
Abstract
Background and Objective
The interleukin-36 signalling pathway is associated with pathogenesis of a number of inflammatory diseases. Spesolimab is a selective, humanised, IgG1 antibody that targets the interleukin-36 receptor. We aimed to evaluate the pharmacokinetics, safety and tolerability of single and multiple doses of spesolimab in healthy non-Japanese and Japanese subjects.
Methods
Five phase I clinical studies (three placebo-controlled dose-escalation, two open-label) were conducted in healthy volunteers; single or multiple doses of spesolimab were administered by intravenous infusion or subcutaneous injection. Plasma samples were collected to investigate the pharmacokinetics of spesolimab and evaluate changes with respect to dose, frequency of dosing, formulation and injection site. Immunogenicity, safety and tolerability were also assessed.
Results
Intravenous spesolimab exhibited target-mediated drug disposition at low doses (0.01–0.3 mg/kg) and linear kinetics at doses ≥ 0.3 mg/kg. Steady state was not attained after the fourth weekly dose because of the long half-life (3–5 weeks). Bioavailability of subcutaneous spesolimab increased with increasing dose over the range of 150–600 mg and was higher when administered to the thigh than to the abdomen. The pharmacokinetic profile was consistent between Japanese and non-Japanese subjects. Positive anti-drug antibody responses occurred during the terminal phase of the spesolimab concentration–time profile in 26.7–33.3% and 16.7–37.5% of subjects receiving intravenous and subcutaneous spesolimab, respectively. The impact of anti-drug antibodies on spesolimab pharmacokinetics was low in healthy volunteers, with the impact on spesolimab plasma concentrations only observed in a few subjects at higher titres (≥ 11,400). No serious adverse events were reported; intravenous doses up to 1200 mg were well tolerated in healthy volunteers.
Conclusions
The pharmacokinetic profile and safety data obtained from these phase I clinical studies have been used to guide spesolimab dosing in clinical studies of patients with interleukin-36-mediated diseases.
Clinical Trial Registration
For Studies 1–5, NCT02525679, NCT02852824, NCT03100903, NCT03123094, NCT03617835.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Spesolimab demonstrated linear pharmacokinetics over the dose range of 0.3–20 mg/kg and had a long half-life of 3–5 weeks. |
Spesolimab was well tolerated at clinically relevant doses. |
1 Introduction
Interleukin (IL)-36 belongs to the IL-1 pro-inflammatory cytokine family and includes three pro-inflammatory agonists, IL-36α, IL-36β and IL-36γ, and an IL-36 receptor (IL-36R) antagonist [1,2,3]. The IL-36 signalling pathway is associated with the pathogenesis of several inflammatory diseases, including dermatological disorders such as generalized pustular psoriasis (GPP), palmoplantar pustulosis, allergic contact dermatitis, atopic dermatitis and hidradenitis suppurativa, in addition to inflammatory bowel disease [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18].
Spesolimab (BI 655130) is a selective, humanised IgG1 antibody that blocks agonists binding to IL-36R [2]. Binding of spesolimab to IL-36R modulates the activation of downstream effector pro-inflammatory cytokines implicated in the pathogenesis of GPP, including tumour necrosis factor-α, IL-17, IL-23, IL-1β and IL-8 [2, 18]. Blockade of the IL-36 pathway with spesolimab has a rapid biological effect, normalising cytokine signalling, immune cell activation and neutrophil recruitment, and inhibiting hyperkeratosis to restore epithelial barrier function in the skin [18]. The therapeutic potential of spesolimab has been demonstrated in two clinical trials in patients with GPP flares [9, 19]. Here, we present data from five phase I clinical studies describing the pharmacokinetics, immunogenicity and safety of spesolimab at escalating intravenous (IV) doses up to 20 mg/kg and subcutaneous (SC) doses up to 600 mg in healthy male and female non-Japanese and Japanese subjects.
2 Methods
2.1 Study Designs
Study designs for the five spesolimab phase I studies are described in Table S1 of the Electronic Supplementary Material (ESM). For Studies 1, 2 and 4, subjects were randomised to treatment or placebo using a validated system to produce a pseudorandom number and supplied seed number. Studies 3 and 5 were open-label. Predefined criteria for stopping dose escalation were used for all studies. For the multiple rising dose part of Study 2, each dose group was treated as two consecutive cohorts. All studies had a screening visit within 4 weeks of the first dose and an end-of-study visit (days 71–74 for Study 1, day 148 ± 3 days for Study 4 and day 176 ± 3 days for Studies 2, 3 and 5).
For Study 1, spesolimab doses were selected to cover the subtherapeutic and therapeutic range and estimated to cover low supratherapeutic doses. Dose escalation did not exceed a factor of two from 3 mg/kg onwards. For Study 2, spesolimab doses were selected based on data from Study 1. For Study 3, a maximum dose of 300 mg was selected to provide a dose < 10 mg/kg, which was well tolerated in Studies 1 and 2. For Study 4, IV doses of 300, 600 and 1200 mg were selected to cover the potential therapeutic dose range and provide a safety margin for future clinical development. For Study 5, the 300-mg dose was selected to correspond to the potential maintenance dose of patients in clinical studies, and the 600-mg dose was included to widen the SC dose range.
In all studies, spesolimab was administered under fasting conditions, fluids were not permitted from 1 h pre-dose to 1.5 h post-dose, or 2 h post-SC administration in Study 5, and fluids were restricted until 24 h post-dose (except in Study 5). Standardised meals were provided during the dosing periods. Studies 1–3 and 5 were conducted at the SGS Life Sciences Services Clinical Pharmacology Unit, Antwerp, Belgium. Study 4 was conducted at the Department of Pharmacology and Clinical Pharmacology, University Busan Paik Hospital, Busan, Korea. All studies were conducted in compliance with the ethical principles of the Declaration of Helsinki, all International Council for Harmonisation Good Clinical Practice Guidelines, and local regulatory and legal requirements. All subjects provided written informed consent before participating.
2.2 Subjects
All subjects were non-Japanese, except for those enrolled into Study 4: male individuals were enrolled into Studies 1, 2 and 4; male and female individuals were enrolled into Studies 3 and 5. Female subjects were postmenopausal, surgically sterilised, sexually abstinent or practising accepted methods of contraception from ≥ 30 days before administration of spesolimab and had to have negative pregnancy tests before and during the study.
Subjects were excluded if they had evidence of clinically significant disease, allergies or hypersensitivity; clinically significant abnormalities identified by laboratory testing or physical examination; intention to perform excessive physical activity (e.g. competitive sport) within 1 week prior to spesolimab administration; inability to comply with the dietary regime; evidence of drug abuse; excessive alcohol (> 30 g/day) or cigarette (> 10/day) use; or a history of cardiac arrythmias. Additionally, for Study 4, subjects were excluded if they tested positive for active tuberculosis, human immunodeficiency virus or viral hepatitis; had previous exposure to an IL-36 inhibitor; or had received a live vaccine within 6 weeks prior to randomisation. Lactating women were excluded from Studies 3 and 5.
2.3 Pharmacokinetic Assessments
Blood samples (3 mL) for quantification of spesolimab plasma concentrations were collected at pre-determined timepoints (Table S1 of the ESM). Plasma samples were isolated by centrifugation and stored at − 20 °C within 30 min of collection.
Non-compartmental analyses of spesolimab plasma concentration–time data were performed using WinNonlin software (version 6.3). Parameters included: area under the plasma concentration–time curve (AUC) from time zero to infinity (AUC∞), AUC from time zero to time t (AUCt), AUC during a dosing interval (AUCτ), maximum plasma concentration (Cmax), time to reach maximum plasma concentration (tmax), apparent total body clearance of drug from plasma (CL), accumulation ratio calculated from AUCτ after the last dosing and AUCτ after the first dosing (Rac(AUC)), accumulation ratio calculated from Cmax at steady state and Cmax after single dosing (Rac(Cmax)), elimination half-life (t½) and apparent volume of distribution at steady state (Vss). Concentration values in the lag phase (between time zero and the first timepoint with a concentration above the quantification limit) identified as below the limit of quantification were set to zero.
2.4 Immunogenicity Assessments
Blood samples (3 mL) for quantification of spesolimab anti-drug antibodies (ADAs) were collected in dipotassium ethylenediaminetetraacetic acid-anticoagulant tubes at pre-determined timepoints (Table S1 of the ESM). Serum ADAs were examined using a validated electrochemiluminescent method with biotin-spesolimab capture and ruthenium-spesolimab detection (Studies 1 and 2 used first-generation assays performed at Charles River Laboratories, Reno, NV, USA; Studies 3–5 used second-generation assays performed at QPS, Newark, DE, USA) in a three-tiered approach (screening, confirmation and titration analysis). Sensitivity of the first-generation assay was 62.3 ng/mL, with tolerance limits of spesolimab 100 μg/mL in the presence of 250-ng/mL positive controls. Sensitivity of the second-generation assay was 2.5 ng/mL, with a very high tolerance limit of spesolimab 2000 μg/mL in the presence of 100-ng/mL positive controls. Titres were determined by analysis of serial two-fold dilutions of a sample. The reported titre was the highest fold dilution that produced a mean electrochemiluminescent value more than the confirmatory cut point. For studies using second-generation assays, ADA results were only reported if the plasma concentration was within the drug tolerance level (i.e. for all ADA results, the concentration of spesolimab was below the drug tolerance level). Please refer to the ESM for the categorisation of ADA status.
2.5 Safety Assessments
Safety was assessed by monitoring treatment-emergent adverse events (TEAEs; Medical Dictionary for Regulatory Activities, Version 19.0 [Study 1], Version 20.1 [Studies 2–4] and Version 22.0 [Study 5]), physical examinations, vital signs, 12-lead electrocardiograms, continuous electrocardiogram monitoring (except for Study 5), safety laboratory tests (haematology, coagulation including bleeding time, clinical chemistry and urinalysis) and recording oral body temperature (except for Studies 4 and 5). The investigator assessed local tolerability according to ‘swelling’, ‘induration’, ‘heat’, ‘redness’, ‘pain’ or ‘other findings’.
2.6 Statistical Analysis
All safety and pharmacokinetic (PK) results were analysed using descriptive statistics. No formal calculation of sample size was performed. The safety populations included all subjects who had received spesolimab or placebo. The PK populations included all subjects who had received spesolimab and had evaluable PK data without relevant protocol violations. All subjects with valid PK profiles were included in the PK analysis sets. For the non-compartmental analysis (please refer to the ESM for rules and criteria), concentration data identified with no sample available, no valid result or not analysed were generally not considered. Concentration values in the lag phase identified as below the limit of quantification or no peak detectable were set to zero. All other below the limit of quantification/no peak detectable values of the profile were ignored. The lag phase was defined as the period between timepoint zero and the first timepoint with a concentration above the quantification limit.
For Studies 1, 2 and 4, dose proportionality of spesolimab was explored using a power model based on Cmax, AUCt and AUC∞. The model described the functional relationship between dose and PK endpoints: exp(Yij) = α′*exp(Xi)β + \(\varepsilon_{ij}^{\prime }\). After logarithmic transformations, the model was converted to the linear form: Yij = α + β*Xi + εij. Dose proportionality was assumed if the slope of the regression line (β) was equal to one.
For Study 3, AUCt, Cmax and AUC∞ were used to compare (i) the IV versus SC spesolimab 300-mg formulations and (ii) the SC spesolimab 150-mg versus 300-mg formulations, using an analysis of variance with ‘treatment’ as a fixed effect. The corresponding least-squares means and two-sided 90% confidence intervals (CIs) based on the t-distribution were computed. These data were back-transformed to provide a point estimate and a 90% CI for each comparison. For Study 5, relative bioavailability following SC spesolimab administration at different doses (300 and 600 mg), injection volumes (2 and 1 mL) and injection sites (abdomen and thigh) was explored using an analysis of variance with ‘treatment’ as a fixed effect and ‘matched pair’ as a random effect: ykjm = μ + τk + pj + eijkm.
3 Results
3.1 Subjects
Demographic and baseline characteristics for all phase I studies are summarised in Table 1. For Study 1, two subjects had a body mass index exceeding the upper limit of the target range by < 2%, but this was considered irrelevant for interpretation of the study results. For Study 2, one subject (12.5%) from the multiple-dose placebo group withdrew consent after receiving all four planned doses, and two subjects (33.3%) in the spesolimab 20-mg/kg multiple-dose group discontinued treatment because of adverse events (AEs), one after the first dose and one during the third infusion. For Study 3, one subject (8.3%) from the IV spesolimab 300-mg group discontinued treatment because of an AE of moderate panic attack beginning shortly after the start of IV infusion. For Study 4, four subjects (12.5%) discontinued treatment for personal reasons (informed consent withdrawn; one subject receiving placebo, two subjects receiving IV spesolimab 600 mg and one subject receiving IV spesolimab 1200 mg). No subjects discontinued spesolimab treatment in Study 5. Within each study, treatment groups were similar with respect to demographic and baseline characteristics (Table 1), except for age in Study 1 (mean age 25.5 and 40.0 years in the 0.001- and 0.1-mg/kg groups, respectively) and Study 3 (mean age 42.3, 38.6 and 34.3 years in the SC 150-mg, the IV 300-mg and the SC 300-mg groups, respectively). Between studies, body weight was lower in Studies 3–5, likely reflecting the inclusion of female patients (Studies 3 and 5), and Japanese patients (Study 4).
3.2 Single-Dose and Multiple-Dose Pharmacokinetics Following IV Administration in Non-Japanese Subjects in Studies 1 and 2
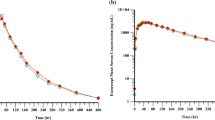
For Studies 1 and 2 in non-Japanese subjects, spesolimab plasma concentrations were below the lower limit of quantification (0.01 μg/mL) for dose groups ≤ 0.003 mg/kg. Spesolimab exhibited target-mediated drug disposition (TMDD) pharmacokinetics after single-dose administration (Fig. 1). A more than dose-proportional increase in Cmax and AUC was noted at low doses but increased linearly within the dose range of 0.3–20 mg/kg (Table 2). Within the dose range of 0.3–20 mg/kg, CL and Vss were not dose dependent (0.147–0.193 L/day and 6.02–7.84 L, respectively) and t½ was 3–5 weeks (Table 2).

Mean (± standard deviation) spesolimab plasma concentration–time profile following single-dose administration to healthy non-Japanese male subjects in Study 1: a linear scale with standard deviation and b semi-log scale. An unplanned group was introduced to accommodate three subjects who were wrongly administered spesolimab 0.05 mg/kg instead of the planned 0.1 mg/kg
Spesolimab exhibited dose proportionality following multiple-dose administration across the dose range of 3–20 mg/kg (Fig. 2), with statistical evaluation in line with these observations (slope values were close to 1 and their 95% CIs included 1). Steady state was not reached for any of the multiple-dose groups. The geometric mean Rac(Cmax) and Rac(AUC) values gradually increased after once-weekly dosing for all dose groups, indicating accumulation of spesolimab. The accumulation ratios after 4 weeks of dosing were similar across treatment groups, and t½ was similar to single-dose administration (Table 3).

Mean (± standard deviation) spesolimab plasma concentration–time profile following multiple-dose administration to healthy non-Japanese male subjects in Study 2: a linear scale with standard deviation and b semi-log scale
3.3 Pharmacokinetics Following SC Administration in Non-Japanese Subjects in Studies 3 and 5
Following SC administration of spesolimab 150 and 300 mg in Study 3, the plasma concentration–time profiles were similar, with a median tmax of 7.02 and 6 days, respectively; as expected, plasma concentrations were higher following IV than SC administration (Fig. 3). The bioavailability of SC spesolimab was dose dependent (approximately 50% with spesolimab 150 mg and approximately 70% with 300 mg; Table 4).

Geometric mean spesolimab plasma concentration–time profile following subcutaneous (SC) or intravenous (IV) administration to healthy non-Japanese subjects in Study 3: a linear scale with standard deviation and b semi-log scale
Following SC administration of spesolimab 150 and 300 mg in Study 5, spesolimab was absorbed with a median tmax of 5.5–7 days across the treatment groups. The apparent t½ was similar across treatment groups, with a geometric mean t½ of approximately 3 weeks. The bioavailability of spesolimab 300 mg administered to the left and right side of the abdomen (two 150-mg injections) was similar to spesolimab 300 mg administered as a single SC injection to the abdomen, with an adjusted geometric mean ratio (90% CI) for AUC and Cmax of 94% (78–112) and 99% (84–117), respectively. The average exposure of spesolimab 300 mg was 40–56% higher following administration to the thigh compared with administration to the abdomen, with an adjusted geometric mean ratio (90% CI) for AUC and Cmax of 140% (117–167) and 156% (132–185), respectively. Spesolimab AUC and Cmax increased more than dose proportionally when the dose was increased from 300 to 600 mg, consistent with the increased bioavailability at higher doses observed in Study 3. The adjusted geometric mean ratio (90% CI) of dose-normalised AUC and Cmax was 122% (102–145) and 138% (117–164), respectively (data on file; BI December 2019; 1368-0029).
3.4 Pharmacokinetics in Japanese Subjects in Study 4
Spesolimab AUC and Cmax following IV administration over the dose range of 300–1200 mg increased in a dose-proportional manner and were consistent (comparable AUC and Cmax) with the spesolimab PK profiles observed in non-Japanese subjects in Studies 1 and 2. No dose dependency was observed for CL and Vss (0.153–0.170 L/day and 6.19–6.97 L, respectively) and t½ ranged from 4 to 5 weeks in all dose groups (Table 2). Spesolimab AUC∞ and Cmax following SC 300-mg administration were 73.5 and 32.3%, respectively, compared with IV 300-mg administration. Median tmax was 5.5 days and t½ was approximately 4 weeks (data on file; BI September 2018; 1368-0009).
3.5 Immunogenicity
3.5.1 Immunogenicity Following IV Administration in Studies 1 and 2
For Studies 1 and 2, the first-generation assay was used to assess ADAs. Following IV administration in Study 1, six spesolimab-treated subjects (10%) had treatment-induced or boosted ADAs (median onset time 4 weeks). In Study 2, one subject (17%) following single-dose administration and one subject (4%) following multiple-dose administration was ADA-positive (onset time of 17 and 21 weeks, respectively). The impact of immunogenicity on spesolimab pharmacokinetics was evaluated by visually examining individual plasma concentration–time profiles overlaid with ADA status and titre. In Study 1, ADA formation had a minimal impact on spesolimab plasma concentrations, with shortened t½ and lower spesolimab concentration observed in one subject with a high ADA titre; there was no apparent impact in Study 2 (Fig. 4a, b, respectively). All hypersensitivity events reported (4/58 subjects [6.9%] in Study 1 and 7/30 subjects [23.3%] in Study 2) were non-serious and prior to ADA formation. Two of the four subjects in Study 1 and one of the seven subjects in Study 2 with hypersensitivity reactions later developed treatment-related ADAs.

Individual spesolimab plasma concentration–time profiles overlaid with anti-drug antibody (ADA) status and titre value following intravenous administration in Studies 1 and 2: a single-dose (SD) intravenous administration in healthy non-Japanese male subjects in Study 1 and b SD and multiple-dose (MD) intravenous administration in healthy non-Japanese male subjects in Study 2
3.5.2 Immunogenicity Following SC Administration in Studies 3 and 5
Following SC administration in Study 3, treatment-induced ADAs were detected in 13 subjects (36%; SC 150 mg, n = 5; SC 300 mg, n = 4; IV 300 mg, n = 4; median onset time 9–10 weeks). Of these subjects, one showed a transient ADA response, and 12 showed a potentially persistent or persistent ADA response. ADA formation in Study 3 had no apparent impact on spesolimab plasma concentrations (Fig. 5a). Hypersensitivity events were reported in 11/36 subjects (30.6%) and were all non-serious and prior to ADA formation; of these subjects, two later developed treatment-related ADAs. In Study 5, 17 subjects (35%) had treatment-induced ADAs (16 were female and one was male; median onset time 8 weeks). Formation of ADAs appeared to lower spesolimab plasma concentrations in some subjects (Fig. 5b) and was mostly observed during the terminal phase of the spesolimab concentration profile, when spesolimab exposure was ≤ 1000 ng/mL. As TMDD begins to dominate at this concentration range, the lower drug concentrations observed in some subjects may not be entirely attributable to ADA formation. Overall, there was no substantial difference in spesolimab exposure (Cmax and AUC) between ADA-negative and ADA-positive subjects. A non-serious hypersensitivity event was reported in 1/48 subjects (2.1%), which occurred after ADA formation.

Individual spesolimab plasma concentration–time profiles overlaid with anti-drug antibody (ADA) status and titre value following subcutaneous (SC) or intravenous (IV) administration in Studies 3–5: a single-dose (SD) SC or IV administration in healthy male and female non-Japanese subjects in Study 3; b SD SC administration in healthy male and female non-Japanese subjects in Study 5; and (c) SD SC or IV administration in healthy male Japanese subjects in Study 4. l + r left + right, pmbl periumbilical
3.5.3 Immunogenicity in Japanese Subjects in Study 4
Four subjects (22%) in Study 4 developed treatment-induced ADAs (median onset time 17 weeks), and formation of ADAs had no impact on spesolimab plasma concentrations (Fig. 5c). No hypersensitivity or local tolerability events were reported (data on file; BI September 2018; 1368-0009).
3.6 Safety
The TEAEs observed during the phase I studies are summarised in Table 5. No serious or severe TEAEs were reported; all TEAEs were mild or moderate in severity and resolved without intervention. For Studies 1, 2 and 4, the proportion of subjects with TEAEs was similar between spesolimab and placebo groups. For all studies, no relevant changes were observed in safety laboratory tests, vital signs or electrocardiograms, except for one subject (8%) in Study 5 receiving spesolimab 600 mg (administered to the left and right side of the abdomen) who had aspartate aminotransferase and alanine aminotransferase values up to 3.1 × the upper limit of normal. Assessments of local tolerability revealed no relevant issues.
In Study 1, no subjects discontinued because of AEs, with the most frequently reported TEAEs being nasopharyngitis (21%), headache (9%) and influenza-like illness (7%). In Study 2, two subjects discontinued study treatment because of AEs. One patient experienced mild pyrexia (associated with dyspnoea, headache, back pain, oropharyngeal pain and fatigue) and because of the concurrent infection was withdrawn from the trial. The other patient received three infusions of spesolimab 20 mg/kg and experienced mild potential infusion-related reactions (associated with anxiety, dizziness, dyspnoea, headache and paraesthesia) after each infusion; these AEs were considered significant according to the International Council for Harmonisation E3 guideline, and treatment was discontinued. The pyrexia event was considered unrelated to treatment. The most frequently reported TEAEs following single-dose and multiple-dose spesolimab administration were nasopharyngitis (33 and 42%, respectively), headache (17 and 42%, respectively) and injection-site erythema (50 and 21%, respectively). Headache was the only drug-related AE that followed a dose-dependent increase.
In Study 3, one subject discontinued study treatment because of an AE of moderate panic attack beginning shortly after the start of infusion, which was considered possibly drug related. The overall incidence of AEs was higher in the 300-mg group (both IV and SC) than in the 150-mg SC group (IV 300 mg 92%, n = 11; SC 300 mg 92%, n = 11; SC 150 mg 67%, n = 8), a difference mainly driven by the TEAE of dermatitis acneiform. The incidence of AEs assessed as possibly drug related was numerically higher following SC than IV administration (SC 150 mg 42%, n = 5; SC 300 mg 42%, n = 5; IV 300 mg 25%, n = 3).
In Study 4, there were no discontinuations because of AEs. The overall incidence of TEAEs was higher in the placebo group (25%) than the spesolimab groups (13%). All TEAEs were observed following IV administration (one subject in each of the 300-mg [mild upper respiratory tract infection], 600-mg [mild contusion] and 1200-mg [mild gastroenteritis and moderate temporomandibular joint disorder] dose groups); no TEAEs were observed following SC administration. None of the TEAEs were assessed as drug related.
In Study 5, there were no discontinuations because of AEs. The overall incidence of TEAEs was similar across treatment groups. The most frequently reported TEAEs were headache (44%, n = 21), acne (38%, n = 18) and injection-site erythema (29%, n = 14).
4 Discussion and Conclusions
Spesolimab is a selective, humanised antibody against IL-36R that has shown clinical efficacy in patients with GPP [9, 11, 19]. The phase I studies were conducted to aid dose selection in an initial proof-of-concept study and the global Effisayil™ 1 study [9, 11, 19]. The initial proof-of-concept study demonstrated rapid and continued efficacy after a single IV spesolimab 10-mg/kg dose [9]. In Effisayil™ 1, a global, randomised, placebo-controlled study, a single IV spesolimab 900-mg dose was associated with rapid pustular and skin clearance within 1 week [19].
Following escalating single-dose IV administration in non-Japanese subjects in Study 1, spesolimab exhibited TMDD at low doses of 0.01–0.3 mg/kg. Saturation of the non-linear elimination pathway occurred at approximately 0.3 mg/kg, with linear kinetics from ≥ 0.3 mg/kg onwards. CL and Vss were not dose dependent. We did not know whether to expect TMDD prior to these studies, as the IL-36R expression level in the blood or tissues is unknown. It should be noted that the nonlinearity in spesolimab pharmacokinetics at very low doses may reflect the TMDD in organs that are easily accessible, such as the blood. In the skin, slow distribution may be the rate-limiting step, and spesolimab concentration in the tissue may be largely determined by the distribution process rather than elimination, i.e. spesolimab pharmacokinetics observed in plasma may not mirror what happens in the skin, and plasma pharmacokinetics may not capture the TMDD in the skin. It is unknown whether IL-36R target occupancy in the skin is saturated at the 0.3-mg/kg dose. Following multiple-dose administration in non-Japanese subjects in Study 2, spesolimab exhibited an approximately linear dose–exposure relationship over the dose range of 3–20 mg/kg. Steady state was not attained after 4-weekly doses because of the long t½ (3–5 weeks).
Following SC administration in Study 3, Cmax was attained in approximately 7 days. At 300 mg, the relative bioavailability of spesolimab following SC administration in non-Japanese subjects in Study 3 was approximately two-thirds of the total IV exposure (AUC). The relative bioavailability in non-Japanese subjects in Study 5 following SC spesolimab administration to different injection sites indicated that (1) exposure was similar following a single or double (left and right side) administration to the abdomen; (2) bioavailability was 40–56% higher following administration to the thigh versus the abdomen; and (3) there was a trend towards higher bioavailability with an increasing dose. Based on pooled analyses across studies, bioavailability (90% CI) was 56% (46–68), 65% (59–71) and 74% (64–85) following abdominal SC administration of 150 mg, 300 mg and 600 mg, respectively. The reason for higher spesolimab bioavailability following administration to the thigh versus the abdomen is unclear. As tmax following subcutaneous injection into the thigh and abdomen was similar, lymph flow is unlikely to account for the PK differences. In the abdomen, the lower bioavailability (with similar tmax) suggests higher pre-systemic catabolism (elimination), but there are no target density data in different parts of the body to support this hypothesis.
The observed PK profile was consistent in Japanese and non-Japanese subjects. Therefore, no adjustments to spesolimab dosing recommendations are warranted for future clinical studies.
In Studies 1 and 2, ADAs developed in few subjects (9%) after single-dose IV administration, and in even fewer (4%) after multiple-dose IV administration. The lower ADA incidence rates observed in these two studies may be related to the use of first-generation ADA methods. Whilst first-generation ADA methods did not impact Study 1 ADA results (all spesolimab concentrations were below the drug tolerance level after week 3), it may have impacted Study 2 ADA results, particularly for the 10-mg/kg and 20-mg/kg multi-dose groups. Consequently, the ADA incidence could be higher than reported for Study 2, although we did not observe any impact on plasma concentration. ADA samples from the other studies were analysed using improved second-generation methods; all spesolimab concentrations were below the drug tolerance level, with no impact on ADA results that detected ADAs in 27 and 35% of subjects who received IV and SC spesolimab, respectively. Overall, the ADA incidence was similar between IV and SC administration. The ADA incidence appeared to be higher in female subjects than male subjects; although the maximum titre was comparable between male and female subjects, following single-dose administration of SC or IV spesolimab, the ADA incidence rate was 39–52% in female subjects compared with 19–24% in male subjects. This sex difference in ADAs may account for some of the observed inter-study variations in the observed ADA rate. For example, Study 4 was conducted in Japanese male volunteers and had a lower total ADA incidence of 21%. The majority of volunteers were female in Study 3 (n = 33/36) and Study 5 (n = 40/48), and had higher overall ADA incidences. In general, the impact of ADAs on spesolimab pharmacokinetics was low in healthy volunteers. In all studies, the impact of positive ADAs on spesolimab plasma concentrations was observed in a few subjects (range 0–17%) at higher titres (≥ 11,400) and occurred during the terminal phase of the spesolimab plasma concentration–time profile, when the exposure was low or depleted. At this concentration range, TMDD begins to dominate and may complicate the interpretation of the impact of ADAs (i.e. lower spesolimab concentrations observed in some subjects may also be related to TMDD instead of entirely due to ADA formation). There was no association between ADA formation and the route of administration or hypersensitivity reactions.
Spesolimab was well tolerated at doses up to 1200 mg, with nasopharyngitis and headache being the most common TEAEs. All TEAEs were described as mild to moderate in nature. Generally, the proportion of subjects with TEAEs was similar between spesolimab and placebo groups. The overall incidence of TEAEs was similar when spesolimab exposure (Cmax and AUC) was substantially lower following SC versus IV administration, and when spesolimab exposure was substantially higher following spesolimab 600-mg administration to different injection sites. Spesolimab was well tolerated in healthy non-Japanese and Japanese subjects.
Limitations of our studies include the small number of subjects per dose group, which limited the safety data. We used a non-compartmental analysis to gain an initial understanding of the pharmacokinetics of spesolimab, and a population PK model (including data from healthy volunteers, patients with GPP and all other potential indications) will be the subject of another publication. Use of a very conservative dose-escalation approach to account for limitations in preliminary PK data was a strength of the study.
Two other IL-36R antibodies are undergoing clinical development. In a phase I trial of 72 healthy subjects, IV imsidolimab (10–750 mg) had a median t½ range between 3 and 4 weeks, bioavailability was approximately 90% and exposure (Cmax and AUC) increased in a dose-dependent manner [20]. Another IL-36R monoclonal antibody, HB-0034, is currently undergoing phase I clinical development, with data due at the end of 2022.
Overall, PK data from the phase I clinical studies suggest that spesolimab exhibits linear kinetics at doses ≥ 0.3 mg/kg and has a long t½ of 3–5 weeks. Following SC administration, spesolimab bioavailability was dependent on dose and injection site, with higher bioavailability observed in the thigh compared with the abdomen. Spesolimab exposure in Japanese subjects was within the range of non-Japanese subjects. The impact of ADAs on spesolimab pharmacokinetics was overall low in healthy volunteers; however, additional ADA data will need to be collected in future studies to strengthen the data for ADAs in target patient populations. Spesolimab was well tolerated at clinically relevant doses, with a similar proportion of subjects reporting TEAEs in the spesolimab and placebo groups. Data from these phase I clinical studies have been used to guide spesolimab dosing in clinical studies of patients with GPP.
References
Queen D, Ediriweera C, Liu L. Function and regulation of IL-36 signaling in inflammatory diseases and cancer development. Front Cell Dev Biol. 2019;7:317.
Ganesan R, Raymond EL, Mennerich D, et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. MAbs. 2017;9:1143–54.
Towne JE, Renshaw BR, Douangpanya J, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286:42594–602.
Marrakchi S, Guigue P, Renshaw BR, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365:620–8.
Gabay C, Towne JE. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J Leukoc Biol. 2015;97:645–52.
Yi G, Ybe JA, Saha SS, et al. Structural and functional attributes of the interleukin-36 receptor. J Biol Chem. 2016;291:16597–609.
Johnston A, Xing X, Wolterink L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017;140:109–20.
Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev. 2018;281:169–78.
Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380:981–3.
Mrowietz U, Burden AD, Pinter A, et al. Spesolimab, an anti-interleukin-36 receptor antibody, in patients with palmoplantar pustulosis: results of a phase IIa, multicenter, double-blind, randomized, placebo-controlled pilot study. Dermatol Ther. 2021;11:571–85.
Choon SE, Lebwohl MG, Marrakchi S, et al. Study protocol of the global Effisayil 1 phase II, multicentre, randomised, double-blind, placebo-controlled trial of spesolimab in patients with generalized pustular psoriasis presenting with an acute flare. BMJ Open. 2021;11: e043666.
Otobe S, Sugaya M, Nakajima R, et al. Increased interleukin-36γ expression in skin and sera of patients with atopic dermatitis and mycosis fungoides/Sézary syndrome. J Dermatol. 2018;45:468–71.
Suárez-Fariñas M, Ungar B, Correa da Rosa J, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol. 2015;135:1218–27.
Hessam S, Sand M, Gambichler T, et al. Interleukin-36 in hidradenitis suppurativa: evidence for a distinctive proinflammatory role and a key factor in the development of an inflammatory loop. Br J Dermatol. 2018;178:761–7.
Medina-Contreras O, Harusato A, Nishio H, et al. Cutting edge: IL-36 receptor promotes resolution of intestinal damage. J Immunol. 2016;196:34–8.
Scheibe K, Backert I, Wirtz S, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut. 2017;66:823–38.
Russell SE, Horan RM, Stefanska AM, et al. IL-36α expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. 2016;9:1193–204.
Baum P, Visvanathan S, Garcet S, et al. Pustular psoriasis: molecular pathways and effects of spesolimab in generalized pustular psoriasis. J Allergy Clin Immunol. 2022;149:1402–12.
Bachelez HCS, Marrakchi S, Burden AD, Tsai TF, Morita A, Navarini AA, Effisayil 1 Trial Investigators, et al. Trial of spesolimab for generalized pustular psoriasis. N Engl J Med. 2021;385:2431–40.
Londei M, Khanskaya I, Pinkstaff J, et al. A phase 1 study of ANB019, an anti-IL-36 receptor monoclonal antibody, in healthy volunteers. In: Poster presented at the European Academy of Allergy & Clinical Immunology (EAACI) Congress 2018 [Abstract 0527].
Acknowledgements
Michèle Underhill, of OPEN Health Communications (London, UK), provided medical writing, editorial and formatting support, which was contracted and funded by Boehringer Ingelheim.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Boehringer Ingelheim.
Conflict of interest
David Joseph and Xiujiang Li are employees of Boehringer Ingelheim Pharmaceuticals Inc., Ridgefield, CT, USA. Christian Thoma is an employee of Boehringer Ingelheim International GmbH, Biberach, Germany. Thomas Haeufel is an employee of Boehringer Ingelheim International GmbH, Ingelheim, Germany.
Ethics approval
These studies were conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Council for Harmonisation Good Clinical Practice Guidelines, and all local regulatory requirements were followed.
Consent for participation
All subjects were advised on the risks and benefits of participation in these studies and submitted written informed consent.
Consent for publication
All subjects signed informed consent regarding publication of their study data.
Availability of data and material
To ensure independent interpretation of clinical study results and enable authors to fulfil their role and obligations under the International Committee of Medical Journal Editors criteria, Boehringer Ingelheim grants all external authors access to clinical study data pertinent to the development of the publication. In adherence with the Boehringer Ingelheim Policy on Transparency and Publication of Clinical Study Data, scientific and medical researchers can request access to clinical study data when it becomes available on Vivli-Center for Global Clinical Research Data, and earliest after publication of the primary manuscript in a peer-reviewed journal, regulatory activities are complete and other criteria are met. Please visit Medical & Clinical Trials | Clinical Research | MyStudyWindow for further information.
Code availability
Not applicable.
Author contributions
All authors equally contributed to the concept, design and drafting of the manuscript. All authors read and approved the final manuscript. All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Joseph, D., Thoma, C., Haeufel, T. et al. Assessment of the Pharmacokinetics and Safety of Spesolimab, a Humanised Anti-interleukin-36 Receptor Monoclonal Antibody, in Healthy Non-Japanese and Japanese Subjects: Results from Phase I Clinical Studies. Clin Pharmacokinet 61, 1771–1787 (2022). https://doi.org/10.1007/s40262-022-01176-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01176-5