
Abstract
Background and Objectives
YLB113 is being developed as a biosimilar of the antitumor necrosis factor-alpha antagonist etanercept, which is approved for the treatment of moderate-to-severe rheumatoid arthritis (RA) and other chronic immune-mediated inflammatory diseases. An open-label, crossover, pharmacokinetic study was conducted to compare the relative bioavailability and safety of YLB113 and the etanercept reference product (RP) Enbrel®.
Methods
Healthy male subjects aged 18–50 years were randomized to receive a single subcutaneous dose of YLB113 in one period and the etanercept RP in another period. A washout period of 28 days separated the two treatment periods. Blood samples were collected for pharmacokinetic analysis predose and until 480 h postdose during both periods.
Results
Overall, 52 subjects were enrolled, including 51 subjects who completed the first period and 43 subjects who completed the second period. The 90% confidence intervals for the least squares means derived from an analysis of the log-transformed pharmacokinetic parameters maximum serum concentration (Cmax), area under the serum concentration–time curve (AUC) from 0 to the last measurable concentration (AUC(0-t)) and AUC from 0 to infinity (AUC(0-∞)) for etanercept were between the limits of 80 and 125%. Thus, YLB113 met the bioequivalence criterion. YLB113 and the etanercept RP were well tolerated, with 24 subjects reporting 53 adverse events, including 42 mild and 11 moderate events. Treatment-emergent adverse events were reported by 14 and 16 subjects following the administration of YLB113 and the etanercept RP, respectively.
Conclusions
A single dose of YLB113 exhibited pharmacokinetic and safety profiles comparable with those of the etanercept RP in healthy adult male subjects. Therefore, YLB113 and the etanercept RP can be considered bioequivalent. These findings support the continued development of YLB113 for use in patients with RA.
Jordan Food & Drug Administration unique trial number
31/Clinical/2018.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
YLB113 is being developed as a biosimilar of the antitumor necrosis factor-alpha antagonist etanercept, which is approved for the treatment of moderate-to-severe rheumatoid arthritis (RA) and other chronic immune-mediated inflammatory diseases |
Pharmacokinetic and safety data demonstrate the bioequivalence of YLB113 and the etanercept RP in healthy adult male subjects, supporting the continued development of YLB113 for use in patients with RA. |
1 Introduction
Etanercept was one of the first of a group of modern drugs called biologics or biologicals to be developed. Unlike synthetic drugs, biologics are complex molecules produced by living cells. Biologics are designed to mimic or change processes in the human body and are used to treat various diseases ranging from cancer to rheumatoid arthritis (RA).
RA is an autoimmune disease in which the body’s immune system—which normally maintains health by attacking foreign substances such as bacteria and viruses—mistakenly attacks the joints, leading to pain and irreversible deformation and destruction of the joints if not treated adequately [1]. Approximately 1.5 million people in the United States have RA. In European countries, between 20 and 50 cases per 100,000 inhabitants are newly diagnosed each year [1]. Worldwide, 0.24% of the adult population have RA.
Etanercept is a tumor necrosis factor-alpha (TNF-α) antagonist used in the treatment of adult and juvenile RA, psoriasis, psoriatic arthritis, and ankylosing spondylitis [2]. Adults with moderate-to-severe RA are given etanercept in combination with methotrexate when the response to disease-modifying antirheumatic drugs has been inadequate [2, 3]. It can be given as monotherapy in patients who cannot tolerate methotrexate or when continued methotrexate treatment is not appropriate. Etanercept is a recombinant, fully human, soluble, TNF-α fusion protein consisting of two copies of the extracellular ligand-binding domain of the human 75-kDa TNF-α receptor linked to the Fc portion of human immunoglobulin G1 [3]. The structure of etanercept makes it 50- to 100-fold more potent at binding to TNF-α than the endogenous unconjugated soluble TNF-α receptor [4].
High treatment costs restrict access to high-quality biological medicines. In the case of etanercept, biosimilar versions of this recombinant protein have been developed to provide high-quality alternatives that are more economically attractive than their present recombinant TNF-α fusion protein counterparts [4]. A biosimilar is a biological agent that is highly similar but not identical to a reference product (RP) and is considered for separate marketing approval after the expiration of the RP’s patent [5]. Biosimilars meet high standards for comparability to the reference product and are approved for use in the same indications [4].
The current study was performed to examine the pharmacokinetic bioequivalence of a 50 mg solution of the etanercept biosimilar YLB113 to a 50 mg solution of the marketed product Enbrel® (the etanercept RP) after single subcutaneous (SC) administration in healthy adult male volunteers. In addition, the safety profiles of YLB113 and the RP were compared.
2 Methods
2.1 Study Design
This was an open-label, randomized, two-period, two-treatment, two-sequence, crossover, balanced, single-dose, comparative pharmacokinetic study (Jordan Food & Drug Administration unique trial number: 31/Clinical/2018). The study protocol was prepared in line with the requirements set in the European Medicines Agency (EMA) guidance for conducting bioequivalence studies [6]. Subjects were randomized to receive a single dose of YLB113 in one period and the etanercept RP in the other period. A washout period of 28 days separated the two doses. In sequence 1, subjects received YLB113 or the etanercept RP followed by a 28-day washout period. In sequence 2, subjects were crossed over to either the etanercept RP or YLB113, depending on what they received in sequence 1.
Each period consisted of 22 days, including the following:
-
Day −1: admission of subjects to the clinical site (approximately 24 h prior to study drug administration)
-
Day 0: study drug administration, starting with dosing at approximately 8 am and subsequent scheduled blood sampling until the 12 h postdose sample
-
Days 1–5 (confinement period): pharmacokinetic blood sample collection and safety monitoring
-
Days 6–20 (144–480 h): pharmacokinetic blood sample collection and safety monitoring.
2.2 Subjects
Subjects were healthy male adults selected from the Jordanian population who were physically and mentally healthy, aged 18–50 years, and had a body weight of at least 63 kg, a body mass index (BMI) between 18.5 and 30 kg/m2, results for all red blood cell indices and hemoglobin within the normal ranges or ± 5% of the medical laboratory reference ranges at screening, white blood cells within the medical laboratory reference range at screening and admission to period 1, a normal electrocardiogram (ECG) at screening, kidney and liver function tests within their medical laboratory reference ranges, and vital signs within the following ranges: systolic blood pressure, 90–140 mm Hg; diastolic blood pressure, 60–90 mm Hg; body temperature, 36.1–37.8 °C; pulse, 60–100 beats per minute; and respiratory rate, 12–18 breaths per minute at screening and admission.
Subjects were excluded based on the identification of significant diseases or clinically abnormal findings during screening, skin abnormalities at the injection site, a history of difficulty in donating blood, a known allergy to etanercept or any of its ingredients, sensitivity to latex, a history of or a current tuberculosis infection, major illness within 1 week of the first dosing, a history of or current compulsive abuse of alcohol, moderate smoking (> 10 cigarettes/day), a history of hepatitis B, or a history of cancer. Subjects were also excluded if they participated in a bioavailability/bioequivalence study within 80 days of the first dosing, donated blood within 80 days of the first dosing, would have donated (through to the completion of this study) > 900 mL of blood in 20 weeks, or had a positive test for HIV I and II, hepatitis B, or hepatitis C. Considering that there is no apparent etanercept-related pharmacokinetic difference between male and female subjects [7], only males were included in this study.
2.3 Treatment
Patients received 1 mL of a YLB113 50 mg solution (AY Pharmaceuticals Co., Japan) or 1 mL of an Enbrel® 50 mg solution (the etanercept RP; Pfizer Limited, UK).
Subjects fasted overnight for at least 10 h and received a standard breakfast 30 min before dosing (subjects started their breakfast within 30 min prior to the administration of the drug and ate their breakfast within 30 min). After the subjects had finished breakfast and within 30 min of starting the meal, the study drugs were administered slowly via the SC route in the abdomen with the subjects adopting a supine posture, according to the randomization schedule.
With a quick, short motion, the needle was pushed all the way into the skin at an angle between 45° and 90° without pushing the needle into the skin too slowly or with great force. When the needle was completely inserted into the skin, the skin was released with the free hand and the syringe was held near to its base to stabilize it. The plunger was then pushed to inject all the solution at a slow, steady rate. After emptying the syringe, the needle was pulled out of the skin at the same angle as when it was inserted. The subinvestigators pressed a cotton ball or gauze over the injection site for 10 s without rubbing the injection site. The 5-cm area around the navel was avoided.
Subjects were confined to the clinical facility and activities were monitored by clinical staff for 120 h following treatment. Subjects remained in a supine position for 30 min after dosing, with only necessary movement allowed.
2.4 Sample Collection and Analysis
Blood samples (5 mL) were collected in each study period at predose and at 2, 4, 12, 18, 24, 30, 36, 48, 60, 72, 96, 120, 144, 168, 216, 264, 312, 384, and 480 h postdosing. Subjects remained at the clinical facility for 120 h postdosing and returned to the clinical facility for subsequent collection of ambulatory blood samples, compliance assessment, and to have their vital signs and well-being assessed. After collection, whole blood samples were clotted at room temperature for 45 ± 15 min and then spun at 1600 × g for 10 min at 10 °C. Each pharmacokinetic serum sample was separated into two aliquots. Samples were stored at − 70 ± 20 °C until shipment on dry ice to Lupin Bioresearch Center, Pune, India, for analysis.
Temperature loggers were used during shipment to record the internal temperature and ensure that the samples were maintained in a frozen state. The samples were received in good condition and in a frozen state with sufficient dry ice in the package at the analytical site, so they were analyzed for etanercept using a validated sandwich enzyme-linked immunosorbent assay (ELISA) method.
A sensitive and selective method based on antigen–antibody complex formation using sandwich ELISA to quantify etanercept in human serum over the concentration range of 75.000–1100.000 ng/mL was developed and validated at Lupin Bioresearch Center, Pune, India (for further details, see the Electronic supplementary material, ESM).
The serum sample/standard containing etanercept was loaded onto an ELISA plate coated with human anti-etanercept antibody and incubated for 12–72 h at 2–8 °C. Later, a labeled detection antibody (anti-etanercept horseradish peroxidase antibody) specific to etanercept was added and incubated, which caused etanercept to become sandwiched between the capture and detection antibody. This complex was developed for colorimetric detection with tetramethylbenzidine substrate; the reaction was terminated by the addition of stop solution and the endpoint was measured at 450 nm and quantified with a known standard curve. The main objective of the methodology was to evaluate the etanercept that was captured and detected by the antibody pair. The study sample analysis was performed on an ELISA reader (Synergy™ H1 Microplate reader; BioTek Instruments, Inc., Winooski, VT, USA) with absorbance mode color detection (optical density), utilizing an endpoint of 430 nm and a reference of 630 nm.
Pharmacokinetic samples from the pivotal pharmacokinetic study were reanalyzed as part of the incurred sample reproducibility assessment. The results of the incurred sample reanalysis met the acceptance criterion, demonstrating satisfactory reproducibility of the pharmacokinetic assay throughout the sample analysis period.
2.5 Adverse Events
All adverse events (AEs) were graded as follows: (1) mild—causing no limitation of usual activities, the subject may experience slight discomfort; (2) moderate—causing some limitation of usual activities, the subject may experience annoying discomfort; or (3) severe—causing inability to carry out usual activities, the subject may experience intolerable discomfort or pain.
2.6 Statistical Analysis
Missing samples and samples with nonreportable concentrations were excluded during statistical and pharmacokinetic analysis. Concentrations below the lower limit of quantification were considered to be zero during statistical and pharmacokinetic analyses. Analyses of variance (ANOVAs) were performed on the untransformed pharmacokinetic parameters. Log-transformed data were used for analysis. Untransformed and log-transformed pharmacokinetic parameters—the maximum serum concentration (Cmax), the area under the serum concentration–time curve from 0 to the last measurable concentration (AUC0-t), and the area under the serum concentration–time curve from 0 to infinity (AUC0-∞)—were analyzed using ANOVA. The ANOVA model included sequence, formulation, and period as fixed effects and subject (sequence) as a random effect. Pharmacokinetic equivalence of the test product (A) with the RP (B) for etanercept was demonstrated if the 90% confidence interval (CI) of the ratio of the geometric least squares means for each ln-transformed pharmacokinetic parameter Cmax, AUC0-t, and AUC0-∞ fell within the acceptance range of 80.00–125.00% [6].
Data from subjects who completed both study periods were analyzed using Phoenix® WinNonlin® version 6.3 (Certara L.P., Princeton, NJ, USA) for pharmacokinetic analysis and SAS version 9.4 (SAS Institute, Cary, NC, USA) for statistical analysis.
3 Results
A total of 52 healthy Caucasian male subjects aged 19–45 years were enrolled, with a mean age of 31 years. One subject withdrew for personal reasons before dosing in period 1. In period 1, 51 subjects with a mean BMI of 25.0 kg/m2 were dosed and completed the period. In period 2, 43 subjects with a mean BMI of 24.6 kg/m2 were dosed and completed the period. Overall, 8 subjects withdrew before period 2 (due to a medical reason, abnormal laboratory results, or a positive result of a drug of abuse test). Subject demographics are shown in Table 1.
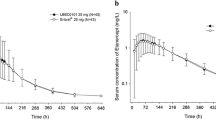
Serum samples from 51 subjects were analyzed, with data from 43 subjects included in the final etanercept pharmacokinetic analysis and statistical analysis. Mean and individual etanercept concentration profiles are shown in Figs. 1 and 2.

Mean etanercept concentration–time profiles: arithmetic mean (a) and log transformed (b)

Individual etanercept concentration–time profiles of YLB113 and the etanercept reference product (RP): arithmetic mean (a and b) and log transformed (c and d)
Noncompartmental analysis was performed to derive the primary (Cmax, AUC0-t, and AUC0-∞) and secondary (time to reach maximum serum concentration, Tmax; the AUC extrapolated from time t to infinity as a percentage of the total AUC, AUC%extrap obs; half-life, t1/2; elimination rate constant, Kel; volume of distribution, Vd; and clearance, CL) pharmacokinetic parameters (Table 2). The bioequivalence criterion (i.e., the 90% CI of the ratio of the geometric means of YLB113 and RP must be within 80.00–125.00%) was met for Cmax, AUC0-t, and AUC0-∞(Table 3).
Overall, 53 AEs were reported in 24 subjects. Of these, 42 AEs were considered mild and 11 were considered moderate. No AEs were serious. Among the AEs, 26 (49.06%) and 27 (50.94%) were observed following the administration of YLB113 (Table 4) and the etanercept RP (Table 5), respectively. Ten AEs were classified as not related to the administered treatment, 36 were possibly related, 3 were probably related, and 4 were definitely related. Following YLB113 administration, 14 subjects experienced ≥ 1 treatment-emergent AEs (TEAEs), whereas 16 subjects experienced ≥ 1 TEAEs following etanercept RP administration. Headache and sore throat were the most commonly reported AEs (54.72% of the cases). More AEs were reported in period 2 for both treatments. No clinically significant out-of-range values were observed for vital signs or during physical examinations performed throughout the study.
4 Discussion
The purpose of this study was to evaluate the relative bioavailability, safety, and tolerability of YLB113 (an etanercept biosimilar) 50 mg solution for injection and the etanercept RP 50 mg solution for injection following a single SC dose in healthy adult male subjects. Based on the similarity of their pharmacokinetic parameters within the accepted range, YLB113 and the etanercept RP can be considered bioequivalent. Furthermore, based on the observed power and intrasubject coefficient of variation, the sample size selected is sufficient to establish the bioequivalence between YLB113 and the etanercept RP.
The results of the current study indicate that a single dose of YLB113 and the etanercept RP are bioequivalent based on the systemic exposure demonstrated by AUC and Cmax. In support of this key finding, no appreciable differences in Tmax, t1/2, Kel, Vd, or CL for etanercept were detected as per noncompartmental pharmacokinetic analysis. Based on the ANOVA results, a significant period effect was observed for the log-transformed pharmacokinetic parameters Cmax and AUC at the 0.05 level of significance. This significant period effect is caused by the plasma levels (and AUC) being higher or lower in one period than in the other, which could be attributable to various factors such as the postural behavior of the subjects, the timing and degree of physical activity performed during the study, and interindividual physiological differences. However, considering that all of the abovementioned factors were the same in both periods, the observed period effect has no significant influence on the outcome of the study, because both treatments were given in each period in a randomized manner to each of the study participants in a crossover design. The crossover design was planned such that each treatment was given the same number of times in each period. This is most efficient and yields unbiased estimates of treatment differences.
The pharmacokinetic t1/2 of etanercept is approximately 102 ± 30 h, with a clearance of 160 ± 80 mL/h [8]. The EMA recommends a minimum of five half-lives for 96.9% elimination of the initial dose. Two subjects were observed to have predose concentrations; however, the concentration in both cases was < 5% Cmax, so they were included in the pharmacokinetic analysis set, which indicates that a washout period of 28 days was adequate to avoid any carryover effect.
In terms of bioanalysis, the method was validated for parameters such as selectivity, linearity, reproducibility, precision, accuracy, and stability. Incurred sample reanalysis was also performed to demonstrate the reliability and reproducibility of the analytical method employed during study sample analysis. Long-term stability data were appropriately generated to cover the duration needed for the clinical study.
A pharmacokinetic study in healthy volunteers is considered to be a particularly sensitive method of evaluating the similarity of a biosimilar to its RP. A similar strategy involving pharmacokinetic analysis in healthy subjects has been used previously to evaluate other etanercept biosimilars [9,10,11].
No significant safety issues were reported in the safety assessment. No clinically relevant findings related to ECG, vital signs assessment, physical examination, or laboratory examination were observed. The AEs reflect the comparable safety profiles of YLB113 and the etanercept RP.
4.1 Conclusions
The results of the current study indicate that a single dose of YLB113 exhibits pharmacokinetic and safety profiles comparable to those of the etanercept RP in healthy adult male subjects, and the products can therefore be considered bioequivalent. These findings support the continued development of YLB113 for use in patients with RA.
References
Fazal SA, Khan M, Nishi SE, Alam F, Zarin N, Bari MT, et al. A clinical update and global economic burden of rheumatoid arthritis. Endocr Metab Immune Disord Drug Targets. 2018;18(2):98–109. https://doi.org/10.2174/1871530317666171114122417.
Nestorov I. Clinical pharmacokinetics of TNF antagonists: how do they differ? Semin Arthritis Rheum. 2005;34(5 Suppl 1):12–8. https://doi.org/10.1016/j.semarthrit.2005.01.004.
Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor: Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340(4):253–9. https://doi.org/10.1056/NEJM199901283400401.
Chadwick L, Zhao S, Mysler E, Moots RJ. Review of biosimilar trials and data on etanercept in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20(12):84. https://doi.org/10.1007/s11926-018-0799-0.
Combe C, Tredree RL, Schellekens H. Biosimilar epoetins: an analysis based on recently implemented European Medicines Evaluation Agency guidelines on comparability of biopharmaceutical proteins. Pharmacotherapy. 2005;25(7):954–62.
European Medicines Agency. Guideline on the investigation of bioequivalence. London: England; 2010.
Combe B. Update on the use of etanercept across a spectrum of rheumatoid disorders. Biologics. 2008;2(2):165–73. https://doi.org/10.2147/btt.s1379.
Immunex Corporation. Enbrel (etanercept): prescribing information. Thousand Oaks, CA: Immunex Corporation; 2012.
Lee H, Chung H, Lee S, Lee H, Yang SM, Yoon SH, et al. LBEC0101, a proposed etanercept biosimilar: pharmacokinetics, immunogenicity, and tolerability profiles compared with a reference biologic product in healthy male subjects. BioDrugs. 2017;31(4):349–55. https://doi.org/10.1007/s40259-017-0230-9.
von Richter O, Skerjanec A, Afonso M, Sanguino Heinrich S, Poetzl J, Woehling H, et al. GP2015, a proposed etanercept biosimilar: pharmacokinetic similarity to its reference product and comparison of its autoinjector device with prefilled syringes. Br J Clin Pharmacol. 2017;83(4):732–41. https://doi.org/10.1111/bcp.13170.
Lee YJ, Shin D, Kim Y, Kang J, Gauliard A, Fuhr R. A randomized phase l pharmacokinetic study comparing SB4 and etanercept reference product (Enbrel®) in healthy subjects. Br J Clin Pharmacol. 2016;82(1):64–73. https://doi.org/10.1111/bcp.12929.
Acknowledgements
We would like to thank the subjects who participated in the trial, and acknowledge editorial support from Dr. Chirag Shah, study management assistance from Mr. Asif Tamboli, and quality oversight and monitoring help from Ms. Radhika Inapakolla.
Author information
Authors and Affiliations
Contributions
MS, RA-J, SK, RSK, NG, AA-G, PV, and DB made substantial contributions to the conception or design of the manuscript or the acquisition, analysis, or interpretation of data for the manuscript, and all of the authors were involved in drafting the manuscript or revising it critically for important intellectual content. The authors were fully responsible for all content and editorial decisions and received no financial support or other form of compensation related to the development of this manuscript. All authors gave final approval of the manuscript and are accountable for all aspects of the work in ensuring the accuracy and integrity of this manuscript.
Corresponding author
Ethics declarations
Funding
Funding for the phase 1 study was provided by Lupin Ltd. Editorial assistance was provided under the direction of the authors by The Lynx Group LLC. Funding support for the manuscript was provided by Mylan Inc.
Conflict of Interest
Mustafa Shennak is employed as Principal Investigator at Triumpharma. Rana Al-Jaouni is employed as Chief Operating Officer at Triumpharma. Santhosh Kshirasagar is employed as Assistant Director at Lupin Bioresearch Center in the Biopharmaceutics Department. Ravi Sekhar Kasibhatta is employed as Senior Vice President at Lupin Bioresearch Center. Neelima Godse is employed as a Research Scientist in the Medical Research Department of Lupin Limited (Research Park). Ahmad Al-Ghazawi is employed as President and Chief Executive Officer at Triumpharma. Praveen Vittala is employed as Assistant Director at Lupin Bioresearch Center in the Bioanalytic Research Department. Dhananjay Bakhle is employed as Executive Vice President—Medical Research, Lupin Limited.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Review Board of Triumpharma and were conducted in accordance with international clinical research guidelines and the principles enunciated in the 1964 Declaration of Helsinki and its later amendments, Good Clinical Practice, the Committee for Medicinal Products for Human Use guideline on the investigation of bioequivalence, and the local regulations and laws set by the Jordan Food and Drug Administration. The protocol of the study was approved by the IRB and the Ethical Committee of Triumpharma Research Institution of Amman, Jordan. The unique Jordan Food & Drug Administration trial number is 31/Clinical/2018.
Informed Consent
Written informed consent was obtained from all individual participants included in the study.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shennak, M., Al-Jaouni, R., Kshirasagar, S. et al. An Open-Label, Randomized, Single-Dose, Crossover, Comparative Pharmacokinetics Study of YLB113 and the Etanercept Reference Product in Healthy Adult Male Subjects. Eur J Drug Metab Pharmacokinet 45, 467–475 (2020). https://doi.org/10.1007/s13318-020-00613-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-020-00613-9