
Abstract
Treatment of multidrug-resistant Gram-positive infections continues to challenge clinicians as the emergence of new resistance mechanisms outpaces introduction of novel antimicrobial agents. Tedizolid phosphate is a next-generation oxazolidinone with activity against both methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus spp. Tedizolid has consistently shown potency advantages over linezolid against Gram-positive microorganisms including those with reduced susceptibility to linezolid. Of particular significance, minimum inhibitory concentrations of tedizolid appear to be largely unaffected by the chloramphenicol–florfenicol resistance (cfr) gene, which has been implicated in a number of published linezolid-resistant organism outbreaks. Tedizolid phosphate also has been found to have a favorable pharmacokinetic profile allowing for once-daily dosing in both oral and intravenous forms. Potency and pharmacokinetic advantages have allowed for lower total daily doses of tedizolid, compared to linezolid, being needed for clinical efficacy in the treatment of acute bacterial skin and skin structure infections (ABSSSI). The decreased total drug exposure produced may in part be responsible for a decrease in the observed adverse effects including thrombocytopenia. Tedizolid phosphate is currently indicated for the treatment of ABSSSI and under investigation for the treatment of nosocomial pneumonia. Although much of the role of tedizolid remains to be defined by expanding clinical experience, tedizolid is likely a welcomed addition to the mere handful of agents available for the treatment of multidrug-resistant Gram-positive infections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multidrug-resistant Gram-positive organisms are commonly causative of nosocomial infections and are associated with significant morbidity and mortality [1]. Of particular concern to clinicians, methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE) remain sensitive to few antimicrobial agents available for the treatment of serious infections [2–4]. Daptomycin and linezolid have been increasingly utilized over the past 15 years, and reduced susceptibility to each of these drugs has emerged among staphylococci and enterococci [5–8]. Though resistance to these agents is currently rare, expanding the antibiotic armamentarium available for the treatment of serious MRSA and VRE infections has become increasingly important.
Oxazolidinones are a synthetic class of antimicrobials, developed over the past 30 years by numerous pharmaceutical companies [9]. Linezolid, the first oxazolidinone approved for use in clinical practice, has activity against both multidrug-resistant staphylococci and enterococci. Currently indicated for the treatment of complicated and uncomplicated skin and skin structure infections (cSSSI), community-acquired and nosocomial pneumonia, as well as infections caused by VRE, linezolid has found widespread utilization in clinical practice [10].
Tedizolid phosphate (formerly torezolid; Cubist Pharmaceuticals) is the first of the next generation of oxazolidinones to obtain Food and Drug Administration (FDA) approval. A number of similarities exist between tedizolid and its predecessor linezolid. However, initial experience with tedizolid has shown advantages in antimicrobial potency against key organisms including those with reduced susceptibility to linezolid, lower incidence of adverse effects over short courses of therapy, and favorable pharmacokinetics. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Structure and Mechanism of Antimicrobial Activity
Throughout the development of the oxazolidinones, the class pharmacophore was increasingly refined by a series of structure–activity relationship studies [9]. Linezolid demonstrates several structural features strongly associated with enhanced antimicrobial potency. The addition of the N-aryl group (B-ring) to the oxazolidinone ring (A-ring) is essential for activity and is further enhanced by both a meta-fluorine and para-oriented electron withdrawing or unsaturated group (C-ring). Linezolid, as well as a number of oxazolidinones currently undergoing clinical investigation, features an acetamidomethyl sidechain on C-5 of the oxazolidinone ring. The incorporation of this sidechain has displayed potency benefits among both Gram-positive organisms and mycobacterium [11].
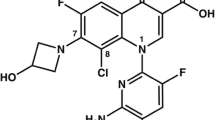
The approval of tedizolid phosphate marks not only the first of a new generation of oxazolidinone antimicrobials, but also a departure from previously held structure–activity relationships (Fig. 1). While earlier studies found a hydroxymethyl sidechain in the C-5 position offered decreased potency, it was later shown that this could be circumvented with the addition of a fourth para-oriented ring structure (D-ring) [12]. It is believed the D-ring structure adds additional sites for hydrogen bonding and further stabilizes interactions with the target site. Interestingly, with these structural features bookending the structure of tedizolid, an antimicrobial activity several fold above that of linezolid was achieved. The utilization of the phosphorylated prodrug, tedizolid phosphate, enables significantly improved solubility in water and excellent oral bioavailability while also masking the C-5 hydroxymethyl from interactions with monoamine oxidase (MAO) [13]. The phosphate group is readily cleaved in blood by serum phosphatase and does not impair antimicrobial potency [9, 14].

Structures of tedizolid, tedizolid phosphate, and linezolid
Oxazolidinones exert antimicrobial activity through inhibition of protein synthesis, binding to the V-domain of the 23S rRNA component of the 50S ribosomal subunit. This inhibition produces primarily bacteriostatic antimicrobial activity at clinically relevant concentrations. Extensive interactions between oxazolidinones and the A-site of the peptidyl-transferase center (PTC) block the alignment of incoming aminoacyl-tRNA and halt peptide elongation [15]. Although the hypothesized interaction sites for oxazolidinones are at least partially unique to this class of protein synthesis inhibitors, there appears to be a degree of overlap with other antimicrobials such as chloramphenicol, clindamycin, and streptogramin A [16]. As with other protein synthesis inhibitors such as clindamycin, some studies have suggested that oxazolidinones may decrease the production of certain toxins among staphylococci and streptococci [17, 18]. However, further investigation will be required to identify the clinical utility of this effect.
Susceptibility
Linezolid has demonstrated significant activity against a number of Gram-positive organisms as well as mycobacterium. The most recent data from the Zyvox® (Pfizer) Annual Appraisal of Potency and Spectrum (ZAAPS) reports the susceptibility of S. aureus, coagulase-negative staphylococci (CoNS), enterococci, and streptococci, totaling 7972 isolates, collected across the five continents in 2012 (Table 1). In this study, the 90% minimum inhibitory concentration (MIC90) for methicillin-susceptible S. aureus (MSSA), MRSA, Enterococcus faecium (including vancomycin-resistant isolates), and Enterococcus faecalis was found to be 2 mg/L. CoNS and β-hemolytic streptococci had MIC90 of 1 mg/L. Linezolid non-susceptible isolates were reported but occurred very rarely, with the largest proportion (0.9%) among CoNS [19].
Tedizolid phosphate exhibits a spectrum of activity similar to that of linezolid. However, the potency of tedizolid against Gram-positive organisms has been reported to be two- to eightfold higher than that of linezolid throughout development. Recently, tedizolid susceptibility among 6884 isolates from the USA and 11 European countries was examined. In general, tedizolid displayed MIC90 approximately fourfold lower than those of linezolid. Organisms with decreased susceptibility to linezolid also commonly have elevated tedizolid MIC. Among isolates non-susceptible to linezolid, tedizolid MIC (ranging from 0.5 to 8 mg/L) were 8- to 16-fold lower than those of linezolid, varying with specific resistance mechanisms (Table 2). Interestingly, for three of the four S. aureus isolates harboring cfr-mediated resistance, tedizolid MIC remained at 0.5 mg/L, with the fourth isolate having an MIC of 1 mg/L.
The current FDA approved clinical breakpoint for tedizolid susceptibility is ≤0.5 mg/L.
Mechanisms of Resistance
Although it was originally anticipated that the fully synthetic nature of the oxazolidinone class would circumvent preexisting pools of resistance, the first isolates of Gram-positive organisms with resistance to linezolid appeared during preclinical trials [20]. Additionally, a clinical isolate of S. aureus was found to be resistant to linezolid after acquisition of a previously known and naturally occurring resistance mechanism [21]. Emergence of clinical isolates with reduced susceptibility to linezolid has nonetheless remained relatively rare for over a decade. Resistance rates vary slightly by region, but remain under 1% across staphylococci and enterococci. Resistance is most frequently reported in individual patients following repeated or extended exposures to linezolid, although horizontal transfer has led to outbreaks of organisms with reduced linezolid susceptibility at institutions with higher linezolid utilization [8, 22]. Reduced susceptibility to oxazolidinones is most frequently associated with either point mutations within the ribosomal complex or the acquisition of the chloramphenicol–florfenicol resistance (cfr) gene.
Point mutations within either the 23S rRNA or ribosomal proteins L3 and L4 were the first widely recognized mechanisms of reduced oxazolidinone susceptibility. As staphylococci and enterococci typically possess five to six and four to six copies of 23S rRNA genes, respectively, multiple mutations often must be acquired before MIC climb above clinical breakpoints. The G2576T mutation is a notable exception, where a single copy mutation can create resistant isolates [23]. This genetic redundancy may at least in part be responsible for the sustained activity of linezolid. However, single copy mutations have been shown to be relatively stable and possess low fitness cost, making additional mutations and more rapid emergence of resistant isolates possible with multiple exposures to linezolid [24]. This more rapid emergence of resistance has been reported clinically after as few as two courses of linezolid [25].
L3 and L4 ribosomal proteins are located proximally to the 23S rRNA, and mutations in these proteins appear to disturb the interactions between oxazolidinones and the PTC. This mechanism of resistance may be less common than others among staphylococci and enterococci; however, a recent study of the Center for Disease Control and Prevention’s Active Bacterial Core surveillance found L4 mutations to be the only mechanism detected among linezolid-resistant pneumococcus (though only present in 3 of the 45,099 isolates tested) [26]. In staphylococci and enterococci, modifications to L4 in particular appear to have a clinically significant impact on linezolid MIC, leading to a fourfold increase when added to previously susceptible isolates [27]. Additionally, L3 mutations have been shown to restore fitness to organisms with multiple 23S rRNA mutations, as well as dramatically inflate MIC in the presence of the cfr gene [28, 29].
Perhaps, the most worrisome of these resistance mechanisms is the acquisition of the cfr gene. This gene, previously best known for leading to phenicol-resistant infections in livestock, is often carried on mobile genetic elements and has been shown to be horizontally transferrable [30]. In fact, cfr has now been detected in both Gram-positive and Gram-negative organisms ranging from enterococci to Enterobacteriaceae [31]. The cfr gene encodes for the RNA methyltransferase Cfr, which adds a second methyl group at A2503 of the 23S rRNA. This additional methylation is known to occupy a portion of the PTC binding pocket which leads to decreased affinity for a number of protein synthesis inhibitors. CM05, the first clinical isolate observed to possess Cfr-mediated linezolid resistance, also possessed the ermB methyltransferase gene. The combination of these genes under regulation of a single promoter became known as the mlr operon and essentially abolishes the activity of all protein synthesis inhibitors common to clinical practice today [32, 33]. To date, the mlr operon has not been reported to be present in other clinical isolates.
Cfr-mediated A2503 methylation, in the case of the oxazolidinones, overlaps with the site normally occupied by the C-5 sidechain. As such, linezolid MIC have been observed to increase two- to fourfold in organisms acquiring the cfr gene [29]. Conversely, tedizolid MIC are typically stable in the presence of Cfr methylation [34–36]. This is believed to be due to the fact that the hydroxymethyl C-5 sidechain is smaller and more flexible than acetamidomethyl sidechain of linezolid [36]. The preserved potency of tedizolid among cfr isolates suggests it may remain useful against some linezolid-resistant organisms. However, clinical experience has yet to verify this in vitro advantage as clinically significant.
Pharmacokinetics
The pharmacokinetic profile of tedizolid phosphate has been well evaluated in Phase I and Phase II studies. When 200 mg of tedizolid phosphate is administered intravenously, conversion to the active form by serum phosphatase begins immediately. The maximum serum concentration (C max) of tedizolid, 2.6 mg/L, is observed at approximately 1 h following initiation of infusion. Clearance in healthy adults is about 5 L/h, with an elimination half-life of 12 h. With daily administration, the total area under the curve at 24 h (AUC0–24) is roughly 30 mg h/L. Seventy-five to 80% of tedizolid is bound to serum proteins [37, 38].
When administered orally, tedizolid phosphate has an absolute bioavailability of 91%. C max and AUC0–24 are only slightly decreased compared to intravenous administration (2 mg/L and 26 mg h/L, respectively, under fasting conditions). The time to C max is 2 h when administered under fasting conditions, but increases to 8 h in patients having recently eaten a full meal. The fed state also appears to blunt C max values, but AUC0–24 remains unchanged [37, 39].
Tedizolid pharmacokinetic parameters have also been studied in patients with various degrees of renal or hepatic dysfunction. When intravenously administered to patients with severe renal impairment, 200 mg tedizolid phosphate produced C max and AUC0–24 minimally changed from the matched control group. C max and AUC0–24 were however reduced by approximately 15% and 25%, respectively, in patients undergoing hemodialysis. Four-hour hemodialysis sessions using a high-flux filter were observed to remove less than 10% of tedizolid doses administered intravenously immediately prior. Among patients with moderate and severe hepatic dysfunction, 200 mg of orally administered tedizolid produced similar C max values, but 22% and 34% higher AUC0–24, respectively [38]. Currently, no dosage adjustment is recommended for patients with either renal or hepatic dysfunction.
Much like linezolid, the volume of distribution for tedizolid is relatively large. Central compartment volume of distribution is approximately 70 L, with the volume of distribution for the periphery ranging from 13 to 25 L [37, 40, 41]. C max in soft tissues including muscle and adipose approximate free drug concentrations seen in plasma, with modestly elevated free AUC (fAUC) compared to plasma [42]. Studies examining the pulmonary distribution of tedizolid in both various murine and healthy volunteer populations have reported significant accumulation of tedizolid. While AUC0–24 for linezolid in epithelial lining fluid (ELF) have been reported to be around fivefold those seen in plasma, tedizolid was found to have an ELF AUC0–24 40-fold higher (approximately 100 mg h/L for a 200 mg dose) than plasma. Alveolar macrophages also appear to accumulate tedizolid tenfold higher than linezolid relative to respective plasma concentrations [43]. Given the concentration-dependent activity of oxazolidinones, this increased pulmonary exposure of tedizolid may be therapeutically advantageous for the treatment of pulmonary infections.
Pharmacodynamics
Oxazolidinones, including both linezolid and tedizolid, appear to have pharmacodynamic profiles best described by the fAUC0–24 to MIC ratio (fAUC0–24:MIC). Initial modeling with linezolid suggested a pharmacodynamic target of 58 mg h/L, which was subsequently echoed by pharmacodynamic evaluation of linezolid clinical outcomes in the treatment of both skin and soft tissue infections and pneumonia [44, 45]. While early work with tedizolid suggested pharmacodynamic targets of approximately 47 and 20 mg h/L for bacterial stasis in classic neutropenic models of murine thigh infections and pneumonia, respectively, the dose of tedizolid phosphate selected for late-stage clinical trials was significantly lower than that required to meet these targets [46, 47]. This was in large part due to the clinical response observed with 200 mg daily dosing in Phase II trials, and later supported by pharmacodynamic evaluations investigating the impact of granulocytes on the activity of tedizolid. In an assessment of the pharmacodynamics of tedizolid in a non-neutropenic murine thigh infection model, a 16-fold lower dose to achieve bacterial stasis at 24 h was required compared to neutropenic mice. This dose would be estimated to achieve an fAUC0–24:MIC of approximately 3 mg h/L [48]. It was also observed that in the presence of granulocytes, tedizolid reduced bacterial burden of MRSA by 3.5 log10CFU and 4.7 log10CFU at 48 and 72 h, respectively, when accounting for the direct activity of granulocytes without tedizolid therapy [49]. This seemingly bactericidal activity contrasts the normal expectation of bacteriostasis with oxazolidinone treatment. These data, along with the non-inferiority of tedizolid compared to linezolid in Phase II and III clinical trials, support the use of the 200 mg daily dosing of tedizolid for acute bacterial skin and skin structure infections (ABSSSI).
Early Clinical Experience
A single Phase II randomized, double-blind, dose-ranging clinical trial was completed to evaluate the efficacy, safety, and pharmacokinetic profile of tedizolid for cSSSI [41]. Patients enrolled were aged 18–75 years with deep extensive cellulitis, surgical or post-traumatic wounds, or an abscess, caused by Gram-positive pathogens with systemic signs/symptoms of infection. Patients were randomized 1:1:1 to receive 200, 300, or 400 mg of oral tedizolid phosphate dosed once daily for 5–7 days as determined by the investigator based upon clinical response. The primary outcome evaluated was clinical response in the clinically evaluable (CE) and clinically modified intent-to-treat (cMITT) populations at the 7–14 days post-treatment test-of-cure (TOC) visit. Clinical cure was defined as resolution or improvement of cSSSI symptoms such that no further treatment was required.
Baseline characteristics were similar between groups, with the majority of patients being Caucasian (76%), male (65%), and with a mean age of 36 years [41]. The most common infection type was abscess (77%) with the majority of lesions between 10 and 20 cm (45%) in size. S. aureus was the most common isolated pathogen (90%) with 81% of these isolates being MRSA. The mean duration of therapy was 6.4 days, and 95% of patients completed treatment.
Primary outcomes were similar between groups for each population, with clinical cure rates of 88.9%, 88.9%, and 85.5% in cMITT, 98.2%, 94.4%, and 94.4% in CE, and 100%, 93.2%, and 95.7% in the microbiologically evaluable populations for the 200, 300, and 400 mg groups, respectively [41]. Similar cure rates were also seen among the infection type subgroups and by baseline pathogen. Microbiologic eradication rates were also similar, ranging from 92.6% to 100% for MRSA in all three dosing groups, and 100% eradication in the severe infection group specifically. Adverse events were reported similarly between groups with any adverse event reported in 66.7% of those taking 200 mg, 69.8% on 300 mg, and 71.0% on 400 mg doses. The most common adverse events were nausea, secondary abscess, headache, and vomiting, and no patients discontinued therapy due to adverse events. The pharmacokinetic data collected resulted in a model displaying linear kinetics with an estimated total clearance of 8.28 L/h, distribution clearance of 2.95 L/h, a central compartment volume of 71.4 L, and a peripheral compartment volume of 27.9 L.
Tedizolid phosphate studied at 200, 300, and 400 mg once daily was found to be effective, safe, and well tolerated when used for 5–7 days for cSSSI [41]. Clinical cure rates were similar between treatment groups, and the 200 mg daily dose was considered to be the lowest efficacious dose, supporting the use of this dose for future trials involving skin and skin structure infections.
Two Phase III clinical trials, ESTABLISH-1 (ClinicalTrials.gov #NCT01170221) and ESTABLISH-2 (ClinicalTrials.gov #NCT01421511), were conducted to compare a 5-day course of tedizolid phosphate to a 10-day course of linezolid in patients with ABSSSI [50–52]. The ESTABLISH studies were randomized, double-blind, double-dummy, multicenter, multi-national, and non-inferiority trials with patients stratified by clinical syndrome, geographic region, and fever (ESTABLISH-1 only). Patients were randomized 1:1 to either receive tedizolid phosphate 200 mg daily or linezolid 600 mg twice daily. ESTABLISH-1 evaluated oral therapy only for this indication, while patents in ESTABLISH-2 received two or more doses of intravenous therapy prior to having the option of being switched to oral therapy. Patients included were aged 18 years or older in ESTABLISH-1 and 12 years or older in ESTABLISH-2, who had an ABSSSI (cellulitis/erysipelas, major cutaneous abscess, or wound infection) caused by a Gram-positive pathogen, and whose lesion area was at least 75 cm2. Early clinical response in the intention-to-treat (ITT) population at 48–72 h was selected as the primary end point, but was defined slightly differently in the two trials due to recommendations presented in the 2013 FDA guidance for ABSSSI. For the pooled analysis, the primary efficacy outcomes measured were a reduction of ≥20% in lesion area, receipt of concomitant systemic antibiotics with Gram-positive activity, and all-cause mortality within 72 h [52]. Secondary end points included clinical response at day seven, end of therapy (EOT) visit between days 11–13, and post-therapy evaluation (PTE) visit at 7–14 days after the EOT (days 18–25), as well as safety outcomes assessed throughout.
Baseline characteristics were similar between studies and treatment groups with the majority of patients being men (63%) with a median age of 44 years [50–52]. Cellulitis/erysipelas was the most common type of ABSSSI (46%) and was most frequently located on either lower (41%) or upper extremities (34%). The pathogen isolated most frequently was S. aureus (82%), with 43% of those being MRSA and 57% being MSSA. A numerically higher percentage of patients enrolled in ESTABLISH-1 were from North America (81%) compared to those enrolled in ESTABLISH-2 (47%), leading to higher percentages of patients with MRSA isolated in ESTABLISH-1 than ESTABLISH-2 (43% compared to 28%). Tedizolid met the criteria for non-inferiority in both trials, with similar early clinical response in the pooled ITT population of 81.6% for tedizolid and 79.4% for linezolid (95% CI −2.0 to 6.5). This similarity between groups at early clinical response was also seen between all pre-specified subgroups, including key causative pathogen, with a response of 85.1% and 80.7% observed against S. aureus for tedizolid and linezolid, respectively. Additionally, secondary end points of clinical response at EOT were also similar between groups with 87.0% for tedizolid and 87.9% for linezolid (95% CI 14.4–2.7), as well as investigator-assessed clinical response at PTE, with a response of 86.7% for tedizolid and 86.8% for linezolid (95% CI −3.8 to 3.6).
Treatment-emergent adverse events (TEAE) were reported in 43% of patients overall, with the most common events being nausea, headache, and abscess [50–52]. Gastrointestinal adverse events were the most frequently reported and were more likely to occur in the linezolid group (23% compared to 16%; P = 0.0015). A difference in the occurrence of hematologic laboratory abnormalities between groups was also reported. The most prominent difference was seen in the incidence of thrombocytopenia (defined as platelets <150,000 cells/mm3), with 3.7% versus 5.6% (P = 0.585) at 7–9 days, and 4.9% versus 10.8% (P = 0.0003) at 11–13 days for tedizolid and linezolid, respectively. Other hematologic laboratory abnormalities notable were absolute neutrophil counts less than the lower limit of normal reported in 1.9% of those taking tedizolid and 3.3% of those taking linezolid at EOT, and hemoglobin levels less than the lower limit of normal in 28.9% and 31.1% of patients at EOT for tedizolid and linezolid, respectively. In general, TEAEs were mild with only 0.5% of patients receiving tedizolid and 0.9% of those receiving linezolid discontinuing therapy due to adverse events.
Based on these data, once-daily tedizolid for 6 days was observed to be non-inferior to twice-daily linezolid for 10 days for the treatment of ABSSSI. Although both agents were well tolerated, significantly fewer gastrointestinal adverse events and less thrombocytopenia occurred in the tedizolid group.
Tedizolid phosphate is also currently under investigation for use in the treatment of nosocomial pneumonia due to Gram-positive microorganisms. Over 230 institutions across six continents will be enrolling patients through the end of 2016. Seven days of tedizolid phosphate 200 mg daily will be compared to 10 days of linezolid 600 mg twice daily in this randomized, double-blind, Phase III trial. Therapy with either agent will be extended to 14 days if concomitant bacteremia is present. To date, no results from this study have been reported.
Safety
The current safety profile of tedizolid phosphate is limited to experience in approximately 1000 patients [41, 50–53]. Hematologic laboratory abnormalities have repeatedly been reported with the oxazolidinone class. Thrombocytopenia is the most notable hematologic effect associated with linezolid and can be treatment limiting. A Phase I trial examining the effect of 200, 300, or 400 mg of tedizolid phosphate daily compared to 600 mg of linezolid twice daily for 21 days in healthy volunteers found less thrombocytopenia in the 200 mg tedizolid group (mean reduction 15% versus 22% for tedizolid and linezolid, respectively; data presented in poster form by Prokocimer and colleagues at the 48th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy) Higher doses of tedizolid, however, were not found to be different when compared to linezolid. In the Phase III ESTABLISH studies, more patients receiving linezolid (12.6%) exhibited platelet counts <150,000 cells/mm3 than in the tedizolid group (6.4%) through last dose of study drug (P = 0.0016) [50–52]. Other hematologic laboratory abnormalities were similar between groups at the EOT.
Linezolid has also been reported to be associated with peripheral and optic neuropathies. This effect, typically reported following longer courses of therapy, is hypothesized to be associated with inhibition of mitochondrial protein synthesis (MPS) [54] While there currently are no published cases of tedizolid therapy extending beyond 21 days, Phase III trials have observed similar rates of neuropathies for tedizolid and linezolid (peripheral: 1.2% versus 0.6%, optic: 0.3% versus 0.2%, respectively) given in shorter courses. A recent study extensively examined in vitro and in vivo effects of tedizolid relevant to neuropathy [55]. In this study, tedizolid was administered to rats for 9 months and dosed to achieve AUC around eightfold higher than that observed in human subjects receiving the approved 200 mg daily dosing. No neuropathy was detected in any of the rats at the conclusion of therapy. In vitro data, however, showed a significantly lower MPS 50% inhibitory concentration (IC50) for tedizolid compared to that of linezolid (0.3 versus 6 µM respectively), suggesting increased risk of neurotoxicity. The investigators utilized human pharmacokinetic data and Monte Carlo simulations to further evaluate this discrepancy in findings. Interestingly, significant periods of systemic-free concentrations of tedizolid, but not linezolid, below the respective IC50 were observed. While free linezolid concentrations drop below the measured MPS IC50 in only 38% of patients for less than 1 h, free tedizolid concentrations fall below this level for just under 8 h in more than 80% of patients. This sub-IC50 period is hypothesized to allow for mitochondrial recovery and may account for the lack of neuropathies observed in vivo. As with linezolid, the potential of neuropathies associated with tedizolid therapy will largely be defined by Phase IV experience.
An additional safety concern among the oxazolidinone class is the ability of these agents to inhibit MAO, resulting in interactions with other medications and certain foods, and potentially leading to the development of serotonin syndrome. The FDA issued a warning on the label of linezolid to caution clinicians of this possible effect if given concurrently with other agents that increase serotonin including selective serotonin reuptake inhibitors and serotonin–norepinephrine reuptake inhibitors. To evaluate the effects of tedizolid on MAO, IC50 for MAO-A and MAO-B enzymes were determined in vitro [13]. Tedizolid was found to be a weak, reversible inhibitor of MAO-A and MAO-B activity with only minimal effects observed with the prodrug, tedizolid phosphate. Additionally, tedizolid was studied in 30 human subjects concomitantly given tyramine to test the dose needed to increase blood pressure by 30 mmHg compared to placebo. The median dose needed to elicit this effect was 325 mg of tedizolid phosphate compared to 425 mg of placebo, resulting in a clinically irrelevant sensitivity ratio of 1.33 (≥2 considered clinically relevant) [13]. Finally, tedizolid was compared to linezolid in a murine serotonergic model to evaluate the effect on mouse head twitching seen when these oxazolidinones were given with other medications known to increase serotonin. This model showed that tedizolid did not increase head twitch response even when dosed at 25 times the normal human dose. Based on these studies, tedizolid is thought to have a low probability of causing MAO-related adverse events when given in combination with serotonin-increasing medications. However, clinical experience with this potential adverse effect of tedizolid is currently lacking as patients on medications such as these were excluded from the Phase III trials.
Conclusion
Tedizolid phosphate is the first of the second-generation oxazolidinones to receive FDA approval and is currently indicated for the treatment of ABSSSI and under investigation for the treatment of hospital acquired pneumonia. While greatly similar to linezolid, tedizolid phosphate possesses more favorable pharmacokinetics, enhanced antimicrobial potency, and lower incidence of adverse effects including thrombocytopenia. Clinical experience with tedizolid, however, is largely limited to Phase II and III clinical trials, and careful consideration of therapeutic alternatives should be recommended. It remains to be seen if the in vitro activity of tedizolid against some linezolid-resistant isolates will prove clinically useful.
References
Sievert DMP, Ricks PP, Edwards JRMS, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol. 2013;34(1):1–14.
Kaku N, Yanagihara K, Morinaga Y, et al. Influence of antimicrobial regimen on decreased in-hospital mortality of patients with MRSA bacteremia. J Infect Chemother. 2014;20(6):350–5.
Haas EJ, Zaoutis TE, Prasad P, Li M, Coffin SE. Risk factors and outcomes for vancomycin-resistant Enterococcus bloodstream infection in children. Infect Control Hosp Epidemiol. 2010. (Epub ahead of print).
Chou CH, Lee NY, Lee HC, Chang CM, Lee CC, Ko WC. Emergence of vancomycin-resistant Enterococcus bloodstream infections in southern Taiwan. J Microbiol Immunol Infect. 2012;45(3):221–7.
Wudhikarn K, Gingrich RD, de Magalhaes Silverman M. Daptomycin nonsusceptible enterococci in hematologic malignancy and hematopoietic stem cell transplant patients: an emerging threat. Ann Hematol. 2013;92(1):129–31.
Gasch O, Camoez M, Dominguez MA, et al. Emergence of resistance to daptomycin in a cohort of patients with methicillin-resistant Staphylococcus aureus persistent bacteraemia treated with daptomycin. J Antimicrob Chemother. 2014;69(2):568–71.
Miyazaki M, Nagata N, Miyazaki H, et al. Linezolid minimum inhibitory concentration (MIC) creep in methicillin-resistant Staphylococcus aureus (MRSA) clinical isolates at a single Japanese center. Biol Pharm Bull. 2014;37(4):679–82.
Sanchez Garcia M, De la Torre MA, Morales G, et al. Clinical outbreak of linezolid-resistant Staphylococcus aureus in an intensive care unit. JAMA. 2010;303(22):2260–4.
Shaw KJ, Barbachyn MR. The oxazolidinones: past, present, and future. Ann N Y Acad Sci. 2011;1241:48–70.
Walker S, Dresser L, Becker D, Scalera A. An assessment of linezolid utilization in selected Canadian provinces. Can J Infect Dis Med Microbiol. 2006;17(3):177–82.
Brickner SJ, Hutchinson DK, Barbachyn MR, et al. Synthesis and antibacterial activity of U-100592 and U-100766, two oxazolidinone antibacterial agents for the potential treatment of multidrug-resistant gram-positive bacterial infections. J Med Chem. 1996;39(3):673–9.
Im WB, Choi SH, Park JY, Choi SH, Finn J, Yoon SH. Discovery of torezolid as a novel 5-hydroxymethyl-oxazolidinone antibacterial agent. Eur J Med Chem. 2011;46(4):1027–39.
Flanagan S, Bartizal K, Minassian SL, Fang E, Prokocimer P. In vitro, in vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob Agents Chemother. 2013;57(7):3060–6.
Bae SK, Yang SH, Shin KN, Rhee JK, Yoo M, Lee MG. Pharmacokinetics of DA-7218, a new oxazolidinone, and its active metabolite, DA-7157, after intravenous and oral administration of DA-7218 and DA-7157 to rats. J Pharm Pharmacol. 2007;59(7):955–63.
Wilson DN, Schluenzen F, Harms JM, Starosta AL, Connell SR, Fucini P. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc Natl Acad Sci. 2008;105(36):13339–44.
Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 2014;12(1):35–48.
Coyle EA, Cha R, Rybak MJ. Influences of linezolid, penicillin, and clindamycin, alone and in combination, on streptococcal pyrogenic exotoxin a release. Antimicrob Agents Chemother. 2003;47(5):1752–5.
Diep BA, Equils O, Huang DB, Gladue R. Linezolid effects on bacterial toxin production and host immune response: review of the evidence. Curr Ther Res Clin Exp. 2012;73(3):86–102.
Mendes RE, Hogan PA, Streit JM, Jones RN, Flamm RK. Zyvox(R) Annual Appraisal of Potency and Spectrum (ZAAPS) program: report of linezolid activity over 9 years (2004–2012). J Antimicrob Chemother. 2014;69(6):1582–8.
Stevens DL, Smith LG, Bruss JB, et al. Randomized comparison of linezolid (PNU-100766) versus oxacillin-dicloxacillin for treatment of complicated skin and soft tissue infections. Antimicrob Agents Chemother. 2000;44(12):3408–13.
Toh SM, Xiong L, Arias CA, et al. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol Microbiol. 2007;64(6):1506–14.
Schulte B, Heininger A, Autenrieth IB, Wolz C. Emergence of increasing linezolid-resistance in enterococci in a post-outbreak situation with vancomycin-resistant Enterococcus faecium. Epidemiol Infect. 2008;136(8):1131–3.
Besier S, Ludwig A, Zander J, Brade V, Wichelhaus TA. Linezolid resistance in Staphylococcus aureus: gene dosage effect, stability, fitness costs, and cross-resistances. Antimicrob Agents Chemother. 2008;52(4):1570–2.
Tsakris A, Pillai SK, Gold HS, et al. Persistence of rRNA operon mutated copies and rapid re-emergence of linezolid resistance in Staphylococcus aureus. J Antimicrob Chemother. 2007;60(3):649–51.
Tazi A, Chapron J, Touak G, et al. Rapid emergence of resistance to linezolid and mutator phenotypes in Staphylococcus aureus isolates from an adult cystic fibrosis patient. Antimicrob Agents Chemother. 2013;57(10):5186–8.
Dong W, Chochua S, McGee L, Jackson D, Klugman KP, Vidal JE. Mutations within the rplD gene of linezolid-nonsusceptible Streptococcus pneumoniae strains isolated in the United States. Antimicrob Agents Chemother. 2014;58(4):2459–62.
Wolter N, Smith AM, Farrell DJ, et al. Novel mechanism of resistance to oxazolidinones, macrolides, and chloramphenicol in ribosomal protein L4 of the pneumococcus. Antimicrob Agents Chemother. 2005;49(8):3554–7.
Billal DS, Feng J, Leprohon P, Legare D, Ouellette M. Whole genome analysis of linezolid resistance in Streptococcus pneumoniae reveals resistance and compensatory mutations. BMC Genom. 2011;12:512.
Locke JB, Morales G, Hilgers M, et al. Elevated linezolid resistance in clinical cfr-positive Staphylococcus aureus isolates is associated with co-occurring mutations in ribosomal protein L3. Antimicrob Agents Chemother. 2010;54(12):5352–5.
Kehrenberg C, Aarestrup FM, Schwarz S. IS21-558 insertion sequences are involved in the mobility of the multiresistance gene cfr. Antimicrob Agents Chemother. 2007;51(2):483–7.
Shen J, Wang Y, Schwarz S. Presence and dissemination of the multiresistance gene cfr in Gram-positive and Gram-negative bacteria. J Antimicrob Chemother. 2013;68(8):1697–706.
Smith LK, Mankin AS. Transcriptional and translational control of the mlr operon, which confers resistance to seven classes of protein synthesis inhibitors. Antimicrob Agents Chemother. 2008;52(5):1703–12.
Locke JB, Rahawi S, Lamarre J, Mankin AS, Shaw KJ. Genetic environment and stability of cfr in methicillin-resistant Staphylococcus aureus CM05. Antimicrob Agents Chemother. 2012;56(1):332–40.
Sahm DF, Deane J, Bien PA, et al. Results of the Surveillance of Tedizolid Activity and Resistance Program: in vitro susceptibility of Gram-positive pathogens collected in 2011 and 2012 from the United States and Europe. Diagn Microbiol Infect Dis. 2015;81(2):112–8.
Locke JB, Zurenko GE, Shaw KJ, Bartizal K. Tedizolid for the management of human infections: in vitro characteristics. Clin Infect Dis. 2014;58(Suppl 1):S35–42.
Shaw KJ, Poppe S, Schaadt R, et al. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob Agents Chemother. 2008;52(12):4442–7.
Flanagan S, Fang E, Munoz KA, Minassian SL, Prokocimer PG. Single-and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacotherapy. 2014;34(9):891–900.
Flanagan S, Minassian SL, Morris D, et al. Pharmacokinetics of tedizolid in subjects with renal or hepatic impairment. Antimicrob Agents Chemother. 2014;58(11):6471–6.
Flanagan SD, Bien PA, Munoz KA, Minassian SL, Prokocimer PG. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy. 2014;34(3):240–50.
Flanagan S, Passarell J, Lu Q, Fiedler-Kelly J, Ludwig E, Prokocimer P. Tedizolid population pharmacokinetics, exposure response, and target attainment. Antimicrob Agents Chemother. 2014;58(11):6462–70.
Prokocimer P, Bien P, Surber J, et al. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2011;55(2):583–92.
Sahre M, Sabarinath S, Grant M, et al. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int J Antimicrob Agents. 2012;40(1):51–4.
Housman ST, Pope JS, Russomanno J, et al. Pulmonary disposition of tedizolid following administration of once-daily oral 200-milligram tedizolid phosphate in healthy adult volunteers. Antimicrob Agents Chemother. 2012;56(5):2627–34.
Andes D, van Ogtrop ML, Peng J, Craig WA. In vivo pharmacodynamics of a new oxazolidinone (linezolid). Antimicrob Agents Chemother. 2002;46(11):3484–9.
Rayner CR, Forrest A, Meagher AK, Birmingham MC, Schentag JJ. Clinical pharmacodynamics of linezolid in seriously ill patients treated in a compassionate use programme. Clin Pharmacokinet. 2003;42(15):1411–23.
Louie A, Liu W, Kulawy R, Drusano GL. In vivo pharmacodynamics of torezolid phosphate (TR-701), a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(7):3453–60.
Lepak AJ, Marchillo K, Pichereau S, Craig WA, Andes DR. Comparative pharmacodynamics of the new oxazolidinone tedizolid phosphate and linezolid in a neutropenic murine Staphylococcus aureus pneumonia model. Antimicrob Agents Chemother. 2012;56(11):5916–22.
Lodise TP, Drusano GL. Use of pharmacokinetic/pharmacodynamic systems analyses to inform dose selection of tedizolid phosphate. Clin Infect Dis. 2014;58(Suppl 1):S28–34.
Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300–5.
Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8):696–705.
Prokocimer P, De Anda C, Fang E, Mehra P, Das A. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA. 2013;309(6):559–69.
Shorr AF, Lodise TP, Corey GR, et al. Analysis of the phase 3 ESTABLISH trials: tedizolid versus linezolid in acute bacterial skin and skin structure infection. Antimicrob Agents Chemother. 2015;59(2):864–71.
Prokocimer P, Bien P, Munoz KA, Aster R. Hematological effects of TR-701, linezolid and placebo administered for 21 days in healthy subjects. In: Proceedings of the 48th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); Washington, DC. 2008.
Javaheri M, Khurana RN, O’Hearn TM, Lai MM, Sadun AA. Linezolid-induced optic neuropathy: a mitochondrial disorder? Br J Ophthalmol. 2007;91(1):111–5.
Flanagan S, McKee EE, Das D, et al. Nonclinical and pharmacokinetic assessments to evaluate the potential of tedizolid and linezolid to affect mitochondrial function. Antimicrob Agents Chemother. 2015;59(1):178–85.
Brown SD, Traczewski MM. Comparative in vitro antimicrobial activities of torezolid (TR-700), the active moiety of a new oxazolidinone, torezolid phosphate (TR-701), determination of tentative disk diffusion interpretive criteria, and quality control ranges. Antimicrob Agents Chemother. 2010;54(5):2063–9.
Locke JB, Finn J, Hilgers M, et al. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations. Antimicrob Agents Chemother. 2010;54(12):5337–43.
Acknowledgments
We thank Kayleigh Marx and Michael J. Rybak for reviewing the manuscript and providing feedback. No sponsorship or funding was received for this publication. None of the affiliate authors received support, financial or otherwise, for this manuscript. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Conflict of interest
Jeffrey M. Rybak and Karrine Roberts have no conflict of interest to declare.
Compliance with ethics guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rybak, J.M., Roberts, K. Tedizolid Phosphate: a Next-Generation Oxazolidinone. Infect Dis Ther 4, 1–14 (2015). https://doi.org/10.1007/s40121-015-0060-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-015-0060-3