosing as well as after discontinuation of pentobarbital infusion. However, we observed a large Vd (142L) reflecting the high lipophilicity of pentobarbital. Interestingly, we did observe Vd differences between lean and obese children of the same age (defined as weight-for-age ≥ 2 standard deviations), with the Vd 71% higher in the latter group, probably secondary to the lipophilic nature of pentobarbital. Moffet et al developed a population PK model for children receiving phenobarbital for seizure treatment and demonstrated that the model with allometrically scaled CL and Vd using fat-free mass was superior to body weight [29]. This observation could also be applicable for pentobarbital, despite not finding a superior PK model fit with two-compartment analysis. Further exploration of body composition effect on pentobarbital PK, using body mass index (BMI) or standardised weight-for-height growth curves, is warranted in a prospective manner.
Finally, the dosing simulations yielded important and clinically applicable findings. (1) From a safety point of view, it could be necessary to prescribe a lower continuous infusion rate for increasing body weight (Fig. 2). This is to prevent exceeding the upper limit of the desired pentobarbital range. (2) Further dose reductions of the continuous pentobarbital infusion are required in patients with elevated creatinine and CRP as these patients fail to attain a steady state and progress to toxic levels. Overall, the effect of elevated creatinine and CRP is more pronounced than the effect of body weight. Therefore, elevations of these values should alert the clinician and clinical pharmacist to perform timely dose reductions in an effort to prevent toxic pentobarbital levels. (3) The actual clinical effect of the loading dose (i.e., rapid control of epileptic activity or raised intracranial pressure) was not evaluated in this study, although simulations with loading doses demonstrated a more rapid achievement of pentobarbital concentrations within desired safety margins. It is conceivable that titration to clinical effect could be achieved by additional loading doses on top of a fixed continuous infusion rate determined by body weight during concurrent therapeutic drug monitoring (TDM) and pharmacodynamic monitoring by EEG and/or ICP trends.
To our knowledge, this is the largest study on pentobarbital PK in critically ill children admitted to the PICU. In addition, our PK model was comprehensively validated with several methods as well as an external validation. Finally, it is the first PK study to describe a significant association between creatinine, CRP and pentobarbital CL. These findings were translated to bedside dosing adjustment recommendations in an effort towards safer pentobarbital dosing practices. The main study limitation is its retrospective nature leading to inherent challenges in data collection. Especially challenging was obtaining reliable markers of renal function whereby anthropometric data (weight and height) often have to be estimated upon admission of the patient due to clinical instability. Furthermore, estimating equations, such as the Schwartz formula, are often associated with limitations when estimating eGFR due to the k values that deviate based on age, gender and creatinine assay [18]. Our clinic used a single k value for all ages, which may not properly reflect developmental and gender variability [30]. Another limitation is that our observations reflect a single-centre experience. Also, drug–drug interactions were not tested as variables that influence pentobarbital pharmacokinetics in our model. Finally, we studied two distinct patient groups who received pentobarbital for different indications with different dosing protocols. The rationale to combine these groups was from a power perspective, providing a wide spectrum for age and variability in dosing practice. Although the pentobarbital model incorporates patient-specific covariates, it must be noted that this might not cover all disease-specific factors of influence on pentobarbital PK. Nonetheless, the pentobarbital PK model described in this study is applicable to different disease conditions and for a wide age range as using the diagnosis itself as a covariate was not significant. As such, this PK model is practical in its clinical use to provide guidance in maintaining pentobarbital levels within known safety margins.
Our observations in the development and subsequent dosing simulations of our pentobarbital PK model for paediatric SE and sTBI suggest high PK variability. We recommend that future studies aim at improving this PK model by prospective data collection in combination with multicentre involvement to enhance patient and sample numbers. Also, dosing simulations with prospective data will further improve model informed precision dosing. Furthermore, the incorporation of other renal markers such as cystatin-C and neutrophil gelatinase-associated lipocalin (NGAL), which are less influenced by factors like muscle mass, age, ethnicity and dietary factors [20, 33], could provide more insight into the role of kidney function in pentobarbital CL. Data regarding degree of overall (critical) illness such as additional markers of inflammation (e.g., interleukin-6) could improve the timeliness of pentobarbital dose adjustments. In addition, illness severity stratification (e.g., Glasgow Coma Scale, Injury Severity Score, Vasoactive-Inotropic Score, and Paediatric Logistic Organ Dysfunction Score) could enable patient subgroup identification at increased risk of altered pentobarbital PK and potential intoxication.
Given the current insights provided by this PK model, we want to raise awareness for potential pentobarbital intoxication when dosing pentobarbital in critically ill patients with elevated creatinine and CRP levels. Due to this high PK variability, we advise adjusting the dose in accordance with our dose recommendations, combined with routine and frequent TDM during pentobarbital treatment. This assists in reaching and maintaining pentobarbital concentrations within the safety margin. Based on our experience, we suggest daily pentobarbital TDM after initiation of pentobarbital infusion and daily creatinine and CRP measurements to promptly identify elevated levels and thus facilitate timely pentobarbital dosing adjustments. In addition, to optimise individualised pentobarbital dosing, defining and correlating pharmacodynamic (PD) endpoints to PK data is necessary. Additional PD studies are warranted to further provide meaningful dosing guidance in this complex population, with special focus on measures of pharmacological efficacy such as EEG or sedation scores/ICP trends and measures of safety, such as haemodynamic stability in terms of vasopressor index score.
5 Conclusion
A population PK model of pentobarbital for paediatric SE and sTBI was developed that successfully describes the concentration-time profile of pentobarbital. Serum creatinine and CRP were significantly correlated with pentobarbital clearance. Dosing simulations incorporating bodyweight, creatinine and CRP yielded significant dose adjustments regarding pentobarbital maintenance infusion. Thus, our PK model demonstrates high pentobarbital PK variability, which can be explained for 84% by creatinine and CRP. This underlines the importance of model-informed precision dosing and TDM. Further prospective paediatric pentobarbital PK/PD studies, including dosing simulations, are required that will incorporate renal biomarkers, markers of inflammation and illness severity scores depending on the underlying disease. These covariates in combination with end points of efficacy and safety will advance our understanding of pentobarbital pharmacology and guide individualised dosing.
References
López-Muñoz F, Ucha-Udabe R, Alamo C. The history of barbiturates a century after their clinical introduction. Neuropsychiatr Dis Treat. 2005;1(4):329–43.
Zuppa AF, et al. Population pharmacokinetics of pentobarbital in neonates, infants, and children after open heart surgery. J Pediatr. 2011;159(3):414-419 e1-3.
Hensler DM, et al. Pentobarbital removal during continuous venovenous hemofiltration: case report and review of the literature. J Pharm Pract. 2018;31(6):682–6.
Ketharanathan N, et al. Analgosedation in paediatric severe traumatic brain injury (TBI): practice, pitfalls and possibilities. Childs Nerv Syst. 2017;33(10):1703–10.
Barberio M, et al. Continuous infusion pentobarbital for refractory status epilepticus in children. J Child Neurol. 2012;27(6):721–6.
Humble RM, et al. Therapeutic drug monitoring of pentobarbital: experience at an Academic Medical Center. Ther Drug Monit. 2015;37(6):783–91.
Pérez-Bárcena J, et al. Pentobarbital versus thiopental in the treatment of refractory intracranial hypertension in patients with traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12(4):R112.
Kim SJ, Lee DY, Kim JS. Neurologic outcomes of pediatric epileptic patients with pentobarbital coma. Pediatr Neurol. 2001;25(3):217–20.
Brophy GM, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012;17(1):3–23.
Tasker RC. Emergency treatment of acute seizures and status epilepticus. Arch Dis Child. 1998;79(1):78–83.
Pugin D, et al. Is pentobarbital safe and efficacious in the treatment of super-refractory status epilepticus: a cohort study. Crit Care. 2014;18(3):R103.
Erbguth F. [Management of refractory and super-refractory status epilepticus] Management des refraktären und superrefraktären Status epilepticus. Med Klin Intensivmed Notfmed. 2019;114(7):628–34.
Schaible DH, et al. High-dose pentobarbital pharmacokinetics in hypothermic brain-injured children. J Pediatr. 1982;100(4):655–60.
Marshall GT, et al. Pentobarbital coma for refractory intra-cranial hypertension after severe traumatic brain injury: mortality predictions and one-year outcomes in 55 patients. J Trauma. 2010;69(2):275–83.
Mellion SA, et al. High-dose barbiturates for refractory intracranial hypertension in children with severe traumatic brain injury. Pediatr Crit Care Med. 2013;14(3):239–47.
Vavilala MS, et al. Acute care clinical indicators associated with discharge outcomes in children with severe traumatic brain injury. Crit Care Med. 2014;42(10):2258–66.
Vinks AA. Important role of population pharmacokinetic/pharmacodynamic modeling in pediatric therapeutics. J Pediatr. 2011;159(3):361–3.
Vet NJ, et al. Inflammation and organ failure severely affect midazolam clearance in critically ill children. Am J Respir Crit Care Med. 2016;194(1):58–66.
Lenoir C, et al. Impact of inflammation on cytochromes P450 activity in pediatrics: a systematic review. Clin Pharmacokinet. 2021;60(12):1537–55.
Schwartz GJ, Work DF. Measurement and estimation of GFR in children and adolescents. Clin J Am Soc Nephrol. 2009;4(11):1832–43.
Selewski DT, et al. Validation of the KDIGO acute kidney injury criteria in a pediatric critical care population. Intensive Care Med. 2014;40(10):1481–8.
Sharma V, McNeill JH. To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol. 2009;157(6):907–21.
Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82(1):17–20.
Joerger M. Covariate pharmacokinetic model building in oncology and its potential clinical relevance. Aaps J. 2012;14(1):119–32.
Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res. 1998;15(9):1463–8.
Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37(6):486–95.
Bergstrand M, et al. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. Aaps J. 2011;13(2):143–51.
Vet NJ, et al. The effect of critical illness and inflammation on midazolam therapy in children. Pediatr Crit Care Med. 2012;13(1):e48-50.
Moffett BS, et al. Phenobarbital population pharmacokinetics across the pediatric age spectrum. Epilepsia. 2018;59(7):1327–33.
De Souza VC, et al. Schwartz formula: is one k-coefficient adequate for all children? PLoS ONE. 2012;7(12): e53439.
Filler G, et al. How should we assess renal function in neonates and infants? Acta Paediatr. 2021;110(3):773–80.
Filler G, Lee M. Educational review: measurement of GFR in special populations. Pediatr Nephrol. 2018;33(11):2037–46.
Filho LT, et al. Accuracy of neutrophil gelatinase-associated lipocalin for acute kidney injury diagnosis in children: systematic review and meta-analysis. Pediatr Nephrol. 2017;32(10):1979–88.
Acknowledgements
The authors thank Joke Dunk for her assistance in data acquisition.
Author information
Authors and Affiliations
Contributions
NK, AL, JMPdV, MdH, BCPK and BCMdW: conceptualization, methodology, statistical analysis, writing, reviewing and editing. EW: reviewing and editing.
Corresponding author
Ethics declarations
Conflicts of interest and source of funding
None.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Ethics approval
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
40262_2023_1249_MOESM1_ESM.tif
Goodness-of-fit plots of the final model. Observed concentrations plotted against population predicted concentrations (A) and individual predicted concentrations (B) of pentobarbital (mg/L). Dashed lines represent the line of identity (TIF 1406 KB)
40262_2023_1249_MOESM2_ESM.tif
Interpatient variability of pentobarbital clearance. The variability in pentobarbital clearance (Y-axis) in relation to creatinine values (µmol/L) or C-Reactive Protein (CRP) values (mg/L) respectively (X-axis), decreased with model refinement from base to final model (figures 3A and figures 3B for serum creatinine and CRP respectively). The final model shows how increased values for Creatinine and CRP decrease pentobarbital clearance (L/hr) (figures 3A and figures 3B upper and lower right plots) (TIF 1687 KB)
40262_2023_1249_MOESM5_ESM.tif
Visual predictive check (VPC) of the final model. The VPC’s illustrate how the average trend (solid red line) and variability (two dashed red lines) of the pentobarbital observed concentrations fall within the model-based simulations average trend (red semi-transparent area) variability (blue semi-transparent areas) represented as a 95% confidence interval (CI). A VPC of the final model per diagnosis for Creatinine B VPC of the final model per diagnosis for C-Reactive Protein (TIF 76 KB)
40262_2023_1249_MOESM6_ESM.tif
Effect of pentobarbital loading dose simulations on achieving steady state per diagnosis including 90-percentile range. Solid lines represent the median concentration of the described dose regimen, the 90-percentile range is represented by the dashed lines. The target concentration of 25 mg/L is indicated by the black dotted line. CRP: C-Reactive Protein; SE: Status Epilepticus; TBI: Traumatic Brain Injury (TIF 913 KB)
40262_2023_1249_MOESM7_ESM.docx
KDIGO acute kidney injury guidelines staging system based on serum creatinine change from baseline and urine production. Legend table 1 eGFR: estimated glomerular filtration rate; KDIGO: Kidney Disease Improving Global Outcomes [21] (DOCX 18 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ketharanathan, N., Lili, A., de Vries, J.M.P. et al. A Population Pharmacokinetic Model of Pentobarbital for Children with Status Epilepticus and Severe Traumatic Brain Injury. Clin Pharmacokinet 62, 1011–1022 (2023). https://doi.org/10.1007/s40262-023-01249-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01249-z