
Abstract
Background
Glepaglutide is a novel, ready-to-use, long-acting, glucagon-like peptide-2 (GLP-2) analog intended for the treatment of patients with short bowel syndrome (SBS). This study investigated the impact of renal function on the pharmacokinetics and safety of glepaglutide.
Methods
In this 3-site, non-randomized, open-label study, 16 subjects were enrolled: 4 with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2), 4 with end stage renal disease (ESRD) not on dialysis (eGFR < 15 mL/min/1.73 m2), and 8 matching controls with normal renal function (eGFR ≥ 90 mL/min/1.73 m2). Blood samples were collected over a 14-day period following a single subcutaneous (SC) dose of glepaglutide 10 mg. Safety and tolerability were assessed throughout the study. The primary pharmacokinetic parameters were area under the curve between dosing and 168 h (AUC0–168 h) and the maximum plasma concentration (Cmax).
Results
There was no clinically relevant difference between subjects with severe renal impairment/ESRD and normal renal function with respect to total exposure (AUC0–168 h) and peak plasma concentrations (Cmax) of glepaglutide following a single SC dose. A single SC dose of glepaglutide 10 mg appeared safe and well tolerated in subjects with normal renal function and subjects with severe renal impairment or ESRD. No serious adverse events were reported, and no safety issues were identified.
Conclusions
No difference in glepaglutide pharmacokinetics was seen between renal impaired and normal subjects. Based on this trial, dose adjustment appears not to be warranted in SBS patients with renal impairment.
Trial Registration
The trial is registered at http://www.clinicaltrials.gov (NCT04178447) and has the EudraCT number: 2019-001466-15.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Chronic and acute renal impairment are common comorbidities of short bowel syndrome (SBS), with approximately 30–50% of SBS patients reported to have decreased renal function. |
Severe renal impairment/end-stage renal disease (ESRD) had no clinically relevant impact on the pharmacokinetic properties of glepaglutide, a long-acting GLP-2 analog intended for the treatment of SBS. Glepaglutide had a similar safety profile in subjects with normal renal function and those with renal impairment/ESRD. |
Based on these results, dose adjustment does not appear to be warranted in SBS patients with renal impairment or ESRD. |
1 Introduction
Short bowel syndrome (SBS) is a rare, heterogenous, and potentially life-threatening malabsorption disorder resulting from surgical resection of the small intestine or a loss of functional intestinal length [1,2,3,4]. The condition is characterized by a decreased absorptive capacity of the gut, resulting in reduced uptake of fluids, nutrients, electrolytes, vitamins, and trace elements, which can lead to dehydration, malnutrition, impaired growth, and weight loss if left untreated. In more severe cases, SBS can result in intestinal failure where the patient is reliant on long-term parenteral support (PS) [5]. Although PS is life sustaining, it is associated with potentially fatal complications including sepsis (mainly catheter-related bloodstream infections) [6], central vein thrombosis, intestinal failure associated liver disease [7] and decreased renal function [8, 9]. Therefore, a major advancement in the treatment of SBS in recent years has been the introduction of glucagon-like peptide-2 (GLP-2) analog therapy to reduce or wean the patient off PS by enhancing the absorptive capacity of the remnant bowel [10, 11].
Native GLP-2 is a 33-amino acid intestinal growth factor secreted in response to nutrient intake that plays a key role in enhancing small intestinal mucosal morphology, function, and integrity under both normal and pathophysiological conditions. Because the short half-life of native GLP-2 (5–7 min) precludes clinical use, GLP-2 analogs have been developed for the treatment of SBS that have greater resistance towards in vivo degradation. Teduglutide is the first GLP-2 analog on the market. With a half-life of approximately 2 h, teduglutide is approved for once-daily treatment of patients with SBS and is provided as a lyophilized powder that requires reconstitution prior to subcutaneous (SC) injection [12, 13].
Glepaglutide is a novel, long-acting GLP-2 analog under development for the treatment of SBS [14]. Glepaglutide comprises 39 amino acids, differing from native GLP-2 by the inclusion of nine amino acid substitutions and a C-terminal tail of six lysines that enable glepaglutide to be formulated as a stable, ready-to-use liquid formulation for SC injection. Following injection, two main glepaglutide metabolites of 35 and 34 amino acids (M1 and M2, respectively) are formed in the SC depot via C-terminal proteolytic cleavage. Both metabolites are pharmacologically active and have similar GLP-2 receptor potency to that of the parent drug [15]. Thus, the pharmacokinetic/pharmacodynamic profile of glepaglutide is viewed as a combined effect of unmetabolized parent drug and its two main metabolites. M2 is the predominant metabolite, accounting for approximately 90% of overall exposure at steady state, with the slow release of M1 and M2 into the circulation from the SC depot resulting in an effective half-life for glepaglutide of 88.3 h [15]. Based on this extended half-life, glepaglutide is under development as a once- or twice-weekly treatment.
Renal impairment is known to affect the metabolization and excretion of many drugs, which may impact exposure and, consequently, efficacy and/or safety. The present study was therefore conducted in accordance with regulatory guidance [16, 17] to compare the pharmacokinetics of glepaglutide in subjects with renal impairment versus subjects with normal renal function to assess whether renal impairment affects drug exposure.
2 Methods
2.1 Study Design
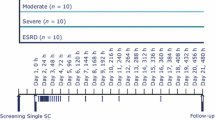
This non-randomized, open-label, two-part Phase 1 trial, designed to compare the pharmacokinetics and safety of glepaglutide in adults with varying degrees of renal function, was conducted at two sites in Hungary (Pharmaceutical Research Associates Hungary Research and Development Ltd, Budapest and St. Imre Teaching Hospital, Budapest) and one site in Poland (SCM Spółka z o.o., Krakow) between 29 November 2019 and 14 July 2020.
The trial consisted of two parts. Part A had two groups: subjects with normal renal function and subjects with severe renal impairment or end stage renal disease (ESRD) not requiring dialysis. Based on the results from Part A, the trial was either terminated or progressed to Part B, consisting of 3 groups: subjects with normal renal function, mild renal impairment, and moderate renal impairment.
The trial protocol, consent form and other information provided to subjects were approved by independent ethics committees (Egészségügyi Tudományos Tanács Klinikai Farmakológiai Etikai Bizottsága, Budapest, Hungary, and Komisja Bioetyczna; przy Okręgowej Izbie Lekarskiej w Krakowie, Krakow, Poland) and by competent authorities. The trial was conducted according to the Declaration of Helsinki and Good Clinical Practice with written informed consent obtained from subjects before trial enrollment. The trial is registered at http://www.clinicaltrials.gov (NCT04178447) and has the EudraCT number: 2019-001466-15.
2.2 Study Population
Eligible subjects were males or females aged between 18–70 years (both inclusive) with a body mass index (BMI) of 20–30 kg/m2. In line with guidance for studies in subjects with renal impairment [16, 17], subjects with renal impairment were recruited and then matched on a group level with subjects with normal renal function with comparable demographics (sex, age, and BMI). Subjects were categorized into a normal renal function group and renal impairment groups (mild, moderate, and severe/ESRD) according to their estimated glomerular filtration rate (eGFR) at screening, calculated using the Modification of Diet in Renal Disease (MDRD) equation [18]: normal renal function (eGFR ≥ 90 mL/min/1.73 m2), mild impairment (eGFR 60 to < 90 mL/min/1.73 m2), moderate impairment (eGFR 30 to < 60 mL/min/1.73 m2), severe impairment (eGFR 15 to < 30 mL/min/1.73 m2), and ESRD (eGFR < 15 mL/min/1.73 m2) not on dialysis.
Subjects with renal impairment were to have a stable disease, with no clinically significant changes in disease status (including disease(s) associated with renal impairment) within 3 months of screening. No changes in medications for the treatment of renal impairment were allowed in the 30 days prior to administration of the study drug.
Key exclusion criteria for all subjects included any clinically relevant medical history likely to interfere with the trial objectives or safety of the participant, uncontrolled hypertension, and current or documented history of repeated clinically significant hypotension or severe episodes of orthostatic hypotension. In addition, subjects with renal impairment were excluded if they had acute renal failure (as judged by the investigator), renal impairment requiring dialysis, a history of kidney transplant regardless of functionality, a serum albumin concentration < 25 g/L, a hemoglobin concentration < 100 g/L, or were taking medications within 60 days prior to study drug administration that were competitors of renal tubular secretion or known to affect the elimination of serum creatinine.
2.3 Procedures
Eligible subjects were admitted to the trial site the day before dosing. On the dosing day (Day 1; morning), subjects received a single SC dose of glepaglutide 10 mg in the abdomen. Subjects stayed at the trial site for the first 3 days, after which they attended daily outpatient visits up to and including Day 8, and a follow-up visit on Day 11. Blood samples for determination of glepaglutide parent drug and the two main metabolites, M1 and M2, were drawn pre-dose and at 30 min and 1, 2, 4, 8, 12, 16, 20, 24, 36, 48, 60, 72, 96, 120, 144, 168 and 240 h post-dose. After Part A of the study, the pharmacokinetics of glepaglutide were compared between subjects with severe renal impairment/ESRD and those with normal renal function before a decision was made to proceed to Part B of the study. This decision was made by a pre-specified Data Review Board comprising the sponsor, the principal investigators, and representatives from the Contract Research Organization (PRA Health Services) that coordinated the trial on behalf of the sponsor. Since no clinically relevant difference in glepaglutide pharmacokinetics was found between the two renal function groups of Part A, the study was stopped according to the study protocol without progressing to Part B.
2.4 Bioanalysis
Venous blood samples were drawn in K2EDTA tubes and stored at − 80 °C until analyzed. Parent drug, M1 and M2 were quantified at the Charles River Laboratories Edinburgh Ltd (UK) using immunoaffinity extraction and subsequent analysis by liquid chromatography–tandem mass spectroscopy (LC–MS/MS); see electronic supplementary material for further details. The LC–MS/MS assay was validated according to current guidelines for bioanalysis of plasma samples and had a lower limit of quantification of 25.0 pmol/L (parent drug and M1) and 50.0 pmol/L (M2), and an upper limit of detection of 2500 pmol/L (parent drug and M1) and 5000 pmol/L (M2).
2.5 Endpoints and Statistical Analyses
The objective of the trial was to compare the pharmacokinetic profile of glepaglutide in subjects with renal impairment relative to the corresponding pharmacokinetic profile obtained in subjects with normal renal function. The primary endpoints were maximum plasma concentration (Cmax) and area under the plasma concentration–time curve from time zero to 168 h (AUC0–168 h) for the constructed analyte (glepaglutide), which was calculated as the sum of parent drug, M1 and M2. Secondary pharmacokinetic endpoints included time to maximum concentration (Tmax), AUC from time zero to infinity (AUC0–∞) and AUC from time zero to time t (AUC0–t), where t is the last time point with concentrations above the lower limit of quantitation (LLOQ). No formal sample size calculations were performed because there was no prior information on the pharmacokinetic variability of glepaglutide in subjects with renal impairment. In line with guidance for the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function [16, 17], approximately 8 subjects were planned to be enrolled per renal function group to obtain at least 6 evaluable subjects for the primary analysis.
Analyses of pharmacokinetic endpoints were based on subjects who were exposed to trial product and had sufficient bioanalytical assessments to calculate reliable estimates of the pharmacokinetic parameters. Pharmacokinetic parameters were estimated via noncompartmental methods using Phoenix WinNonlin® Version 6.4 (Certara USA, Princeton, NJ, USA). Values below LLOQ were set to “missing” for endpoint derivations. Late positive concentration values were set to “missing” if following at least 2 consecutive values below LLOQ. Nominal times were used where actual times were missing.
The primary pharmacokinetic endpoints (Cmax and AUC0–168 h) were log-transformed and evaluated using an analysis of variance (ANOVA), with renal function as fixed effect; ratios of geometric least-squares means (impaired renal function/normal renal function) and corresponding 95% confidence intervals (CIs) were estimated from the model.
Safety and tolerability were assessed throughout the study by means of adverse event (AE) recording (MedDRA preferred terms are presented), vital signs measurements, 12-lead ECGs, clinical laboratory evaluations, physical examinations, and monitoring for injection site reactions.
3 Results
3.1 Subject Disposition and Characteristics
In total, 29 adults were screened, of whom 16 eligible subjects were included in the study: 4 with severe renal impairment, 4 with ESRD not on dialysis, and 8 matching controls with normal renal function. No subjects were withdrawn, and none dropped out during the study; all 16 subjects received a single SC dose of glepaglutide 10 mg, completed the study, and were included in the pharmacokinetic analysis set. All subjects were White and non-Hispanic, and treatment groups were well matched with respect to age, gender, and BMI (Table 1).
3.2 Pharmacokinetics
Geometric mean plasma concentration–time profiles for the constructed glepaglutide analyte (the sum of parent drug, M1 and M2) are shown for subjects with severe renal impairment/ESRD and those with normal renal function in Fig. 1, with pharmacokinetic parameters summarized in Table 2. See electronic supplementary material for geometric mean plasma concentration–time profiles for the glepaglutide parent drug, and the two main metabolites (M1 and M2).

Geometric mean concentration time-profiles for glepaglutide. Vertical bars indicate 95% CIs; glepaglutide = parent + M1 + M2; normal renal function (eGFR ≥ 90 mL/min/1.73 m2); severe/ESRD: severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2) or end stage renal disease (eGFR < 15 mL/min/1.73 m2); parent: unmetabolized glepaglutide; M1: glepaglutide1–35; M2: glepaglutide1–34. CI confidence interval, eGFR estimated glomerular filtration rate, ESRD end-stage renal disease
For both subjects with normal renal function and those with severe renal impairment/ESRD, M2 accounted for approximately 85% of overall glepaglutide exposure in terms of AUC0–∞ and AUC0–168 h, whereas the parent drug contributed less than 2% (Table 2).
As shown in Fig. 2, no difference was found between subjects with severe renal impairment/ESRD and those with normal renal function with respect to the primary pharmacokinetic endpoints of glepaglutide AUC0–168 h (geometric mean ratio [90% CI]: 0.96 [0.69–1.35]) and Cmax (geometric mean ratio [90% CI]: 0.90 [0.62–1.29]). Similar findings were made for M1 and M2, whereas mean values for the parent drug were approximately two-fold higher in subjects with renal impairment (Table 2 and Fig. 2). This difference was small in absolute terms because the parent compound constituted only a small proportion of the overall glepaglutide exposure (Table 2). Renal impairment had no effect on the Tmax of parent drug, M1 and M2 (Table 2).

Forest plots of Cmax and AUC0–168 h for glepaglutide. The primary pharmacokinetic endpoints (Cmax and AUC0–168 h) were log transformed and evaluated using an analysis of variance (ANOVA), with renal function as fixed effect. Ratios of geometric least-squares means (impaired renal function/normal renal function) and corresponding 90% confidence intervals (CIs) were estimated from the model. Glepaglutide = parent + M1 + M2; normal: normal renal function; severe/ESRD: severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2) or end stage renal disease (eGFR < 15 mL/min/1.73 m2); parent: unmetabolized glepaglutide; M1: glepaglutide1–35; M2: glepaglutide1–34. CI confidence interval, eGFR estimated glomerular filtration rate, ESRD end-stage renal disease
3.3 Safety
There were no serious AEs, deaths, or AEs leading to trial withdrawal. A total of 6 AEs (all non-serious and mild) were reported in 3 subjects, of which 2 AEs (2 events of injection site reaction in 1 subject in the severe renal impairment/ESRD group) were considered related to glepaglutide, while the remaining AEs were considered not related (1 event of tinnitus in 1 subject in the severe renal impairment/ESRD group, and 3 events of back pain in 1 subject in the normal renal function group). All AEs had resolved by the end of the study except for the AE of tinnitus, which was ongoing at the last study visit and had stabilized at database closure. There were no clinically significant changes in laboratory evaluations, vital signs, ECG measurements, or physical examinations in the normal renal function or severe renal impairment/ESRD groups.
4 Discussion
Chronic renal impairment is a common comorbidity of SBS, and in particular for patients receiving long-term parenteral nutrition [9]. The prevalence of renal impairment in a broad SBS population has been reported to be approximately 30% [8] compared to approximately 50% of SBS patients receiving long-term PS [8, 9, 19]. For patients who use PS, renal function has been reported to decline much more rapidly than that observed in a healthy population. Patients with short bowel syndrome are also at risk of acute renal dysfunction due to dehydration, diarrhea, kidney stones, and central catheter-related sepsis [20]. Consequently, it is important to establish whether SBS patients with decreased kidney function have altered glepaglutide pharmacokinetics that could warrant dose adjustments.
This Phase 1, non-randomized, open-label trial showed that severe renal impairment/ESRD had no clinically relevant impact on either the exposure or maximum plasma concentrations of glepaglutide compared to healthy matched controls. Similar conclusions were made for the two main metabolites of glepaglutide (M1 and M2), which together accounted for approximately 98% of overall exposure. Consistent with previous findings, the time taken to reach maximum plasma levels of M1 and M2 was considerably longer than for the parent drug [15], reflecting their protracted release from the subcutaneous depot following proteolytic cleavage of the parent drug. Severe renal impairment/ESRD did not affect the time to maximum concentration of glepaglutide, or the relative proportions of parent drug, M1 and M2 in the plasma.
The results of our trial therefore suggest that the glepaglutide dose does not need to be adjusted when treating SBS patients with renal impairment. This differs from teduglutide—the first GLP-2 analog on the market for the treatment for SBS—where it is recommended to reduce the dose by 50% in SBS patients with moderate or severe renal impairment/ESRD due to elevated AUC0–∞ and Cmax [12, 13, 21]. A possible explanation for the absence of an effect of renal impairment on glepaglutide pharmacokinetics is that overall glepaglutide exposure is governed by the slow release of M1 and M2 from the injection site depot into the circulation rather than by clearance via the kidneys as usually observed for peptides. This contrasts with teduglutide, which rapidly enters the circulation [22] and, consequently, renal clearance is expected to play a larger role in determining plasma exposure. Consistent with this, the AUC0–168 h and Cmax of unmetabolized glepaglutide parent drug, which unlike M1 and M2 is rapidly absorbed into the circulation, were approximately two-fold higher in subjects with renal impairment/ESRD. However, since the parent drug constitutes only a small fraction (< 2%) of overall glepaglutide exposure, this was not considered to be clinically relevant.
In conclusion, this trial showed that there was no clinically relevant effect of renal impairment on the pharmacokinetics of glepaglutide. A single SC dose of glepaglutide 10 mg was found to be safe and well tolerated in subjects with normal renal function and those with severe renal impairment or ESRD. These results indicate that glepaglutide can be used without dose adjustment in SBS patients with impaired renal function. Such dosing simplicity is anticipated to be convenient for both patients and healthcare providers.
Change history
13 March 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40262-023-01236-4
References
Jeppesen PB. Spectrum of short bowel syndrome in adults: intestinal insufficiency to intestinal failure. J Parenter Enteral Nutr. 2014;38(1 Suppl):8S–13S.
Mutanen A, Wales PW. Etiology and prognosis of pediatric short bowel syndrome. Semin Pediatr Surg. 2018;27(4):209–17.
Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology. 2003;124(4):1111–34.
Pironi L. Definitions of intestinal failure and the short bowel syndrome. Best Pract Res Clin Gastroenterol. 2016;30(2):173–85.
Pironi L, Arends J, Bozzetti F, Cuerda C, Gillanders L, Jeppesen PB, et al. ESPEN guidelines on chronic intestinal failure in adults. Clin Nutr. 2016;35(2):247–307.
Forbes A. Parenteral nutrition. Curr Opin Gastroenterol. 2005;21(2):192–6.
Goulet O, Joly F, Corriol O, Colomb-Jung V. Some new insights in intestinal failure-associated liver disease. Curr Opin Organ Transplant. 2009;14(3):256–61.
Wang P, Yang J, Zhang Y, Zhang L, Gao X, Wang X. Risk factors for renal impairment in adult patients with short bowel syndrome. Front Nutr. 2020;7: 618758.
Lauverjat M, Hadj Aissa A, Vanhems P, Bouletreau P, Fouque D, Chambrier C. Chronic dehydration may impair renal function in patients with chronic intestinal failure on long-term parenteral nutrition. Clin Nutr. 2006;25(1):75–81.
Cuerda C, Pironi L, Arends J, Bozzetti F, Gillanders L, Jeppesen PB, et al. ESPEN practical guideline: Clinical nutrition in chronic intestinal failure. Clin Nutr. 2021;40(9):5196–220.
Hollanda Martins Da Rocha M, Lee ADW, Marin MLM, Faintuch S, Mishaly A, Faintuch J. Treating short bowel syndrome with pharmacotherapy. Expert Opin Pharmacother. 2020;21(6):709–20.
US Food and Drug Administration (FDA). Gattex (teduglutide): US prescribing information; 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/203441s018lbl.pdf. Accessed 4 Oct 2022.
Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency (EMA). Summary of product characteristics: Revestive (teduglutide); 2022. https://www.ema.europa.eu/documents/product-information/revestive-epar-product-information_en.pdf. Accessed 4 Oct 2022.
Naimi RM, Hvistendahl M, Enevoldsen LH, Madsen JL, Fuglsang S, Poulsen SS, et al. Glepaglutide, a novel long-acting glucagon-like peptide-2 analogue, for patients with short bowel syndrome: a randomised phase 2 trial. Lancet Gastroenterol Hepatol. 2019;4(5):354–63.
Agersnap MA, Sonne K, Knudsen KM, Knudsen CB, Berner-Hansen M. Pharmacokinetics of glepaglutide, a long-acting glucagon-like peptide-2 analogue: a study in healthy subjects. Clin Drug Investig. 2022;42(12):1093–100.
European Medicines Agency (EMA). Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function; 2016. https://www.ema.europa.eu/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf. Accessed 4 October 2022.
US Food and Drug Administration (FDA). Guidance for industry. Pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. Draft guidance; 2020. https://www.fda.gov/media/78573/download. Accessed 4 Oct 2022.
Levey AS, Coresh J, Greene T, Marsh J, Stevens LA, Kusek JW, et al. Expressing the Modification of Diet in Renal Disease Study equation for estimating glomerular filtration rate with standardized serum creatinine values. Clin Chem. 2007;53(4):766–72.
Boncompain-Gerard M, Robert D, Fouque D, Hadj-Aissa A. Renal function and urinary excretion of electrolytes in patients receiving cyclic parenteral nutrition. JPEN J Parenter Enteral Nutr. 2000;24(4):234–9.
Banerjee A, Warwicker P. Acute renal failure and metabolic disturbances in the short bowel syndrome. QJM. 2002;95(1):37–40.
Nave R, Halabi A, Herzog R, Schaffer P, Diefenbach J, Krause S, et al. Pharmacokinetics of teduglutide in subjects with renal impairment. Eur J Clin Pharmacol. 2013;69(5):1149–55.
Marier JF, Beliveau M, Mouksassi MS, Shaw P, Cyran J, Kesavan J, et al. Pharmacokinetics, safety, and tolerability of teduglutide, a glucagon-like peptide-2 (GLP-2) analog, following multiple ascending subcutaneous administrations in healthy subjects. J Clin Pharmacol. 2008;48(11):1289–99.
Acknowledgements
The authors would like to thank all trial subjects, trial staff and investigators. The authors also thank Ebru Ucar Kaarup, M.Sc., (Zealand Pharma A/S) for trial management and Paul G. Drake, PhD, (Zealand Pharma A/S) for medical writing assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Zealand Pharma A/S sponsored the study
Conflict of interest
KMK is employed at Zealand Pharma. MAA and KS were formerly employed at Zealand Pharma. WS has nothing to declare.
Data availability
Provided upon request.
Code availability
Provided upon request.
Author contributions
Zealand Pharma A/S was involved in the design (KS, KMK and MAA designed the trial) and conduct of the study, analysis, and interpretation of the data, including collection, management, and statistical analysis of the data. KMK is an employee of Zealand Pharma. MAA and KS are former employees of Zealand Pharma. The authors had full access to the clinical trial report and associated documents. WS was a trial investigator and helped obtain data. MAA was the medical responsible for the trial and KMK was responsible for the statistical considerations and analyses. All authors were involved in drafting the manuscript or revising it critically for important intellectual content, approved the final version to be published, and take full responsibility for the accuracy and integrity of the content.
Ethics approval
The trial protocol, consent form and other information provided to subjects were approved by independent ethics committees and by competent authorities.
Consent to participate
Written informed consent was obtained from subjects before trial enrollment.
Consent for publication
All authors approved the manuscript for submission.
Additional information
The original online version of this article was revised to correct the images.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Agersnap, M.A., Sonne, K., Knudsen, K.M. et al. Pharmacokinetics, Safety, and Tolerability of Glepaglutide, a Long-Acting GLP-2 Analog, in Subjects with Renal Impairment. Clin Pharmacokinet 62, 645–651 (2023). https://doi.org/10.1007/s40262-023-01215-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01215-9