
Abstract
Tedizolid is an oxazolidinone antibiotic with high potency against Gram-positive bacteria and currently prescribed in bacterial skin and skin-structure infections. The aim of the review was to summarize and critically review the key pharmacokinetic and pharmacodynamic aspects of tedizolid. Tedizolid displays linear pharmacokinetics with good tissue penetration. In in vitro susceptibility studies, tedizolid exhibits activity against the majority of Gram-positive bacteria (minimal inhibitory concentration [MIC] of ≤ 0.5 mg/L), is four-fold more potent than linezolid, and has the potential to treat pathogens being less susceptible to linezolid. Area under the unbound concentration–time curve (fAUC) related to MIC (fAUC/MIC) was best correlated with efficacy. In neutropenic mice, fAUC/MIC of ~ 50 and ~ 20 induced bacteriostasis in thigh and pulmonary infection models, respectively, at 24 h. The presence of granulocytes augmented its antibacterial effect. Hence, tedizolid is currently not recommended for immunocompromised patients. Clinical investigations with daily doses of 200 mg for 6 days showed non-inferiority to twice-daily dosing of linezolid 600 mg for 10 days in patients with acute bacterial skin and skin-structure infections. In addition to its use in skin and skin-structure infections, the high pulmonary penetration makes it an attractive option for respiratory infections including Mycobacterium tuberculosis. Resistance against tedizolid is rare yet effective antimicrobial surveillance and defining pharmacokinetic/pharmacodynamic targets for resistance suppression are needed to guide dosing strategies to suppress resistance development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Tedizolid is a clinically useful antibiotic with activity against Gram-positive bacteria including methicillin-resistant Staphylococci and vancomycin-resistant Enterococci and currently recommended for patients with skin and skin-structure infections. |
The pharmacokinetic (high bioavailability, once-daily dosing, and lack of dosing adjustment in special patient populations) and safety profile as well as high tissue penetration make it a potential candidate for pulmonary infections. |
Further research is needed to optimize its existing usage, explore new therapeutic indications beyond skin and skin-structure infections, and to prevent resistance development. |
1 Introduction
With the increasing prevalence of methicillin-resistant Staphylococcus aureus (MRSA), and the emergence of vancomycin (VAN) less susceptible isolates, the first-line therapy options are shrinking to treat these serious infections [1]. Linezolid (LNZ), an oxazolidinone, was the first in this new class of antimicrobials to be effective against MRSA and less susceptible VAN isolates and widely used as an alternative to treat Gram-positive infection [2, 3]. However, twice-daily dosing, its hematological side effects (thrombocytopenia and myelosuppression), and the emergence of drug resistance, particularly the horizontally transferrable cfr-resistant genes, limit its clinical utility. Tedizolid (TDZ) [formerly called torezolid, TR-700, or DA-7157] is a new oxazolidinone antibiotic with a more favorable pharmacokinetic (PK) and safety profile compared with LZD. It also displayed enhanced antimicrobial activity mainly against Gram-positive bacteria compared with LZD and is currently approved for use in skin and skin-structure infections. In this review, we aim to critically summarize and review the key preclinical and clinical PK and pharmacodynamic (PD) aspects of TDZ as well as identify further directions of research.
2 Methods
A systematic literature search was conducted using terms “tedizolid,” “tedizolid phosphate,” “torezolid,” “TR 700,” and “DA-7157” with or without terms “pharmacokinetics” or “PK” and/or “pharmacodynamics” or “PD” or “resistance” or “immune modulation” or “mycobacterium tuberculosis” or “TB” on PubMed and Web of Science. Original research articles were selected till August 2021.
3 Pharmacokinetics
The pharmacokinetics of TDZ was studied in both healthy subjects and patients. Because of poor water solubility, TDZ is administered as TDZ phosphate, which is rapidly hydrolyzed by plasma phosphatases into the free active moiety TDZ.
3.1 PK Profile in Healthy Subjects
With both oral or intravenous (IV) dosing, TDZ shows a volume of distribution of ~100 L and displays an elimination half-life of ~11 hours, approximately two-fold higher than LNZ, allowing for once-daily administration of TDZ in comparison to the twice-daily dosing of LNZ. Because of the high bioavailability (~91%), no dose adjustment is required while switching between both administration routes. At a dose range from 200 to 1200 mg/day, the area under the concentration–time curve from zero to infinity (AUC0–∞) ranges from 21.6 to 123.1 mg∙h/L while the maximum concentration (Cmax) ranges from 1.8 to 9.5 mg/L. For the standard human dose of 200 mg/day, the Cmax and AUC0–∞ are ~1.8–2.6 mg/L and ~21.6–32.7 mg∙h/L, respectively [4]. After multiple doses of IV 200 mg , at day 7, the area under the concentration–time curve from 0 to 24 hours (AUC0–24h was higher (29.2 mg∙h/L) compared with day 1 (22.3 mg∙h/L) [5]. At 200 mg/day, the clearance (CL) values range from 5.9 to 6.5 L/h. The pharmacokinetics was found to be linear in the dose range of 100–600 mg/day [4,5,6,7]. Food had no effect on the area under the concentration–time curve from 0 to 72 h (AUC0–72), but delayed Cmax by 26% [4]. Reported plasma protein binding ranges from 70 to 90% [8]. Tedizolid is predominantly excreted via the feces (~69%) as an inactive sulfate metabolite while ~10% is excreted via urine [9].
A two-compartment model with sigmoidal absorption and linear elimination was developed that successfully described the plasma disposition kinetics when applied to the pooled data from seven clinical studies (phase I–III) [10]. Neither a clinically important covariate nor ethnic differences affected the PK disposition significantly [11,12,13,14]. The PK characteristics are shown in Table 1.
3.2 Pharmacokinetics in Special Patient Populations
3.2.1 Renal and Hepatic Impairment
Two single-dose (200 mg), phase I, parallel-group studies in patients with renal and hepatic impairment showed no appreciable difference in PK parameters when compared to the control groups. In subjects with severe renal impairment (estimated glomerular filtration rate <30 mL/min/1.73 m2), the mean AUC0–72 (32.02 vs 29.69 mg∙h/L), Cmax (3.11 vs 3.12 mg/L), CL (5.68 vs 6.13 L/h), and half-lives (12.25 vs 12.85 hours) were indifferent to the respective control [15]. In patients undergoing intermittent hemodialysis, a minor impact on CL (< 10%) was reported [15, 16]. In in vitro models of continuous renal replacement therapy, transmembrane clearance of TDZ depended on hemodialysis type, ultrafiltrate rate, and blood flow rate. However, no need for dose adjustment in clinical settings was concluded [17]. However, substantial changes in protein binding, as they might occur in severe hepatic impairment or also in critically ill patients, might increase the CL of TDZ and warrants further investigation.
In patients with moderate and severe hepatic impairment, the CL values were 6.06 and 5.22 L/h while the area under the concentration–time curve from 0 to 96 hours was 29.89 and 34.80 mg∙h/L, which was 22% and 34% higher than the control group (22.80 and 24.37 mg∙h/L), respectively. However, this increased exposure is likely clinically irrelevant and was well tolerated in other multiple-dose clinical studies and hence necessitates no dose adjustment [6, 15].
3.3 Elderly Individuals, Adolescents, and Children
Tedizolid depicted similar PK disposition kinetics after IV/oral administration of 200 mg/day in elderly individuals, adolescents, and adults. The AUC0–24 values for adolescents (25.5 mg∙h/L) were within the range of the previously reported adult values (~30 mg∙h/L). Mean Cmax was similar after oral administration, but was 43% higher than adults after an IV infusion (3.66 mg/L compared with ~2.5 mg/L), suggesting a 200-mg daily dose can be further studied in adolescents [18]. In children aged 2–12 years, once-daily doses of 3–6 mg/kg provided a comparable exposure (AUC0–∞: 17.2–29.6 mg∙h/L) to that reported for adults and adolescents. However, in children (aged 2–6 years), the Cmax was higher than in adults (4.19 vs ~2.5 mg/L), which hints to the splitting of doses as an alternative option [19]. The small sample size (n = 6) of the study indicates that further research might be needed to position TDZ for clinical use in children. Tedizolid displayed similar PK characteristics (200-mg single dose) in elderly subjects when compared to the adult population (AUC0–72 of 34.7 vs 29.9 mg∙h/L) and hence no dose adjustment seems necessary in this patient collective [20].
3.4 Obese Population
In obese patients, both IV and oral administration of the standard 200-mg daily dose, the AUC0–∞, and Cmax geometric mean ratios were 11–18% lower compared with healthy control subjects, indicating no significant difference between both populations [21, 22]. Similarly, no disparity in PK profiles was noted in morbidly obese patients compared to the nonobese subjects [23]. There were no significant differences in PK profiles of patients after bariatric surgery compared to the adult population [24].
3.4.1 Cystic Fibrosis
The CL (mean total and distributional CLs of 9.72 and 4.13 L/h, respectively) was higher in patients with cystic fibrosis compared with values reported earlier (6.69 and 0.959 L/h, respectively) for healthy volunteers and patients with complicated skin and skin-structure infections (pooled data) in one study, but are almost similar to the CL values in patients with skin and skin-structure infections (8.28 and 2.95 L/h, respectively) [25]. Whether these changes in CL (in total or in parts) are due to PK alterations in cystic fibrosis or pathophysiological changes during infection necessitates further investigations to guide an optimal dosing strategy in patients with cystic fibrosis.
3.4.2 Drug Interactions
Although TDZ shows in vitro a weak reversible inhibition of monoamine oxidases, provocative testing of a potential interaction between the therapeutic dose of TDZ and oral pseudoephedrine or tyramine in healthy volunteers in two randomized placebo-controlled trials exhibited no potential serotonergic or hypertensive adverse effects. The results were in line with the murine model studied with a similar objective [26]. Nonetheless, patients treated with TDZ and monoamine oxidases inhibitors should be carefully monitored for potential side effects.
3.4.3 Target Site Exposure
Tedizolid exhibits high tissue penetration into both skin and pulmonary tissues. When investigated in skin and adipose tissue using microdialysis in healthy subjects after a single oral dose of 600 mg, the penetration ratio (measured as free area under the unbound concentration–time curve [fAUC]/minimal inhibitory concentration [MIC]) fAUCtissue/fAUCplasma) for adipose and muscle tissue were 1.1 ± 0.2 and 1.2 ± 0.2, respectively [27], suggesting that unbound plasma represents a reasonable surrogate for tissue concentrations. Similarly, in another microdialysis study in patients with diabetic foot infection receiving 200 mg daily for 3 days, tissue concentrations approximated unbound plasma concentrations: the unbound tissue/unbound plasma concentration ratio was 1.1 (range 0.3–1.6) for patients with diabetic foot infection and 0.8 (range 0.7–1.0) for healthy volunteers, respectively [28]. At a daily dose of 200 mg orally for 3 days in healthy adults, in comparison to the fAUC0–24 in plasma and assuming negligible protein binding in epithelial lining fluid (ELF), TDZ penetration ratios were approximately 40-fold higher in ELF and 20-fold higher in alveolar macrophages compared with plasma. The fAUC0–24 values for ELF and alveolar macrophages were 109.3 and 52.95 mg∙h/L, respectively. This high pulmonary penetration advocates for the potential role of TDZ in the treatment of pulmonary infections [29]. Whether this increased penetration is due to active transport or any other mechanism is still unknown and needs further investigations. Moreover, TDZ shows good sputum penetration with a sputum-to-plasma ratio of 2.88 in patients with cystic fibrosis [30]. Tedizolid-induced suppression of mucin production in alveolar membranes could also contribute to the higher pulmonary penetration of TDZ [31]. Although varying degrees of unbound cerebrospinal fluid-to-unbound plasma penetration ratios are reported in the literature for humans and rats, further studies are needed to confirm these findings to define the potential role of TDZ to treat central nervous system infections [32, 33]. A summary of the tissue penetration of TDZ and LNZ is provided in Table 2.
4 Pharmacodynamics
4.1 Antimicrobial Spectrum
The antimicrobial spectrum of TDZ covers clinically relevant Gram-positive bacteria including methicillin-susceptible Staphylococcus aureus (MSSA), MRSA, methicillin-susceptible S. epidermidis, methicillin-resistant S. epidermidis, VAN-sensitive and VAN-resistant enterococci (VRE), penicillin-susceptible Streptococcus pneumoniae (PSSP) and penicillin-resistant S. pneumoniae (PRSP), and other frequently reported cutaneous and respiratory pathogens (Table 3) [2, 34]. Most Gram-positive bacteria mentioned above are susceptible to TDZ with MIC values of ≤ 0.5 mg/L [35,36,37,38,39]. However, TDZ, compared with Gram-positive bacteria, exhibits lower potencies against Gram-negative bacteria such as Hemophilus influenzae (16 mg/L) and Moraxella catarrhalis (4 mg/L) [35, 40]. Like LNZ, TDZ binds to the ribosomal RNA 50S subunit and inhibits protein synthesis [2].
Staphylococcus aureus is a frequently reported pathogen in many skin, soft-tissue, and respiratory tract infections for which many commonly used antimicrobials gradually lost efficacy, resulting in an increasing number of MRSA, VAN-resistant, and LNZ-resistant isolates [41, 42] and often limiting the available treatment choices of antimicrobials in the underlying infections. Multiple comparative studies repeatedly demonstrated at least a four-fold higher potency of TDZ compared with LNZ. Moreover, TDZ shows efficacy against VAN-resistant, daptomycin (DAP)-resistant, and some LNZ-resistant isolates and hence provides an alternative option to treat the less susceptible isolates of these antimicrobials [35, 37, 43,44,45]. Other studies with further Gram-positive isolates mentioned earlier have almost similar findings [11, 34, 35, 38, 40, 43, 46,47,48,49,50,51,52,53]. However, few isolates that were LNZ resistant (plasmid borne cfr multidrug resistance gene) showed a lower susceptibility (MIC > 0.5 mg/L) [54,55,56,57,58,59]. Hence, isolates above the MIC threshold of >0.5 mg/L are less likely to be successfully treatable with TDZ [10]. A susceptibility breakpoint of 0.5 mg/L is recommended by EUCAST for Staphylococcus species, Streptococci (A, B, C, G) and viridians (Streptococcus anginosus) [60]. The results from the Surveillance of Tedizolid Activity and Resistance Program (STAR program) in Europe showed that the majority of the Enterococci exhibit MIC values of < 0.5 mg/L for TDZ and support the EUCAST breakpoint for Staphylococci [36, 61,62,63,64]. A recent PK/PD simulation-based analysis on existing PK and PD data from the literature reported a similar PK/PD breakpoint of 0.5 mg/L for the majority of Gram-positive isolates [65]. However, no EUCAST susceptibility breakpoints are yet established for Enterococci [60].
With high penetration into human macrophages (TPH-1), TDZ showed good intracellular efficacy against Listeria monocytogenes (MIC 0.125 mg/L) and S. aureus (MIC 0.25–1 mg/L) in infected TPH-1 cells. The intracellular penetration can be potentially exploited to treat intracellular pathogens [66]. Its moderate activity against anaerobes particularly against Bacteroides fragilis (MIC90 of 1 mg/L) can be exploited to treat mixed aerobic and anaerobic infection [67]. Tedizolid expresses good activity against several clinically relevant respiratory pathogens such as PRSP (MIC90 of 0.25 mg/L) and PSSP (MIC90 of 0.125–0.25 mg/L) and has a clinical potential to treat respiratory infections [12, 35, 49, 68].
4.2 In Vitro Pre-Clinical Studies
In vitro time-kill studies depict bacteriostatic activity for TDZ in most Gram-positive isolates. When studied against MRSA (MIC = 0.5 mg/L), methicillin-resistant S. epidermidis (MIC = 0.25 mg/L) and Enterococcus faecalis (MIC = 0.25 mg/L) a bacteriostatic effect was observed at both MIC and 16× MIC for all strains while a bactericidal activity (3 log-kill at 24 hours) at 16× MIC was observed for S. pneumoniae (MIC = 0.25 mg/L) [68]. In line with the above results, a bacteriostatic effect of TDZ was observed against VRE E. faecium (MIC 1 mg/L) and E. faecalis (0.25 mg/L) after 24 hours at 2× MIC [69]. In another time kill study, TDZ exhibited bacteriostatic activity against MRSA and MSSA, while regrowth was observed with LNZ treatment at the MIC and 2× MIC after 24 hours (LNZ MIC: 1 and 2 mg/L for MRSA and MSSA, respectively) [70].
Tedizolid was also evaluated in combination. In a static time kill study using a concentration equal to 0.5× MIC of the respective isolate against MRSA (MIC, 0.25–0.5 mg/L) and S. epidermidis (MIC, 0.125–1 mg/L), TDZ showed synergistic (≥ 2 log10 CFU/mL reduction in combination vs most active single agent) activity with rifampicin and doxycycline, while, in contrast, an antagonistic (≥ 1 log10 CFU/mL growth) activity of TDZ and moxifloxacin (MXF) was observed [71]. However, these findings were not uniform in all the strains (n = 10) and warrant further studies. Against S. aureus with reduced glycopeptide susceptibility, TDZ alone showed bacteriostatic activity, which was comparable to teicoplanin and rifampicin combination therapy in an in vitro static time kill study [48]. Daptomycin and TDZ while active alone against MRSA, in combination were inferior to the respective monotherapy in an in vitro dynamic model [72]. In an in vitro endocarditis model against VAN-resistant S. aureus and VRE (E. faecium and E. faecalis), a step-down therapy (DAP for 3 days followed by TDZ for 2 days) was as effective as DAP for 5 days [72]. However, no clinical study has yet explored these possible combination therapies.
Overall, TDZ exhibits reasonable bacteriostatic activity against Gram-positive pathogens frequently causative of systemic, skin/cutaneous and respiratory infections, especially Staphylococci and Enterococci. However, studies demonstrating the antibiotic effect of TDZ for more than 24 hours in static or dynamic in vitro models are limited. Insights from longer concentration–time studies are critical for better understanding its efficacy, dosing strategies, and the pattern and extent of potential resistance development in a full course of therapy.
4.3 In Vivo Preclinical Studies
4.3.1 Murine Infection Models
Several in vivo studies were conducted in localized and systemic murine infection models to evaluate the pharmacokinetics, pharmacodynamics, and the PK/PD parameters best corelated with efficacy of TDZ against a variety of Gram-positive pathogens. The pharmacodynamics of TDZ was evaluated by Louie et al., where a neutropenic thigh infection model with MRSA and MSSA infections was investigated. Tedizolid was equally effective against both MSSA and MRSA while the fAUC related to MIC (fAUC/MIC) was best correlated with efficacy. The fAUC/MIC ratio related with stasis and 1-log kill relative to the starting inoculum was 49.3 and 105.9, respectively, at 24 hours. A mean dose of 37.6 and 66.9 mg/kg of TDZ was required for stasis and 1 log CFU/g, respectively, at 24 h. In comparison, 150 mg/kg of LNZ failed to induce bacteriostasis, demonstrating the higher potency of TDZ as compared with LNZ and hence confirming the in vitro findings in vivo [73].
When evaluated in a systemic infection model (immunosuppressed [IS]) with MSSA and MRSA infection (MIC: 0.125–0.5 mg/L), TDZ demonstrated a 2–9-fold higher activity than LNZ (MIC: 0.5–8 mg/L) with an ED50 (dose giving half maximal effect) range of 1.5–3.2 to 4.3–7.6 mg/kg vs 7.7–9.6 to 21.4–29.1 mg/kg of TDZ vs LNZ, respectively [34]. Similarly, in a septicemia model induced by LNZ-resistant MRSA (cfr positive), a lower dose of TDZ (20 mg/kg) was superior in activity vs 50 mg/kg of LNZ (100 vs 80% survival). Other studies with Enterococci led to similar results [34, 74].
However, in erythromycin-resistant and clindamycin-resistant Streptococci in a necrotizing infection murine model, TDZ and LNZ were equally effective [75]. Hence, the higher in vitro potency of TDZ as compared with LNZ seems to translate into the in vivo setting.
4.3.2 Role of Immune System Components
Drusano et al. proposed a critical role of granulocytes in immunocompetent (IC) compared with IS mice in a thigh infection model [76]. Despite similar PK profiles, the TDZ activity in IC mice was approximately 25-fold higher than in IS mice at all studied timepoints (24, 48, and 72 hours). A human equivalent dose of ≤ 200 mg/day vs 2300 mg/day produced stasis (24 hours) in IC and IS mice, respectively, whereas a maximal effect was observed at 200 mg/day in IC mice, suggesting that the efficacy of TDZ is grossly mediated by granulocytes. Based on previous results for IS mice, a fAUC/MIC target of 3 was calculated for IC mice for stasis at 24 h as compared with the target of 50 in IS mice [77]. However, when these findings were re-evaluated by Xiao et al. with a similar study design and exposure, contrary to the earlier findings, stasis was achieved in both IC and IS mice after 72 h of TDZ therapy [78]. Moreover, in IS mice, stasis was achieved at 72 h at a lower dose of 166 mg/day compared with the previously reported value (~ 2000 mg/day) by Drusano et al. [78]. The reasons for this discrepancy are unclear. However, based on the findings of Drusano et al., the current label of TDZ limits its use to IC patients [79]. The fAUC/MIC target of 8.9 for stasis at 72 h for MRSA (MIC 0.5 mg/L) in the IS model was close to the clinically reported value of 5–7 in adults [8, 73], and hence correlated well with the human studies [78]. The results also suggest the duration of therapy to be considered (efficacy at 72 h is higher than at 24 h) when comparing the results among murine models. In comparison, in IC mice, a lower target of fAUC/MIC <1.3 for stasis was reported for all studied timepoints [79].
Results in a pulmonary infection murine model, with human equivalent pulmonary exposures, with S. pneumoniae also showed a less pertinent role of granulocytes in TDZ efficacy [80]. In an IC thigh infection model, both TDZ and LNZ (at human equivalent doses) showed similar activity with S. aureus clinical isolate infections [81]. For Enterococci (E. faecalis and E. faecium), in a IC murine model TDZ, despite higher total activity of TDZ than LNZ in vitro, paradoxically, the effect of TDZ was inferior to LNZ in both bacterial killing and relapse prevention at human equivalent dosing [69]. A superior efficacy of LNZ over TDZ contradicts the earlier finding for which a plausible explanation is missing. Keel et al. investigated the role of both infection status and neutropenia and compared PK/PD indices among IC, IS, and noninfected mice at a human equivalent dose of 8.4 mg/kg of TDZ. The fAUC(0–24) was 41% higher in IC and 17% lower in noninfected mice than IS mice. The penetration ratio (bronchoalveolar lavage to blood) was higher for TDZ in infected mice (9.34 and 10.63 for IC and IS) compared with 6.14 in noninfected mice and demonstrates a critical role of infectious status in tissue penetration [82]. A significant decrease of all cytokines (tumor necrosis factor-α, interleukin-1, interleukin-6, and macrophage inflammatory protein-2) was observed at 2 h after the TDZ human equivalent dose as compared with the control in a MRSA-induced pulmonary infection model. Whether this effect was indirectly related to the microbial toxins released by pathogens or induced by TDZ directly remains unclear [83].
An intact immune system is assumed to provide a 2–4 log reduction in AUC/MIC target values [84]; however, the higher granulocyte-mediated effect restricts the clinical utility of TDZ only to IC patients as recommended by the European Medicines Agency [8]. However, the contrasting role of the immune system in some subsequent studies necessitates further investigations to substantiate these findings and define the role of the immune system in relation to TDZ efficacy further. Moreover, PK/PD studies, ideally, covering the full course of therapy, are needed to extend TDZ application in critically ill neutropenic patients where other therapeutic alternatives fail to treat the underlying infection.
4.3.3 Pulmonary Infection Model
While soft-tissue infections are the main therapeutic area for which TDZ is indicated, pulmonary infections represent another major therapeutic area that is frequently investigated and has shown promising potential of clinical application. Choi et al. studied TDZ and LNZ against four PRSP and PSSP in a murine pneumonia model with human equivalent doses. Tedizolid was at least two-fold more potent than LNZ in both PRSP and PSSP systemic infection models, while a lower dose (10 mg/kg/day of TDZ) resulted in a similar outcome (100% survival) as compared to 40 mg/kg/day of LNZ in a PSSP model [85]. In a murine pneumonia model using MRSA and MSSA isolates, the fAUC/MIC target related to stasis for both TDZ and LNZ was 19 and 20, respectively, while roughly doubled values (34.6 and 46.1, respectively) were associated with 1-log kill reduction across all strains. These fAUC/MIC targets were 2–4-fold lower than the reported values for TDZ and LNZ derived from the thigh infection model [73, 86]. This discrepancy might be explainable by the enrichment of TDZ in ELF [87]. When compared with VAN and LNZ at equivalent human ELF exposure against an IC MRSA pneumonia mouse model, TDZ demonstrated similar efficacy to LNZ but was superior to VAN (100% vs 39% survival) [88, 89]. A similar study supports these findings [83]. In another study, interestingly, pulmonary exposure was almost similar in both IC and IS S. pneumoniae-induced pulmonary mice models (~110 mg·h/L) despite unequal dosing (40 mg/kg/day vs 55 mg/kg/day) [80] and further underlines the importance of consideration of target-site drug concentrations when relating systemic exposure to efficacy. These exposures were comparable to humans ELF exposure of 109.30 mg·h/L (200 mg/day), confirming a good correlation of pre-clinical to clinical data. In IS mice, the fAUC0–24/MICs associated with stasis and 1-log reduction in pulmonary bacterial burden relative to initial inoculum were 19.21 and 48.29, respectively, which was in line with the findings of Lepak et al. [87]. In total, the above studies highlight good pulmonary penetration and efficacy of TDZ in all studied isolates and provide a rationale to further explore TDZ for treating pulmonary infections.
4.4 In Vivo Clinical Studies
The exposure–response relationship of TDZ was investigated in several single and pooled clinical studies. In patients with complicated skin and skin-structure infections (main pathogen MRSA, MIC90: 0.25 mg/L), Prokocimer et al. reported a dose of 200 mg/day to be as effective as higher doses when administered for 5–6 days. However, an exposure–response relationship was difficult to establish [6]. In the ESTABLISH 1 and 2 clinical trials, 200 mg/day of TDZ was non-inferior to 600 mg twice-daily dosing of LNZ in patients with ABSSSIs and was subsequently licensed for this indication [90, 91]. Moreover, TDZ reduced skin and soft-tissue infection-related hospital admissions in out-patient settings [92]. Based on pooled PK and PD data from four clinical studies (one phase I, one phase II, and two phase III) where doses of 100–400 mg/day were investigated [10, 15, 91, 91, 93, 94], a PK/PD model was developed to relate exposure to efficacy. At a dose of 200 mg/day, the PK/PD index fAUC/MIC of 3 was defined to relate to clinical outcomes [77]. The developed PK/PD model relating fAUC to MIC was used in a Monte Carlo simulation to predict a therapeutic breakpoint. The fAUC/MIC target of 3 results in a probability of target attainment of ~98% at an MIC value of 0.5 mg/L, while for 1 mg/L the probability of target attainment was ~70% and thus deemed not sufficiently high. Therefore, an MIC of 0.5 mg/L was declared the susceptibility breakpoint for TDZ against S. aureus (MSSA or MRSA) and can be applied to other common bacterial pathogens having a TDZ MIC of ≤0.5 mg/L [10]. This clinical breakpoint was in line with preclinical studies where at a cut-off value of ≤0.5 mg/L, most Gram-positive isolates were sensitive [95]. In addition to clinical studies in skin and skin-structure infections, in the first phase III clinical study in patients with hospital-acquired and ventilator-associated pneumonia, TDZ (200 mg/day for 6 days) was non-inferior to LNZ (600 mg twice daily for 10 days) in the primary outcome (28 days all-cause mortality). However, the non-inferiority of TDZ to LNZ was not established while comparing the secondary outcome (investigator-assessed clinical response at test of cure) [96]. There are no clear reasons for this discrepancy but the complexity of the disease and the subjectivity of defining “clinical cure” by clinicians can lead to this lack of consistency in the clinical outcomes. This is the first clinical investigation of TDZ in patients with pneumonia and further studies are needed to confirm these findings.
4.4.1 Safety Profile
Tedizolid was generally well tolerated in healthy volunteers as well as in multiple phase II and phase III studies and has a superior safety profile compared with LNZ. When evaluated at a daily dose of 200–400 mg in a phase II study, 69% of the patients reported treatment-emergent adverse drug reactions (ADRs) [72.3% mild; 24.6% moderate] with nausea (18.6%), headache (11.2%), and vomiting (10.1%) most predominant. None discontinued therapy because of TDZ-induced ADRs [6]. In the two phase III trials for licensing (ESTABLISH 1 and 2), TDZ showed a superior safety profile compared with LNZ. Gastrointestinal ADRs (nausea, vomiting, diarrhea, and dyspepsia) were frequently reported (16%), hence expanding the observations of phase II studies [29, 97]. When evaluated in pooled data across completed clinical studies (13 phase I, two phase II, and two phase III), drug-related ADRs were 27% of all participants. In line with previous results, gastrointestinal ADRs (13%) and headache (4%) were predominant [98]. However, none of these trials collected long-term (>3 weeks) safety data. While hematological toxicity is a serious concern during prolonged LNZ therapy, TDZ showed a superior hematological safety profile as compared with LNZ with a lower incidence of thrombocytopenia (platelet counts, < 150,000 cells/mm3) at days 7–9: among the TDZ treatment group, thrombocytopenia was 3.2% compared with 5.6% in the LNZ treatment arm at standard human doses [99]. When administered for a longer duration (21 days), thrombocytopenia associated with TDZ administration was dose dependent: at a dose of 200 mg/day, no measurable difference in thrombocytes from baseline was observed while at 400 mg/day, up to a 50% decrease was observed in 12.5% of the patients [99, 100]. Further studies with similar findings suggest a lower impact of TDZ on the hematological profile [101]. However, because of the small sample size (n = 40) of the study, the long-term safety of TDZ necessitates further investigations. Studies with slightly longer durations (mean of 27–29 days) support these findings [102,103,104]. In ESTABLISH 1 and 2 trials, neurological (~ 9%) and dermatological (~6%) toxicities were almost equally often reported for both TDZ and LNZ, which were well supported in other studies [14, 90, 91, 105]. Safety profiles of elderly individuals, adolescents, and children (aged 2–12 years), patients with cystic fibrosis, and renal and hepatically impaired patients were comparable to that of the adult population [15, 19, 20, 25, 106, 107]. The overall safety pattern of TDZ in post-marketing surveillance in the ADR reports (2014–20) in the worldwide US Food and Drug Administration Adverse Events Reporting System was in line with the above results where no serious adverse effect was directly associated with TDZ [108]. For long-term safety, less structured data are available. Yet, in a recent study in patients with bone and joint infections (n = 33) where TDZ was administered for a mean duration of 8 weeks at 200 mg/day, an overall high ADR rate (60%) was reported, and 18% of the patients discontinued the TDZ because of intolerance or severe anemia due to hemorrhage [109]. The results were comparable with overall ADR rates reported in non-tuberculosis mycobacterial infections after an average of 101 days of TDZ administration [110]. A retrospective single-center evaluation of 24 patients with non-tuberculosis mycobacteria infection with an average 7 weeks of standard human doses of TDZ and LNZ showed no differences in hematological safely profiles [111]. Four case reports with long-term use of TDZ in an adolescent patient with pulmonary tuberculosis undergoing a liver transplant (20 months), in a patient with nocardiosis (6 months), in a patient with recurrent MRSA infection (18 months), and in a patient with cutaneous non-tuberculosis mycobacteria infection (8 months) showed no TDZ-induced toxicity [112,113,114,115]. Although animal studies illustrated a lack of any neurological change with long-term TDZ administration, long-term neurological safety studies in humans are lacking [116, 117]. The European Medicines Agency, although acknowledging an overall high safety profile for TDZ, indicate myelosuppression and peripheral neuropathy as potential risks associated with TDZ therapy in their risk management plan [118]. A notable increase in the off-label (particularly in terms of treatment duration) use of TDZ necessitates objective evidence to justify its long-term safety [108]. A clinical summary of TDZ is provided in Table 3.
5 Resistance to TDZ and PK/PD Targets/Magnitudes for Resistant Suppression
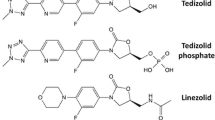
The main mechanisms of resistance against oxazolidinones comprise chromosomal mutations at the 23S ribosomal rRNA target site, mutations in the rplD gene encoding the 50S ribosomal proteins (L3 and L4) [46, 119], plasmid-born chromosomal mutations (cfr methyltransferase gene) [45, 120], and alteration of efflux with ABC transporters (e.g., Opt A) [120, 121]. Because of the presence of a distinct hydroxyethyl group in the molecular structure (Fig. 1), TDZ shows potency against some LNZ-resistant bacteria and stimulates a lower mutation frequency. A comparison of the frequency of resistant mutant selection to TDZ exposed to 2× MIC of S. aureus (MRSA and MSSA) was < 10−10 to < 10−11, respectively, which was approximately two orders of magnitude lower than LNZ (1–5×10−9) [45, 120]. These findings were in line with Jones et al. where a single cell mutation was rare and no growth of S. aureus and E. faecium was observed at 4×, 6×, and 8× MIC of TDZ [46]. Serial passages of MRSA and MSSA to TDZ with a two-fold increasing exposure (started from 4 mg/L) for 30 days resulted in no elevation in the MIC of MSSA. However, reduced susceptibility of MRSA was observed (0.25 vs 2 mg/L) [120]. Chen et al. reported a majority of the S. anginosus group (61.3%) to be non-susceptible to TDZ when the US Food and Drug Administration breakpoint of (≤0.25 mg/L) was applied, which was in contrast to previously reported studies by Prokocimer et al. and Zurenko et al. where S. anginosus was sensitive [122,123,124]. However, the study was conducted in a single center and needs further verification. Enterococcal clinical isolates of E. faecalis from China showed ~6–13 % of the studied isolates to be TDZ non-susceptible (MIC ≥0.5 mg/L) [125]. The non-susceptible isolates displayed an abundant plasmid-mediated ABC transporter optrA [50, 121]. Choudhury et al. also reported two clinical isolates (out of 48) of vanA E. faecium strains as ‘non-susceptible’ to TDZ (MIC 1–2 mg/L) [126]. In a recent study, two isolates of E. faecalis displayed TDZ MIC values of 2 mg/L (23S rRNA G2576T mutation). Tedizolid exhibited lowered sensitivity (MIC >0.5 mg/L) in LNZ-resistant VRE isolates in Germany [127]. However, further systematic investigations are necessary to establish a correlation between previous LNZ resistance and TDZ susceptibility. In addition to the classical resistance mechanism (23S rRNA alteration), PoxtA and OptA gene mutations were also reported from the USA and Turkey from resistant Gram-positive clinical isolates [51].

The chemical structures of tedizolid
Despite few less susceptible isolates that emerged in some studies with the expression of different genetic mutations, the incidence of TDZ resistance is still low in key clinically relevant bacteria. However, the potential for the emergence of resistance and/or cross-resistance between the two oxazolidinones will remain a concern and warrants active surveillance. Moreover, for isolates with undefined EUCAST susceptibility breakpoints, resistant data are lacking. More systematic investigations are required to define the mutant selection window in relation to the TDZ exposure profile for the entire treatment duration, in order to determine which PK/PD index is related to suppression of resistance development [128].
6 TDZ as a Potential Treatment Option Against Mycobacterium tuberculosis
The efficacy against mycobacteria, high pulmonary penetration, and a favorable safety profile compared with LNZ makes TDZ a potential candidate to treat pulmonary tuberculosis caused by M. tuberculosis and in non-tuberculosis mycobacteria pulmonary infections [129]. Moreover, its high intracellular activity makes it an ideal potential candidate to treat intracellular mycobacterium tuberculosis infections. The TDZ MIC values against M. tuberculosis typically range from 0.125 to 1 mg/L [129,130,131,132].
Tedizolid shows superior anti-mycobacterial activity over LNZ and has potential to substitute LNZ in antitubercular combination regimens. When studied in a hollow fiber in vitro infection model against intracellular (disseminated pediatric tuberculosis model) mycobacterium tuberculosis (MIC: 0.5 mg/L) for 28 days (AUC0–24 of 0–139.41 mg · h/L, elimination half-life of 12 h), the EC80 (AUC0–24/MIC ratio associated with 80% maximum bacterial kill) of TDZ associated with an optimal log kill and time to positivity (TTP) was nearly identical (184 vs 189) [133]. In a comparative study with a comparable EC80 (TDZ 238.4 [MIC: 0.5 mg/L] vs LNZ 24.05 [MIC: 1 mg/L]), TDZ showed a >10,000-fold higher activity than LNZ and hence supports TDZ as the preferable choice in intracellular antitubercular therapy in pulmonary cavities and in disseminated tuberculosis in children [133]. This superior activity of TDZ over LNZ was also observed for Mycobacterium avium-intracellular complex in a separate in vitro study [134]. Srivastava et al. further investigated TDZ against semi-dormant mycobacterium tuberculosis (MIC: 0.25 mg/L) in a hollow fiber in vitro infection model, where an EC80 of 200 mg · h/L best related to the sterilizing effect (TTP), which was in line with the earlier reported value (188 mg · h/L). Using Monte Carlo simulations, at a human equivalent dose of 200 mg/day (AUC0–24 mg/L of 31.0 ± 6.6), >90 % of patients achieved EC80 of 200 mg · h/L for an MIC of ≤0.5 mg/L and hence this value was declared as a tentative susceptibility breakpoint [135]. The mitochondrial toxicity, taken as an indirect toxicity predictor, was found lower for TDZ at exposures in the above-mentioned study as compared with LNZ.
The dual combination with TDZ (200 mg/day) and high dose of MXF (800 mg/day) results in sterility (TTP assay) for both log-phase (after 14 days of therapy) and semi-dormant forms (after 42 days of therapy) of mycobacterium tuberculosis in a hollow fiber in vitro infection model [136]. For the non-replicating persisters form, a triple therapy (TDZ 200 mg/day, MXF 800 mg/day, and faropenem twice daily to achieve 66% time above MIC) resulted in sterilization as early as 14 days of therapy compared with 21 days of therapy with the standard regimen (isoniazid 300 mg/day, rifampin 600 mg/day, and pyrazinamide 1.5 g/day) [136].
The above-mentioned results favor TDZ as an alternative to replace LNZ in MDR/XDR-TB therapeutic regimens while a dose of 200 mg/day can be a potential dose taken forward to these clinical studies. Combination therapy in mycobacterium tuberculosis is usually favored for clinical success, better patient compliance, safety, and prevention of drug resistance, and hence the combination regimen, in particularly the triple therapy of TDZ, MXF, and faropenem, might be a favorable antibiotic combination to be further investigated in human studies. Although the in vitro results for a short duration (42 days) propose comparable safety of TDZ, the long-term safety profile of TDZ therapy in patients with tuberculosis is lacking.
7 Conclusions
Tedizolid demonstrates broad in vitro and in vivo efficacy against a number of clinically important Gram-positive pathogens including MRSA and VRE. It is a viable treatment option against skin and skin-structure infections caused by these bacteria. Tedizolid has several advantages over LNZ with regard to dosing frequency and its safety profile. Further research is required to investigate the contribution of the immune system to the efficacy of TDZ. Results from murine pulmonary models, PK studies in healthy volunteers, and a recent comparative clinical trial indicate its potential use in pulmonary infections, but more data on safety in long-term use are needed to establish its role in mycobacterium tuberculosis treatment. Research in further potential clinical applications is warranted.
References
Zhanel GG, Love R, Adam H, Golden A, Zelenitsky S, Schweizer F, et al. Tedizolid: a novel oxazolidinone with potent activity against multidrug-resistant Gram-positive pathogens. Drugs. 2015;75:253–70.
Roger C, Roberts JA, Muller L. Clinical pharmacokinetics and pharmacodynamics of oxazolidinones. Clin Pharmacokinet. 2018;57:559–75.
Pfizer Medical Information. US. Zyvos® indications and usage (linezolid). https://www.pfizermedicalinformation.com/en-us/zyvox/indications-usage. Accessed 27 Aug 2021.
Flanagan SD, Bien PA, Muñoz KA, Minassian SL, Prokocimer PG. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy. 2014;34:240–50.
Flanagan S, Fang E, Muñoz KA, Minassian SL, Prokocimer PG. Single- and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacother J Hum Pharmacol Drug Ther. 2014;34:891–900.
Prokocimer P, Bien P, Surber J, Mehra P, DeAnda C, Bulitta JB, et al. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2011;55:583–92.
Bien P. Human pharmacokinetics of TR-700 after ascending single oral doses of the prodrug TR-701, a novel oxazolidinone antibiotic. IDSA, Washington DC; 2008.
European Medicines Agency. Assessment report Sivextro. https://www.ema.europa.eu/en/documents/assessment-report/sivextro-epar-public-assessment-report_en.pdf. Accessed 14 Jun 2020.
Ong V, Flanagan S, Fang E, Dreskin HJ, Locke JB, Bartizal K, et al. Absorption, distribution, metabolism, and excretion of the novel antibacterial prodrug tedizolid phosphates. Drug Metab Dispos. 2014;42:1275–84.
Flanagan S, Passarell J, Lu Q, Fiedler-Kelly J, Ludwig E, Prokocimer P. Tedizolid population pharmacokinetics, exposure response, and target attainment. Antimicrob Agents Chemother. 2014;58:6462–70.
Chen R, Shen K, Chang X, Tanaka T, Li L, Hu P. Pharmacokinetics and safety of tedizolid after single and multiple intravenous/oral sequential administrations in healthy Chinese subjects. Clin Ther. 2016;38:1869–79.
Wang S, Li Y, Xue F, Liu J, Yang W, Zhang J, et al. Comparative in vitro potency and kill curve activity of tedizolid and linezolid against Gram-positive bacteria isolated from Chinese hospitalized patients in 2013–2016. J Chemother. 2019;31:313–9.
Carvalhaes CG, Sader HS, Flamm RK, Mendes RE. Tedizolid in vitro activity against Gram-positive clinical isolates causing bone and joint infections in hospitals in the USA and Europe (2014–17). J Antimicrob Chemother. 2019;74:1928–33.
Mikamo H, Takesue Y, Iwamoto Y, Tanigawa T, Kato M, Tanimura Y, et al. Efficacy, safety and pharmacokinetics of tedizolid versus linezolid in patients with skin and soft tissue infections in Japan: results of a randomised, multicentre phase 3 study. J Infect Chemother. 2018;24:434–42.
Flanagan S, Minassian SL, Morris D, Ponnuraj R, Marbury TC, Alcorn HW, et al. Pharmacokinetics of tedizolid in subjects with renal or hepatic impairment. Antimicrob Agents Chemother. 2014;58:6471–6.
Flanagan S, Prokocimer P. Reduction in tedizolid plasma exposure among end-stage renal disease patients undergoing dialysis is explained by variations in ideal body weight. Antimicrob Agents Chemother. 2016;60:3246–7.
Lewis SJ, Switaj LA, Mueller BA. Tedizolid adsorption and transmembrane clearance during in vitro continuous renal replacement therapy. Blood Purif. 2015;40:66–71.
Bradley JS, Flanagan SD, Arrieta AC, Jacobs R, Capparelli E, Prokocimer P. Pharmacokinetics, safety and tolerability of single oral or intravenous administration of 200 mg tedizolid phosphate in adolescents. Pediatr Infect Dis J. 2016;35:628–33.
Arrieta AC, Ang JY, Espinosa C, Fofanov O, Tøndel C, Chou MZ, et al. Pharmacokinetics and safety of single-dose tedizolid phosphate in children 2 to <12 years of age. Pediatr Infect Dis J. 2021;40:317–23.
Flanagan SD, Minassian SL, Prokocimer P. Pharmacokinetics, safety, and tolerability of tedizolid phosphate in elderly subjects. Clin Pharmacol Drug Dev. 2018;7:788–94.
Flanagan S, Minassian SL, Passarell JA, Fiedler-Kelly J, Prokocimer P. Pharmacokinetics of tedizolid in obese and nonobese subjects. J Clin Pharmacol. 2017;57:1290–4.
U.S. Food and Drug Administration (FDA). Center for Drug Evaluation and Research. NDA Sivextro, application number 205435Orig1s000. 2013. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205435Orig1s000ClinPharmR.pdf.
Pai MP. Pharmacokinetics of tedizolid in morbidly obese and covariate-matched nonobese adults. Antimicrob Agents Chemother. 2016;60:4585–9.
Grégoire M, Libois JB, Waast D, Gaborit B, Dauty M, Deslandes G, et al. Pharmacokinetics of tedizolid in an obese patient after bariatric surgery. Antimicrob Agents Chemother. 2018;62:e02432-e2517.
Park AYJ, Wang J, Jayne J, Fukushima L, Rao AP, D’Argenio DZ, et al. Pharmacokinetics of tedizolid in plasma and sputum of adults with cystic fibrosis. Antimicrob Agents Chemother. 2018;62(9):e00550-e618.
Flanagan S, Bartizal K, Minassian SL, Fang E, Prokocimer P. In vitro, in vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob Agents Chemother. 2013;57:3060–6.
Sahre M, Sabarinath S, Grant M, Seubert C, DeAnda C, Prokocimer P, et al. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int J Antimicrob Agents. 2012;40:51–4.
Stainton SM, Monogue ML, Baummer-Carr A, Shepard AK, Nugent JF, Kuti JL, et al. Comparative assessment of tedizolid pharmacokinetics and tissue penetration between diabetic patients with wound infections and healthy volunteers via in vivo microdialysis. Antimicrob Agents Chemother. 2018;62:e01880-e1917.
Housman ST, Pope JS, Russomanno J, Salerno E, Shore E, Kuti JL, et al. Pulmonary disposition of tedizolid following administration of once-daily oral 200-milligram tedizolid phosphate in healthy adult volunteers. Antimicrob Agents Chemother. 2012;56:2627–34.
Park AYJ, Wang J, Jayne J, Fukushima L, Rao AP, D’argenio DZ, et al. Pharmacokinetics of tedizolid in plasma and sputum of adults with cystic fibrosis. Antimicrob Agents Chemother. 2018;62:e00500-e518.
Takeda K, Kaku N, Morinaga Y, Kosai K, Uno N, Imamura Y, et al. Tedizolid inhibits MUC5AC production induced by methicillin-resistant Staphylococcus aureus in human airway epithelial cells. J Infect Chemother. 2017;23:598–603.
Wenzler E, Adeel A, Wu T, Jurkovic M, Walde J, Ramasra E, et al. Inadequate cerebrospinal fluid concentrations of available salvage agents further impedes the optimal treatment of multidrug-resistant enterococcus faecium meningitis and bacteremia. Infect Dis Rep. 2021;13:843–54.
Gu L, Ma M, Zhang Y, Zhang L, Zhang S, Huang M, et al. Comparative pharmacokinetics of tedizolid in rat plasma and cerebrospinal fluid. Regul Toxicol Pharmacol. 2019;107:104420.
Bensaci M, Flanagan S, Sandison T. Determination of tedizolid susceptibility interpretive criteria for gram-positive pathogens according to clinical and laboratory standards institute guidelines. Diagn Microbiol Infect Dis. 2018;90:214–20.
Schaadt R, Sweeney D, Shinabarger D, Zurenko G. In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent. Antimicrob Agents Chemother. 2009;53:3236–9.
Bensaci M, Sahm D. Surveillance of tedizolid activity and resistance: in vitro susceptibility of Gram-positive pathogens collected over 5 years from the United States and Europe. Diagn Microbiol Infect Dis. 2017;87:133–8.
Pfaller MA, Flamm RK, Jones RN, Farrell DJ, Mendes RE. Activities of tedizolid and linezolid determined by the reference broth microdilution method against 3,032 Gram-positive bacterial isolates collected in Asia-Pacific, Eastern Europe, and Latin American countries in 2014. Antimicrob Agents Chemother. 2016;60:5393–9.
Pfaller MA, Sader HS, Rhomberg PR, Flamm RK, Mendes RE. In vitro activity of tedizolid in comparison with other oral and intravenous agents against a collection of community-acquired methicillin-resistant Staphylococcus aureus (2014–2015) in the United States. Microb Drug Resist. 2019;25:938–43.
Guo Y, Yang Y, Zheng Y, Wu S, Yin D, Zhu D, et al. Comparative in vitro activities of ceftaroline and tedizolid against clinical strains of Staphylococcus aureus and enterococcus: results from the china antimicrobial surveillance network (CHINET) in 2018. Antimicrob Agents Chemother. 2020;64:e01461-e1520.
Brown SD, Traczewski MM. Comparative in Vitro antimicrobial activities of torezolid (TR-700), the active moiety of a new oxazolidinone, torezolid phosphate (TR-701), determination of tentative disk diffusion interpretive criteria, and quality control ranges. Antimicrob Agents Chemother. 2010;54:2063–9.
World Health Organization. Antimicrobial resistance. https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance. Accessed 15 Jun 2020.
Corey R, Moran G, Goering R, Bensaci M, Sandison T, De Anda C, et al. Comparison of the microbiological efficacy of tedizolid and linezolid in acute bacterial skin and skin structure infections: pooled data from phase 3 clinical trials. Diagn Microbiol Infect Dis. 2019;94:277–86.
Lee Y, Kuk Hong S, Choi S, Yong D, Lee K. Brief communication clinical microbiology in vitro activity of tedizolid against Gram-positive bacteria in patients with skin and skin structure infections and hospital-acquired pneumonia: a Korean multicenter study. Ann Lab Med. 2015;35:523–30.
Im WB, Choi SH, Park JY, Choi SH, Finn J, Yoon SH. Discovery of torezolid as a novel 5-hydroxymethyl-oxazolidinone antibacterial agent. Eur J Med Chem. 2011;46:1027–39.
Shaw KJ, Poppe S, Schaadt R, Brown-Driver V, Finn J, Pillar CM, et al. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob Agents Chemother. 2008;52:4442–7.
Jones RN, Moet GJ, Sader HS, Mendes RE, Castanheira M. TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens. J Antimicrob Chemother. 2009;63:716–20.
Yum JH, Choi SH, Yong D, Chong Y, Im WB, Rhee D-K, et al. Clinical isolates of aerobic and anaerobic bacteria in South Korea. Antimicrob Agents Chemother. 2010;54:5381–6.
Betts JW, Abdul Momin HF, Phee LM, Wareham DW. Comparative activity of tedizolid and glycopeptide combination therapies for the treatment of Staphylococcus aureus infections: an in vitro and in vivo evaluation against strains with reduced susceptibility to glycopeptides. J Med Microbiol. 2018;67:265–71.
Rolston KVI, Reitzel R, Vargas-Cruz N, Shelburne SA, Raad II, Prince RA. In vitro activity of tedizolid and comparator agents against clinical Gram-positive isolates recovered from patients with cancer. Diagn Microbiol Infect Dis. 2018;91:351–3.
Bai B, Hu K, Li H, Yao W, Li D, Chen Z, et al. Effect of tedizolid on clinical Enterococcus isolates: in vitro activity, distribution of virulence factor, resistance genes and multilocus sequence typing. FEMS Microbiol Lett. 2018;91:365.
Carvalhaes CG, Sader HS, Flamm RK, Streit JM, Mendes RE. Assessment of tedizolid in vitro activity and resistance mechanisms against a collection of Enterococcus spp. causing invasive infections, including isolates requiring an optimized dosing strategy for daptomycin from U.S. and European Medical Centers, 2016 to 2018. Antimicrob Agents Chemother. 2020;64:e00175-e220.
Choudhury S, Mun LK, Ng E, Xuan C, Jia LS, Vasoo S, et al. Evaluation of in vitro susceptibility of Gram-positive pathogens from a tertiary care hospital in Singapore to a novel oxazolidinone, tedizolid, by a gradient diffusion method and broth microdilution. J Clin Pathol. 2019;72:181–4.
Barber KE, Smith JR, Raut A, Rybak MJ. Evaluation of tedizolid against Staphylococcus aureus and enterococci with reduced susceptibility to vancomycin, daptomycin or linezolid. J Antimicrob Chemother. 2016;71:152–5.
Livermore DM, Mushtaq S, Warner M, Woodford N. Activity of oxazolidinone TR-700 against linezolid-susceptible and -resistant staphylococci and enterococci. J Antimicrob Chemother. 2009;63:713–5.
Rodríguez-Avial I, Culebras E, Betriu C, Morales G, Pena I, Picazo JJ. In vitro activity of tedizolid (TR-700) against linezolid-resistant staphylococci. J Antimicrob Chemother. 2012;67:167–9.
Sahm DF, Deane J, Bien PA, Locke JB, Zuill DE, Shaw KJ, et al. Results of the surveillance of tedizolid activity and resistance program: in vitro susceptibility of gram-positive pathogens collected in 2011 and 2012 from the United States and Europe. Diagn Microbiol Infect Dis. 2015;81:112–8.
Silva-Del Toro SL, Greenwood-Quaintance KE, Patel R. In vitro activity of tedizolid against linezolid-resistant staphylococci and enterococci. Diagn Microbiol Infect Dis. 2016;85:102–4.
Roch M, Varela MC, Taglialegna A, Rosato AE. Tedizolid is a promising antimicrobial option for the treatment of Staphylococcus aureus infections in cystic fibrosis patients. J Antimicrob Chemother. 2019;75:126–34.
Chen KH, Huang YT, Liao CH, Sheng WH, Hsuehd PR. In vitro activities of tedizolid and linezolid against Gram-positive cocci associated with acute bacterial skin and skin structure infections and pneumonia. Antimicrob Agents Chemother. 2015;59:6262–5.
EUCAST. Clinical breakpoints and dosing of antibiotics. http://www.eucast.org/clinical_breakpoints/. Accessed 19 Sep 2019.
Carvalhaes CG, Sader HS, Flamm RK, Streit JM, Mendes RE. Assessment of tedizolid in vitro activity and resistance mechanisms against a collection of Enterococcus spp. causing invasive infections including isolates requiring an optimized dosing strategy for daptomycin from United States and European Medical Centers, 2016 to 2018. Antimicrob Agents Chemother. 2020;64:e00175-e220.
Carvalhaes CG, Sader HS, Rhomberg PR, Castanheira M, Mendes RE. 1547. Activity of tedizolid and comparator agents against Gram-positive bacterial isolates causing skin and skin structure infections in pediatric patients during 2015–2019 in the US. Open Forum Infect Dis. 2020;7:S773.
Carvalhaes CG, Sader HS, Rhomberg PR, Mendes RE. Tedizolid activity against a multicentre worldwide collection of Staphylococcus aureus and Streptococcus pneumoniae recovered from patients with pneumonia (2017–2019). Int J Infect Dis. 2021;107:92–100.
Tsai HY, Lee YL, Liu PY, Lu MC, Shao PL, Lu PL, et al. Antimicrobial susceptibility of bacteremic vancomycin-resistant Enterococcus faecium to eravacycline, omadacycline, lipoglycopeptides, and other comparator antibiotics: results from the 2019–2020 nationwide Surveillance of Multicenter Antimicrobial Resistance in Taiwan (SMART). Int J Antimicrob Agents. 2021;58:106353.
Rodríguez-Gascón A, Aguirre-Quiñonero A, Aspiazu MAS, Canut-Blasco A. Pharmacokinetic/Pharmacodynamic Analysis of Tedizolid Phosphate Compared to Linezolid for the Treatment of Infections Caused by Gram-Positive Bacteria. Antibiot. 2021;10:755.
Lemaire S, Van Bambeke F, Appelbaum PC, Tulkens PM. Cellular pharmacokinetics and intracellular activity of torezolid (TR-700): studies with human macrophage (THP-1) and endothelial (HUVEC) cell lines. J Antimicrob Chemother. 2009;64:1035–43.
Goldstein EJC, Vreni Merriam C, Citron DM. In vitro activity of tedizolid compared to linezolid and five other antimicrobial agents against 332 anaerobic isolates, including bacteroides fragilis group, Prevotella, Porphyromonas, and Veillonella species. Antimicrob Agents Chemother. 2020;64:e01088–20.
Barman TK, Kumar M, Mathur T, Chaira T, Ramkumar G, Kalia V, et al. In Vitro and In Vivo Activities of a Bi-Aryl Oxazolidinone, RBx 11760, against Gram-Positive Bacteria. Antimicrob Agents Chemother. 2016;60:7134–45.
Abdelhady W, Mishra NN. Comparative efficacies of linezolid vs. tedizolid in an experimental murine model of vancomycin-resistant enterococcal (VRE) bacteremia. Front Med. 2019;6:31.
Bayer AS, Abdelhady W, Li L, Gonzales R, Xiong YQ. Comparative efficacies of tedizolid phosphate, linezolid, and vancomycin in a murine model of subcutaneous catheter-related biofilm infection due to methicillin-susceptible and -resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2016;60:5092–6.
Werth BJ. Exploring the pharmacodynamic interactions between tedizolid and other orally bioavailable antimicrobials against Staphylococcus aureus and Staphylococcus epidermidis. J Antimicrob Chemother. 2017;72:1410–4.
Smith JR, Yim J, Rice S, Stamper K, Kebriaei R, Rybak MJ. Combination of Tedizolid and Daptomycin against Methicillin-Resistant Staphylococcus aureus in an In Vitro Model of Simulated Endocardial Vegetations. Antimicrob Agents Chemother. 2018;62:e00101–e218.
Louie A, Liu W, Kulawy R, Drusano GL. In vivo pharmacodynamics of torezolid phosphate (TR-701), a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55:3453–60.
US FDA. Sivextro (tedizolid) tablets: prescribing information. Kenilworth (NJ): Merck & Co., Inc., 2016. www.fda.gov/medwatch. Accessed 12 Dec 2021.
Bryant AE, Bayer CR, Aldape MJ, McIndoo E, Stevens DL. Emerging erythromycin and clindamycin resistance in group A streptococci: efficacy of linezolid and tedizolid in experimental necrotizing infection. J Glob Antimicrob Resist. 2020;22:601–7.
Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55:5300–5.
Lodise TP, Drusano GL. Use of pharmacokinetic/pharmacodynamic systems analyses to inform dose selection of tedizolid phosphate. Clin Infect Dis. 2014;58(Suppl. 1):S28-34.
Xiao J, Gill C, Liang L, Liu J, Wu J, Feng HP, et al. Use of Translational Pharmacokinetic/Pharmacodynamic Infection Models To Understand the Impact of Neutropenia on the Efficacy of Tedizolid Phosphate. Antimicrob Agents Chemother. 2018;63:e00822–e918.
European Medicines Agency. Sivextro. https://www.ema.europa.eu/en/medicines/human/EPAR/sivextro. Accessed 28 Jul 2021.
Abdelraouf K, Nicolau DP. Comparative in vivo efficacies of tedizolid in neutropenic versus immunocompetent murine Streptococcus pneumoniae lung infection models. Antimicrob Agents Chemother. 2016;61(1):e01957-e2016.
Keel RA, Tessier PR, Crandon JL, Nicolau DP. Comparative efficacies of human simulated exposures of tedizolid and linezolid against Staphylococcus aureus in the murine thigh infection model. Antimicrob Agents Chemother. 2012;56:4403–7.
Keel RA, Crandon JL, Nicolau DP. Pharmacokinetics and pulmonary disposition of tedizolid and linezolid in a murine pneumonia model under variable conditions. Antimicrob Agents Chemother. 2012;56:3420–2.
Kaku N, Morinaga Y, Takeda K, Kosai K, Uno N, Hasegawa H, et al. Antimicrobial and immunomodulatory effects of tedizolid against methicillin-resistant Staphylococcus aureus in a murine model of hematogenous pulmonary infection. Int J Med Microbiol. 2016;306:421–8.
Nielsen EI, Friberg LE. Pharmacokinetic-pharmacodynamic modeling of antibacterial drugs. Pharmacol Rev. 2013;65(3):1053–90.
Choi S, Im W, Bartizal K. Activity of tedizolid phosphate (TR-701) in murine models of infection with penicillin-resistant and penicillin-sensitive Streptococcus pneumoniae. Antimicrob Agents Chemother. 2012;56:4713–7.
Andes D, Craig WA. Pharmacodynamics of the new des-f(6)-quinolone garenoxacin in a murine thigh infection model. Antimicrob Agents Chemother. 2003;47:3935–41.
Lepak AJ, Marchillo K, Pichereau S, Craig WA, Andes DR. Comparative pharmacodynamics of the new oxazolidinone tedizolid phosphate and linezolid in a neutropenic murine Staphylococcus aureus pneumonia model. Antimicrob Agents Chemother. 2012;56:5916–22.
Tessier PR, Keel RA, Hagihara M, Crandon JL, Nicolau DP. Comparative in vivo efficacies of epithelial lining fluid exposures of tedizolid, linezolid, and vancomycin for methicillin-resistant Staphylococcus aureus in a mouse pneumonia model. Antimicrob Agents Chemother. 2012;56(5):2342–6.
Kidd JM, Abdelraouf K, Nicolau DP. Comparative efficacy of human-simulated epithelial lining fluid exposures of tedizolid, linezolid and vancomycin in neutropenic and immunocompetent murine models of staphylococcal pneumonia. J Antimicrob Chemother. 2019;74:970–7.
Prokocimer P, De Anda C, Fang E, Mehra P, Das A. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA. 2013;309:559–69.
Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14:696–705.
Kullar R, Puzniak LA, Swindle JP, Lodise T. Retrospective real-world evaluation of outcomes in patients with skin and soft structure infections treated with tedizolid in an outpatient setting. Infect Dis Ther. 2020;9(1):107–17.
Shorr AF, Lodise TP, Corey GR, De Anda C, Fang E, Das AF, et al. Analysis of the phase 3 ESTABLISH trials of tedizolid versus linezolid in acute bacterial skin and skin structure infections. Antimicrob Agents Chemother. 2015;59:864–71.
Sandison T, De Anda C, Fang E, Das AF, Prokocimer P. Clinical response of tedizolid versus linezolid in acute bacterial skin and skin structure infections by severity measure using a pooled analysis from two phase 3 double-blind trials. Antimicrob Agents Chemother. 2017;61:864–71.
Lv X, Alder J, Li L, O’Riordan W, Rybak MJ, Ye H, et al. Efficacy and safety of tedizolid phosphate versus linezolid in a randomized phase 3 trial in patients with acute bacterial skin and skin structure infection. Antimicrob Agents Chemother. 2019;63:2252–70.
Wunderink RG, Roquilly A, Croce M, Rodriguez Gonzalez D, Fujimi S, Butterton JR, et al. A phase 3, randomized, double-blind study comparing tedizolid phosphate and linezolid for treatment of ventilated Gram-positive hospital-acquired or ventilator-associated bacterial pneumonia. Clin Infect Dis. 2021;73:e710–8.
Burdette SD, Trotman R. Tedizolid: The First Once-Daily Oxazolidinone Class Antibiotic. Clin Infect Dis. 2015;61:1315–21.
Hardalo C, Lodise TP, Bidell M, Flanagan S, De Anda C, Anuskiewicz S, et al. Clinical safety and tolerability of tedizolid phosphate in the treatment of acute bacterial skin and skin structure infections. Expert Opin Drug Saf. 2018;17:359–67.
Lodise TP, Bidell MR, Flanagan SD, Zasowski EJ, Minassian SL, Prokocimer P. Characterization of the haematological profile of 21 days of tedizolid in healthy subjects. J Antimicrob Chemother. 2016;71:2553–8.
Lodise TP, Fang E, Minassian SL, Prokocimer PG. Platelet profile in patients with acute bacterial skin and skin structure infections receiving tedizolid or linezolid: findings from the phase 3 establish clinical trials. Antimicrob Agents Chemother. 2014;58:7198–204.
Das D, Tulkens PM, Mehra P, Fang E, Prokocimer P. Tedizolid phosphate for the management of acute bacterial skin and skin structure infections: safety summary. Clin Infect Dis. 2014;58 Suppl 1:S51–57.
Vendrell MM, Pitarch MT, Lletí MS, Muñoz EC, Ruiz LM, Lao GC, et al. Safety and Tolerability of More than Six Days of Tedizolid Treatment. Antimicrob Agents Chemother. 2020;64:e00356–e420.
York JA, Adams K, Cullen L, Delahay J, Ivan M, Lillie PJ, et al. Tedizolid: a service evaluation in a large UK teaching hospital. Eur J Clin Microbiol Infect Dis. 2021;40:397–405.
Benavent E, Morata L, Escrihuela-Vidal F, Reynaga EA, Soldevila L, Albiach L, et al. Long-term use of tedizolid in osteoarticular infections: benefits among oxazolidinone drugs. Antibiotics. 2021;10:1–10.
Fang E, Muñoz KA, Prokocimer P. 916: Neurologic and ophthalmologic safety results with 10 day dosing of tedizolid phosphate. Crit Care Med. 2013;41:A229–30.
Bradley JS, Antadze T, Ninov B, Tayob MS, Broyde N, Butterton JR, et al. Safety and efficacy of oral and/or intravenous tedizolid phosphate from a randomized phase 3 trial in adolescents with acute bacterial skin and skin structure infections. Pediatr Infect Dis J. 2021;40:238–44.
Senneville E, Dinh A, Ferry T, Beltrand E, Blondiaux N, Robineau O. Tolerance of prolonged oral tedizolid for prosthetic joint infections: results of a multicentre prospective study. Antibiotics. 2021;10:1–12.
Gatti M, Fusaroli M, Raschi E, Moretti U, Poluzzi E, De Ponti F. Serious adverse events with tedizolid and linezolid: pharmacovigilance insights through the FDA adverse event reporting system. Expert Opin Drug Saf. 2021;20:1421–31.
Senneville E, Dinh A, Ferry T, Beltrand E, Blondiaux N, Robineau O. Tolerance of prolonged oral tedizolid for prosthetic joint infections: results of a multicentre prospective study. Antibiotics (Basel). 2020;10:1–12.
Kim T, Wills A, Markus A, Prevots DR, Olivier KN. Safety and tolerability of long term use of tedizolid for treatment of nontuberculous mycobacterial infections. Open Forum Infect Dis. 2016;3:577.
Poon YK, La HRM, Hinan LS, Sanders J, Monogue ML. Tedizolid vs linezolid for the treatment of nontuberculous mycobacteria infections in solid organ transplant recipients. Open forum Infect Dis. 2021;8:1–8.
Yuste JR, Serrano-Alonso M, Carmona-Torre F, Del Pozo JL, Herrero JI. Efficacy and safety of long-term use of tedizolid after liver transplantation in an adolescent with pulmonary tuberculosis. J Antimicrob Chemother. 2019;74:2817–9.
Nigo M, Luce AM, Arias CA. Long-term use of tedizolid as suppressive therapy for recurrent methicillin-resistant Staphylococcus aureus graft infection. Clin Infect Dis. 2018;66:1975–6.
Shaw TD, Smyth M, Turner G, Hunter M. Prolonged tedizolid use in cutaneous non-tuberculous mycobacterial infection. J Clin Tuberc Other Mycobact Dis. 2021;24:100261.
Soueges S, Triffault-Fillit C, Roux S, Labussière-Wallet H, Lebeaux D, Dumitrescu O, et al. Long-term use of liposomal nebulized amikacin and tedizolid for the treatment of disseminated nocardiosis after allogeneic hematopoietic stem cell transplantation. Eur J Clin Microbiol Infect Dis. 2021;40:2033–6.
Yuste JR, Serrano-Alonso M, Carmona-Torre F, Del Pozo JL, Herrero JIH. Efficacy and safety of long-term use of tedizolid after liver transplantation in an adolescent with pulmonary tuberculosis. J Antimicrob Chemother. 2019;74(9):2817–9.
Schlosser MJ, Hosako H, Radovsky A, Butt MT, Draganov D, Vija J, et al. Lack of Neuropathological changes in rats administered tedizolid phosphate for nine months. Antimicrob Agents Chemother. 2015;59(1):475–81.
European Medicines Agency. Risk management plans. https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/pharmacovigilance/risk-management/risk-management-plans. Accessed 26 Sep 2021.
Meka VG, Pillai SK, Sakoulas G, Wennersten C, Venkataraman L, DeGirolami PC, et al. Linezolid resistance in sequential Staphylococcus aureus isolates associated with a T2500A mutation in the 23S rRNA gene and loss of a single copy of rRNA. J Infect Dis. 2004;190:311–7.
Locke JB, Hilgers M, Shaw KJ. Novel ribosomal mutations in Staphylococcus aureus strains identified through selection with the oxazolidinones linezolid and torezolid (TR-700). Antimicrob Agents Chemother. 2009;53:5265–74.
Wang Y, Lv Y, Cai J, Schwarz S, Cui L, Hu Z, et al. A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J Antimicrob Chemother. 2015;70:2182–90.
Chen K-H, Huang Y-T, Liao C-H, Sheng W-H, Hsueh P-R. In vitro activities of tedizolid and linezolid against Gram-positive cocci associated with acute bacterial skin and skin structure infections and pneumonia. Antimicrob Agents Chemother. 2015;59(10):6262–5.
Prokocimer P, Bien P, Deanda C, Pillar CM, Bartizal K. In vitro activity and microbiological efficacy of tedizolid (TR-700) against Gram-positive clinical isolates from a phase 2 study of oral tedizolid phosphate (TR-701) in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2012;56(9):4608–13.
Zurenko G, Bien P, Bensaci M, Patel H, Thorne G. Use of linezolid susceptibility test results as a surrogate for the susceptibility of Gram-positive pathogens to tedizolid, a novel oxazolidinone. Ann Clin Microbiol Antimicrob. 2014;13:46.
Ma X, Zhang F, Bai B, Lin Z, Xu G, Chen Z, et al. Linezolid resistance in enterococcus faecalis associated with urinary tract infections of patients in a tertiary hospitals in china: resistance mechanisms, virulence, and risk factors. Front Public Heal. 2021;9:570650.
Choudhury S, Mun LK, Xuan ENC, Jia LS, Vasoo S, Wickramasinghe SS, et al. Evaluation of in vitro susceptibility of Gram-positive pathogens from a tertiary care hospital in Singapore to a novel oxazolidinone, tedizolid, by a gradient diffusion method and broth microdilution. J Clin Pathol. 2019;72:181–4.
Klupp EM, Both A, Belmar Campos C, Büttner H, König C, Christopeit M, et al. Tedizolid susceptibility in linezolid- and vancomycin-resistant Enterococcus faecium isolates. Eur J Clin Microbiol Infect Dis. 2016;35:1957–61.
Iqbal K, Broeker A, Nowak H, Rahmel T, Nussbaumer-Pröll A, Österreicher Z, et al. A pharmacometric approach to define target site-specific breakpoints for bacterial killing and resistance suppression integrating microdialysis, time–kill curves and heteroresistance data: a case study with moxifloxacin. Clin Microbiol Infect. 2020;26(9):1255.e1-8.
Ruiz P, Causse M, Vaquero M, Casal M. In vitro activity of tedizolid against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2019;63(4):e01939-e2018.
Molina-Torres CA, Barba-Marines A, Valles-Guerra O, Ocampo-Candiani J, Cavazos-Rocha N, Pucci MJ, et al. Intracellular activity of tedizolid phosphate and ACH-702 versus Mycobacterium tuberculosis infected macrophages. Ann Clin Microbiol Antimicrob. 2014;13:13.
Vera-Cabrera L, Gonzalez E, Rendon A, Ocampo-Candiani J, Welsh O, Velazquez-Moreno VM, et al. In vitro activities of DA-7157 and DA-7218 against Mycobacterium tuberculosis and Nocardia brasiliensis. Antimicrob Agents Chemother. 2006;50:3170–2.
Srivastava S, Deshpande D, Nuermberger E, Lee PS, Cirrincione K, Dheda K, et al. The sterilizing effect of intermittent tedizolid for pulmonary tuberculosis. Clin Infect Dis. 2018;67(Suppl. 3):S336–41.
Deshpande D, Srivastava S, Nuermberger E, Koeuth T, Martin KR, Cirrincione KN, et al. Multiparameter responses to tedizolid monotherapy and moxifloxacin combination therapy models of children with intracellular tuberculosis. Clin Infect Dis. 2018;67:S342–8.
Deshpande D, Srivastava S, Pasipanodya JG, Lee PS, Gumbo T. Tedizolid is highly bactericidal in the treatment of pulmonary Mycobacterium avium complex disease. J Antimicrob Chemother. 2017;72:ii30–5.
Srivastava S, Deshpande D, Nuermberger E, Lee PS, Cirrincione K, Dheda K, et al. The sterilizing effect of intermittent tedizolid for pulmonary tuberculosis. Clin Infect Dis. 2018;67:S336–41.
Srivastava S, Cirrincione KN, Deshpande D, Gumbo T. Tedizolid, faropenem, and moxifloxacin combination with potential activity against nonreplicating Mycobacterium tuberculosis. Front Pharmacol. 2021;11.
Park AYJ, Wang J, Jayne J, Fukushima L, Rao AP, D’Argenio DZ, et al. Pharmacokinetics of tedizolid in plasma and sputum of adults with cystic fibrosis. Antimicrob Agents Chemother. 2018;62:e00550-e618.
Dehghanyar P, Bürger C, Zeitlinger M, Islinger F, Kovar F, Müller M, et al. Penetration of linezolid into soft tissues of healthy volunteers after single and multiple doses. Antimicrob Agents Chemother. 2005;49:2367–71.
Housman ST, Samuel J, Russomanno J, Salerno E, Shore E, Kuti JL, et al. Pulmonary disposition of tedizolid following administration of once-daily oral 200-milligram tedizolid phosphate in healthy adult volunteers. Antimicrob Agents Chemother. 2012;56:2627–34.
Boselli E, Breilh D, Rimmelé T, Djabarouti S, Toutain J, Chassard D, et al. Pharmacokinetics and intrapulmonary concentrations of linezolid administered to critically ill patients with ventilator-associated pneumonia. Crit Care Med. 2005;33:1529–33.
Myrianthefs P, Markantonis SL, Vlachos K, Anagnostaki M, Boutzouka E, Panidis D et al (2006) Serum andcerebrospinal fluid concentrations of linezolid in neurosurgical patients. Antimicrob Agents Chemother 50:3971–6. https://pubmed.ncbi.nlm.nih.gov/16982782/
Villani P, Regazzi MB, Marubbi F, Viale P, Pagane L, Cristini F, et al. Cerebrospinal fluid linezolid concentrations in postneurosurgical central nervous system infections. Antimicrob Agents Chemother. 2002;46:936–937.
Traunmüller F, Schintler MV, Spendel S, Popovic M, Mauric O, Scharnagl E, et al. Linezolid concentrations in infected soft tissue and bone following repetitive doses in diabetic patients with bacterial foot infections. Int J Antimicrob Agents. 2010;36:84–6.
Godoy Carvalhaes C. Five years of analysis of the in vitro activity of activity of tedizolid against a worldwide collection of indicated species causing clinical infections: results from the Surveillance of Tedizolid Activity and Resistance (STAR) programme. ESCMID eAcademy. 9; 327519. https://eacademy.escmid.org/escmid/2021/eccmid-2021/327519/cecilia.g.carvalhaes.five.years.of.analysis.of.the.3Cem3Ein.vitro3C.em3E26nbsp.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D2%2Ace_id%3D2029%2Alabel%3D22124%2Amarker%3D1353. Accessed 16 Nov 2021.
Carvalhaes CG, Sader HS, Flamm RK, Streit JM, Mendes RE. Assessment of tedizolid in vitro activity and resistance mechanisms against a collection of Enterococcus spp. causing invasive infections including isolates requiring an optimized dosing strategy for daptomycin from U.S. and European Medical Centers, 2016 to 2018. Antimicrob Agents Chemother. 2020;64:e00175-e220.
Goldstein EJC, Vreni Merriam C, Citron DM. In vitro activity of tedizolid compared to linezolid and five other antimicrobial agents against 332 anaerobic isolates, including bacteroides fragilis group, prevotella, porphyromonas, and veillonella species. Antimicrob Agents Chemother. 2020;64:e01088–e1120.
Brown-Elliott BA, Wallace RJ, Land GA, Bloodcare C. In vitro susceptibility testing of tedizolid against nontuberculous mycobacteria. J Clin Microbiol. 2017;55:1747–54.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL.
Conflicts of interest/competing interests
No conflict of interest to declare.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
KI made a substantial contribution to the conceptualization, literature search, interpretation and analysis of the data, and writing and drafting the original manuscript draft. AM made a substantial contribution to the literature search and drafting the manuscript. SGW substantially contributed to the interpretation of the literature data, editing, reviewing and revising the original manuscript, and approval for the final version of the manuscript for submission.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Iqbal, K., Milioudi, A. & Wicha, S.G. Pharmacokinetics and Pharmacodynamics of Tedizolid. Clin Pharmacokinet 61, 489–503 (2022). https://doi.org/10.1007/s40262-021-01099-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01099-7