
Abstract
The pharmacokinetics of roxadustat are well characterized, with an apparent volume of distribution after oral administration of 22–57 L, apparent clearance of 1.2–2.65 L/h, and renal clearance of 0.030–0.026 L/h in healthy volunteers; the elimination half-life is 9.6–16 h. Plasma binding is 99% and the fraction eliminated by hemodialysis is 2.34%. As an interpretation of the pharmacodynamics of roxadustat, we proposed a concept with a hypothetical cascade of two subsequent effects, first on erythropoetin (EPO) and second on hemoglobin (delta Hb). The primary effect on EPO is observed within a few hours after roxadustat administration and can be modeled using the sigmoidal Hill equation. The concentration at half-maximum effect can be inferred at 10–36 µg/mL, the Hill coefficient at 3.3, and the effect bisection time at 10–17 h, corresponding to EPO half-life. The subsequent effect on hemoglobin (delta Hb) is observed after several weeks and can be interpreted as an irreversible, dose proportional, unsaturable effect, continuing in agreement with the lifespan of red blood cells of 63–112 days.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Roxadustat pharmacokinetics are largely independent from kidney function and hemodialysis, and can be described by a cascade of two subsequent effects, first on erythropoietin (EPO) and second on hemoglobin. |
The effects of roxadustat on EPO can be described using the sigmoidal maximum effect model. |
The effects of roxadustat on hemoglobin are linear, long-lasting, and likely irreversible. |
1 Introduction
In 2019, William Kaelin Jr, Sir Peter Ratcliffe, and Gregg Semenza were awarded the Nobel prize for their work on the hypoxia-inducible factor (HIF) proteins [69]. Under physiological conditions, the HIF proteins are inactivated by enzymes of the prolyl hydroxylase domain (PHD), by targeting HIF proteins for degradation. For the condition of hypoxia, the PHD enzymes are downregulated and the HIF proteins stay high. HIF proteins stimulate the erythropoietin (EPO) gene in kidney parenchymal cells to produce EPO, as well as other genes, including the iron transporter ferroportin. EPO activates red blood cell (RBC) production in bone marrow, and ferroportin facilitates iron absorption in the small intestine. Thus, when oxygen tension decreases, new reticulocytes are released and hemoglobin concentrations rise.
Roxadustat (FG-4592, ASP1517, Anatomical Therapeutic Classification (ATC) code B03XA05) is an orally administered small molecule drug that prevents the enzymatic degradation of HIF-1α by inhibiting the PHD enzymes. In consequence, the hematocrit rises and hemoglobin concentrations increase. The causal chain of events induced by roxadustat can be described by the following sequence: roxadustat inhibits PHD enzymes so that HIF is ‘stabilized’, and EPO is released that stimulates RBC production in parallel with hemoglobin synthesis. What can be measured includes roxadustat, EPO, and RBCs; however, the purpose, and, of foremost interest, to be determined, is the change in hemoglobin (∆Hb).
2 Search Strategy
We conducted a search of the PubMed electronic database using the terms ‘roxadustat’ and ‘pharmacokinetics’ or ‘pharmacodynamics’. For our systematic search and narrative review of the pharmacokinetic and pharmacodynamic literature, an approach similar to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines was used [51]. Publications without an abstract were excluded, and the reference sections in the identified publications were screened for additional articles. At the end of May 2021, 170 results had been obtained (electronic supplementary material (ESM) Fig. A1). The abstracts were viewed for pertinent data, yielding 13 papers specifically relating to pharmacokinetics and 82 more general papers relating to pharmacokinetics and pharmacodynamics. Finally, pharmacokinetic parameters were extracted from 29 papers and pharmacodynamic parameters were extracted from 18 papers.
Values and measures of variability were compiled in tabulated form (Tables 1 and 2; ESM Table A1). The respective estimates of pharmacokinetic and pharmacodynamic parameter values were the mean and standard deviation, or medians with the minimum–maximum range of values as reported in the literature.
When the required parameters were not explicitly stated in the publications but related parameters were documented or figures given, the values for the respective parameter of interest were either predicted using standard equations (see the ESM) or estimated by graphical analysis similar to commercial vectorial software tools [32].
To synthesize the primary data, the range of published mean or median values was used (minimum–maximum) to characterize the drug roxadustat. No primary data analysis was performed. The key pharmacokinetic and pharmacodynamic equations used to predict parameter values are reproduced in the text or outlined in the ESM. Preclinical data were only considered where relevant data in humans have not been published in the scientific literature.
3 Structure and Mechanism of Action
Roxadustat is a small molecule (molecular weight 352.34 g/mol) inhibiting the enzyme prolyl hydroxylase. Its chemical formula is C19H16N2O5 (Fig. 1). Roxadustat stabilizes the hypoxia-inducible intracellular transcription factor (HIF). The HIF proteins mainly activate, among others, the EPO gene. This gene encodes EPO, with its release leading to red cell production in bone marrow. The rise in EPO can be measured in blood. In addition, elevated hepcidin levels are downregulated by roxadustat, leading to improved iron absorption and utilization from iron stores [11]. The effects of roxadustat on hepcidin and iron utilization are independent from its effect on EPO [50].

Chemical structure of roxadustat
EPO is produced in the tubular part of the parenchyma of the kidneys by interstitial cells, and extrarenal production of EPO takes place in the liver [48]. This explains why an EPO response to roxadustat can still be found in anephric hemodialysis patients with no renal parenchyma [6].
4 Dosing and Formulation
Roxadustat is available on the Japanese market in tablet form at 20, 50, and 100 mg doses, and on the European market at 20, 50, 70, 100, and 150 mg doses (Japanese Pharmaceuticals and Medical Devices Agency [34], European Medicines Agency 2021). In clinical trials, the reported dose range is 1.0–4.0 mg/kg. In recent clinical trials, the predominant dose was approximately 1.5 mg/kg, corresponding to 100 mg administered three times weekly. Initially, and in contrast to the once-daily dosing of vadadustat [13], randomization to even twice weekly dosing has been reported with roxadustat [8]. There must be a pharmacodynamic reason for the thrice- or twice-weekly dose interval because it contrasts to the experience that medication adherence would be expected best with once-daily dosing [20]. However, in patients on hemodialysis, administration three times weekly would be suitable on the day of hemodialysis.
Approved initial doses differ between countries (Japanese Pharmaceuticals and Medical Devices Agency [34], European Medicines Agency 2021). Factors to consider include body weight, pretreatment with erythropoesis-stimulating agents (ESAs), average ESA dose in the last 4 weeks, and liver function. Subsequently, the roxadustat dose is adjusted individually, based on actual hemoglobin and change in hemoglobin in the last 2–4 weeks, and considering approved maximum doses.
5 Pharmacokinetic Properties
The pharmacokinetics of roxadustat after oral administration are well characterized (Table 1) and appear to be linear (Yu [71], Groenendaal [26, 27], Groenendaal [28], Shibata [62], Shibata [63], Provenzano [55], Rekic [59], Groenendaal [29, 30], Takada [67]). Roxadustat is readily absorbed after oral administration, has a moderate apparent volume of distribution, and is eliminated largely by metabolism (cytochrome P450 (CYP) 2C8, UDP-glucuronosyltransferase (UGT) 1A9). In vitro data indicate that roxadustat is a substrate of organic anion transporter polypeptide (OATP) 1B1, organic anion transporter (OAT) 1, OAT3, and breast cancer resistance protein (BCRP), and may be an inhibitor of OATP1B1, OATP1B3, OAT3, and BCRP (Japanese Pharmaceuticals and Medical Devices Agency [34], European Medicines Agency 2021).
5.1 Absorption
After an oral dose of 100–200 mg, an average peak roxadustat concentration of 69 ng/mL/mg (range of reported average values, 61–85) in healthy White volunteers and 96 ng/mL/mg (89–106) can be measured at 1–2 h in healthy Japanese volunteers (Table 1). Pharmacokinetics in healthy volunteers were investigated under fasting conditions, unless stated otherwise (Table 1).
The value for absolute bioavailability (F) after oral dosing is unknown. In a mass balance study using 14C-roxadustat in six healthy volunteers, 45.8% of the administered radioactivity was excreted in urine (Japanese Pharmaceuticals and Medical Devices Agency [34], European Medicines Agency 2021), indicating at least moderately high absorption. Whether roxadustat is subject to enterohepatic recirculation is unknown but appears to be a possibility because a preclinical study in monkeys indicated biliary secretion (Japanese Pharmaceuticals and Medical Devices Agency [34], European Medicines Agency 2021) and the main metabolite, O-glucuronide-roxadustat, might be subject to deglucuronidation in the gut.
5.2 Distribution
The average apparent volume of distribution after oral administration (Vd/F) is 39 L (range of reported average values, 30–57) in healthy White volunteers and 23 L (22–26) in healthy Japanese volunteers. The unbound plasma fraction (fu) is low at 0.084 (0.081–0.087) as plasma binding (PB%) is high at approximately 99% (Table 1).
5.3 Metabolism and Elimination
Roxadustat is largely eliminated by metabolism and subsequent excretion of roxadustat and roxadustat metabolites in urine and feces. In the liver, roxadustat undergoes phase I oxidation by CYP enzyme 2C8, and phase II glucuronidation by UGT1A9 [26]. Elimination of unchanged roxadustat in urine was only 1.3% (1.0–1.6) of the orally administered dose (Table 1),correspondingly, renal clearance (CLren) was low at 0.028 L/h (0.03–0.026). In volunteers with normal renal function, 20.3% of an oral dose was recovered in urine as O-glucuronide-roxadustat, 7.21% was recovered as O-glucoside-roxadustat, and 2% was recovered as sulphate of hydroxy-roxadustat [29].
The average elimination half-life (t½) was 11.8 h (9.6–16) in healthy White volunteers and 12 h (10.9–13.1) in healthy Japanese volunteers (Table 1). In comparison, the exogenous and recombinant ESA epoetin-α exhibits a t½ of 6 h after intravenous administration and 24 h subcutaneously [35].
5.4 Population Pharmacokinetics
Population pharmacokinetic models of roxadustat have been developed using a two-compartment model with first-order absorption [59] or a two-compartment model with first-order absorption and lag-time [67]. In one study, no-effect boundaries of − 30% to + 43% were defined, based on pharmacologic reasoning (considering differences between starting doses from previous phase III trials), for covariates as recommended in the US FDA guidance. Using these boundaries, no relevant effects of bodyweight, age, race, sex, dialysis status, dose, albumin, or estimated glomerular filtration rate (eGFR) on PK parameters were found [59]. The estimated apparent clearance (CL/F) was slightly less and the Vz/F, calculated using two-compartment model parameter values, was larger than from non-compartmental pharmacokinetics in chronic kidney disease (CKD) patients (Table 1). In another study, an effect of age on CL/F was reported and the effect of phosphate binders on F was quantified [67].
5.5 Kinetics of Erythropoietin Response
With a roxadustat dose of 1.5 mg/kg, the EPO concentrations rise up to a peak of 150 IU/L [26]. This peak occurs 8–10 h (time to peak concentration (tpeak)) after roxadustat administration (Table 2) and 7 h after maximum concentration (Cmax) of roxadustat [16]. Subsequently, the EPO concentrations decrease again, with a t½ of 13 h (Table 2). The t½ corresponds to the elimination rate constant (ke) of 0.0533 per hour. Based on the Bateman function and tpeak, the EPO proliferation rate constant (kp) can be estimated as 0.20 per hour.
Accordingly, the proliferation t½ can be predicted as 3.46 h for EPO after roxadustat. This compares to a much faster roxadustat absorption t½ of 0.74 or 1.1 h based on the reported absorption rate constants of 0.94 or 0.63 per hour [59, 67]. The area under the EPO concentration-time curve adds up to 1390 h·IU/L [29]. Because the EPO kinetics are induced by roxadustat, the kinetic parameters can be interpreted as pharmacodynamic parameters of the roxadustat effect on EPO (Table 2).
5.6 Modulators
Data on the influence of age, sex, and body weight on roxadustat pharmacokinetics and pharmacodynamics from dedicated pharmacokinetic clinical trials have not yet been published, but population pharmacokinetic analyses did not indicate pronounced effects. Age ≥ 65 years was associated with a 21% reduction in CL/F [67], sex was not associated with drug exposure [59, 67], and body weight of 50 kg was associated with 13% higher and 100 kg with 14% lower drug exposure, compared with a body weight of 70 kg [59]. No data are available on the influence of pharmacogenetics such as single nucleotide polymorphisms (SNPs) on roxadustat pharmacokinetics or pharmacodynamics.
5.6.1 Impact of Food
After food, the area under curve decreases slightly (0.94-fold, 90% CI 90–99), but the difference is judged to be without clinical relevance [63]. Thus, food had no clinically relevant effect on the PK of the drug.
5.6.2 Impact of Renal Impairment
In stage 5 CKD, CL/F decreases slightly from 2.62 to 1.45 L/h, whereas the t½ was unchanged [29]. The Vd/F was lower and the fu higher at 1.1%, reflecting slightly less plasma binding (Table 1). The roxadustat area under the concentration-time curve from time zero to infinity (AUC∞) approximately doubled (1.95-fold, 90% confidence interval (CI) 1.65–2.29). The AUC of the metabolites with unknown activity, O-glucuronide-roxadustat, O-glucoside-roxadustat, and sulphate of hydroxy-roxadustat, increased 2.5-, 2.9-, and 2.3-fold, respectively, whereas their t½ was prolonged from 8.1 to 17.5 h, 8.5 to 14.6 h, and 15.9 to 18.2 h, respectively. These changes in exposure might be of limited clinical relevance because roxadustat is used in patients with renal anemia, who usually have severe renal impairment, and the dose is adjusted to therapeutic and adverse response, but not to renal function. However, if new indications for roxadustat in patients without significant renal impairment should arise, the effects of renal function could be relevant.
During hemodialysis, a very low roxadustat fraction (HD%) of 2.34 ± 1.26% was eliminated and there was no difference when roxadustat was administered 2 h before or 2 h after completion of hemodialysis. O-glucuronide-roxadustat AUC∞ was slightly lower when roxadustat was administered before hemodialysis (0.89-fold, 90% CI 81–99), which is considered clinically not relevant [29]. Similarly, another trial found no difference when roxadustat was administered 2 h before or 1 h after completion of hemodialysis [56]. One patient on continuous ambulatory peritoneal dialysis had similar parameters as patients with severe renal impairment [29]. In a population pharmacokinetic analysis, the type of dialysis, hemodialysis, or peritoneal dialysis had no effect on the pharmacokinetics [67].
The reason for higher roxadustat exposure in patients with renal impairment is unclear. It has been suspected that non-renal elimination of roxadustat could be altered (Groenendaal [29]. Another hypothesis might be that enterohepatic recycling with biliary secretion of O-glucuronide-roxadustat and consecutive intestinal deglucuronidation and reabsorption could play a role. Assuming that reduced renal excretion leads to increased O-glucuronide-roxadustat exposure, this in turn might lead to increased regeneration of roxadustat, for example by biliary secretion, deglucuronidation, and reabsorption.
5.6.3 Impact of Hepatic Impairment
In patients with liver cirrhosis (Child–Pugh B) CL/F was decreased to 1.57 L/h, t½ was slightly longer at 14.7 h, the AUC∞ was 1.23-fold greater (90% CI 0.86–1.75), and PB% slightly decreased from 99.2 to 98.9%, compared with healthy volunteers who were matched for sex, age, and body mass index. Unbound exposure (AUCu) was 1.7-fold greater (90% CI 1.19–2.43), which could be explained by the higher AUC and lower protein binding, but was considered to not be of clinical relevance. Similarly, slightly increased CLren could be explained by lower protein binding [26].
Although pharmacokinetic parameters and exposure increased, the peak effect on EPO (EPOpeak) was reduced in liver patients and only 73% of normal [26],however, the combined effects of advanced liver cirrhosis and severe renal impairment were unclear. The initial dose of roxadustat should be reduced to one half in patients with moderate liver cirrhosis (Child–Pugh B). Roxadustat is not recommended in patients with severe liver cirrhosis (Child–Pugh C) (European Medicines Agency 2021).
5.6.4 Impact of Ethnicity
Roxadustat was developed in Japan and was first approved in Japan and China for clinical use. Pharmacokinetically, the roxadustat AUC∞ was slightly less in Caucasians, but t½ was unchanged and Vd/F was smaller in Japanese patients (Table 1). Meanwhile, studies have been performed worldwide with no signs of a clinically relevant impact of ethnic differences on pharmacokinetics.
Pharmacodynamically, the peak EPO response (EPOpeak) and the area under the effect-time curve of effect E1 on EPO (AUEC1) were threefold higher in Japanese than Caucasian volunteers [71]. The roxadustat dose response appears to be stronger in Japanese compared with American patients. While 36 mg (= 0.6 mg/kg) three times weekly produced a delta hemoglobin of 1.3 g/dL in Japanese patients [3], a nearly threefold higher dose of 140 mg (= 1.68 mg/kg) administered three times weekly produced a rise in hemoglobin of only 1.0 g/dL in American patients [53].
5.6.5 Impact of Drug–Drug Interactions
We found only few data on drug interactions with roxadustat as either a victim or a perpetrator. Gemfibrozil, by CYP2C8 and OATP1B1 inhibition, and probenecid, by UGT and OAT inhibition, can likely reduce roxadustat metabolism and elimination. Scientific publications could not be found. On the other hand, roxadustat, by OATP1B1/B3 inhibition, interacts with statin pharmacokinetics, apparently increasing statin exposure to a clinically relevant extent (Japanese Pharmaceuticals and Medical Devices Agency [34], European Medicines Agency 2021). However, independent from and additive to statin therapy, roxadustat decreases low-density lipoprotein (LDL) cholesterol concentrations [11, 23, 53].
The phosphate binders sevelamer carbonate and calcium acetate reduced roxadustat AUC∞ to 59% and 69%, respectively. This effect was less when roxadustat and phosphate binder administration were separated by ≥ 1 h [30]. In this study, the phosphate binders were administered repeatedly over approximately 34 h. Of note, a faster concentration decline can be observed during phosphate binder administration, whereas after 36 h, roxadustat concentrations declined in parallel to the control group without phosphate binders. This finding could indicate enterohepatic recycling of roxadustat and an effect of phosphate binders on interrupting this process.
Omeprazole had no clinically relevant effect on roxadustat pharmacokinetics [28]. Furthermore, lanthan and spherical carbon adsorbent had no clinically relevant effect on roxadustat pharmacokinetics [62, 63]. Roxadustat did not influence warfarin [27].
6 Clinical Efficacy
Roxadustat can be taken orally in tablet form. Until now, most patients on hemodialysis receive exogenous ESAs administered intravenously, which may be comfortable when on hemodialysis. In stage 3–4 CKD, intravenous dosing of ESAs is uncommon and subcutaneous administration in non-dialysis-dependent CKD (NDD-CKD) patients and peritoneal dialysis patients is uncomfortable. Thus, the oral drug roxadustat has advantages, especially for patients not on hemodialysis and patients on peritoneal dialysis with a 100% anemia response rate [2].
Clinical trials on roxadustat in patients with NDD-CKD demonstrated the therapeutic efficacy on anemia (+ ∆Hb) to be superior to placebo [11]. In CKD patients depending on dialysis, roxadustat was non-inferior to epoetin-α [12]. In dialysis patients, the percentage of cases with hemoglobin response was 88% for roxadustat and 84% for epoetin-α, resulting in Hb >11 g/dL, ∆Hb >1 g/dL or >2 g/dL, respectively, depending on initial Hb [57]. Similar to roxadustat, the HIF stabilizer daprodustat corrects anemia to levels comparable with the ESA darbepoetin [4].
Importantly, the target hemoglobin concentration had already been achieved after 9 weeks with roxadustat but after only 19 weeks with epoetin-α in dialysis patients [12]. While the roxadustat dose needed to maintain the hemoglobin concentration at 11.5 g/dL could be decreased from 5.2 to 3.0 mg/kg per week, the epoetin-α dose had to be increased from 7000 IU to 10,000 IU per week, especially in patients with elevated C-reactive protein (CRP) [12].
Roxadustat treatment leads to physiological levels of EPO. This contrasts to the response to the exogenous ESAs with supraphysiological dosing and high EPO concentrations [40]. More effectively than exogenous ESAs, the HIF stabilizers also improve iron status and transferrin saturation by inhibition of hepcidin, thus leading to increased intestinal iron absorption and better utilization of iron stores in the body [43]. In contrast to short-term studies [7], long-term treatment with roxadustat also shows that an iron supplement should be administered [10]. Nonetheless, it can be seen as an advantage of the HIF stabilizers that oral iron treatment is sufficient and does not need intravenous administration.
Elevated LDL cholesterol decreased with roxadustat [14, 64], as did ferritin and uric acid levels [53]. With roxadustat, the response in hemoglobin levels was independent from elevated CRP, while high CRP levels were associated with resistance to epoetin-α [12]. ESA-resistant patients could be treated with the standard roxadustat 1.5 mg/kg dose without increasing dose size or frequency, as required with darbepoetin [4]. First results indicate that anemia due to myelodysplastic syndrome also responds to usual doses of roxadustat with prolonged transfusion independence [41].
Some additional effects of roxadustat might be of therapeutic interest. Ischemic preconditioning activates HIFs [68],therefore, advantages were also expected for roxadustat on tubular renoprotection in acute kidney injury (AKI), vascular endothelial growth factor, and markers of microinflammation. HIF targeted genes also antagonize apoptosis and oxidative stress, which might explain why roxadustat was found to be protective against doxorubicin-induced cardiotoxicity in a mouse model [45]. HIF activation may also prevent acute cisplatin nephrotoxicity and might thus protect cancer patients from progressing to CKD [52].
HIF stabilizers may protect from AKI and acute kidney disease, as suggested by data from mouse models [60]. However, a trial with roxadustat more frequently found a decline in GFR in 6.3% of patients with stage 4 CKD than in 3.7% of controls, but absolute numbers were very low and the difference was not statistically significant [1].
7 Ongoing Clinical Trials
In the ROAD study, major adverse cardiovascular events (MACE) will be investigated in patients receiving roxadustat [72]. Trials on long-term cancer risk and tumor growth are important but are still awaited.
8 Pharmacodynamics and Exposure–Response Associations
The effect of EPO on hemoglobin is apparently stronger and lasts longer after administration of HIF stabilizers, such as roxadustat, than the transient effect after exogenous ESAs [56]. The physiologically induced rise in EPO more efficiently stimulates the hematopoetic stem cells than exogenous ESAs. Much higher doses of exogenous ESAs are needed than with endogenous EPO, however, we found no explanation for this observation. Pharmacokinetic/pharmacodynamic models describing the effects of roxadustat on EPO and hemoglobin in humans have not been published to date.
In the following sections, we attempt to provide an interpretation of the published observations on roxadustat pharmacodynamics. We propose to characterize the action of roxadustat by an effect cascade of two different but subsequent events. The pharmacokinetics of roxadustat with one single concentration (C) at time (t) induces the pharmacodynamic sequence with two effects (E1 and E2). E1 on EPO can be distinguished from the subsequent E2 on hemoglobin (ΔHb) levels [8]. Similar to the two-step cascade from roxadustat to EPO and hemoglobin, a cascade from roxadustat to hepcidin and iron utilization could be modeled. The effect on hepcidin is measured to decrease to normal values within 7 days, inversely to the rise in hemoglobin [8, 50].
Roxadustat exerts at least two subsequent effects; first, the transient effect on EPO (E1 => EPO), and second, the persistent downstream effect on hemoglobin (E2 => ΔHb). In this concept, the target effect is on hemoglobin (E2 => ΔHb) and this effect could be interpreted as an irreversible effect, different from reversible roxadustat action on erythropoesis, because newly produced hemoglobin will remain as long as erythrocytes survive.
8.1 Reversible Effect (E1) on Erythropoetin (EPO)
A roxadustat dose of 1.0 mg/kg will produce a Cmax of 6 µg/mL after 2 h, and a peak EPO response is observed at a dose of 120 IU/L at 8 h after an oral dose [8]. This delay in peaks (Fig. 2) corresponds to a counter-clockwise hysteresis loop. In Japanese patients, EPO concentrations in the high physiological range of up to 300 IU/L are expected with a dose of 2.0 mg/kg [71]. Higher roxadustat doses of 3 mg/kg, or even 4 mg/kg, are rarely recommended since they could produce supraphysiological EPO levels of up to > 1000 IU/L (Table 2). These high EPO levels indicate that PHD inhibition, HIF stabilization, and EPO stimulation follow capacity limited dynamics only under physiological conditions.

Reversible effect–concentration correlation between roxadustat and EPO. When roxadustat concentrations rise, EPO levels start increasing, with a 5 h delay. When roxadustat concentrations decrease, the EPO levels decline faster here in animals than in humans [16]. EPO erythropoetin, tmax time to reach maximum concentration, Cmax maximum concentration, AUC area under the concentration-time curve, tpeak time to peak concentration, AUEC area under the effect-time curve
The supraphysiological peak concentration of the exogenous ESA epoetin-α is tenfold higher at > 2000 IU/L, corresponding to ‘overshooting’ recombinant erythropoietin concentrations [56]. A comparable hemoglobin response needs tenfold higher concentrations of exogenous ESAs compared with the more physiological concentrations of endogenous EPO after HIF stabilization.
The effect of roxadustat on prolyl hydroxylase enzymes, and thus on HIF-1α, appears to be receptor-mediated and capacity limited; therefore, the signal transduction on EPO follows reversible pharmacodynamics (Fig. 2).
The pharmacodynamics of the roxadustat E1 on EPO is likely capacity limited and saturable. It can be described by the classical capacity limited maximum effect (Emax) model with the sigmoidal Hill equation. This corresponds to the receptor-mediated reversible inhibition of prolyl hydroxylase enzymes [17]. Such a sigmoidal Emax model was also used by others to describe the effect of the new HIF stabilizer PHI-1 on erythropoesis in mice [66].
For the condition where the drug concentration equals the concentration producing the half-maximum to maximum effect (C = CE50), the Hill coefficient no longer matters since it holds (1.0H = 1.0). Thus, the concentration producing the half-maximum effect (CE50) can be determined from, or even found in, published figures and estimated from measured data without knowing the Hill coefficient.
Therefore, based on physiological considerations, the roxadustat concentration producing the CE50 can be determined independent from the Hill coefficient. As far as the PHD enzyme and HIF enzyme activities are not distinguished from EPO generation, the roxadustat CE50 represents both the half-maximum PHD enzyme inhibition and the half-maximum EPO production. Otherwise, an indirect response model would be needed.
The usual dose of 1.5 mg/kg or 100 mg will lead to a roxadustat Cmax of 10 µg/mL and a peak EPO response (E1 peak) of 150 IU/L. A 2.0 mg/kg dose will produce a peak EPO response (E1 peak) of 240 IU/L (Table 2). Without pharmacological intervention, the physiological range of endogenous EPO levels is up to 300 IU/L [18, 56]. Therefore, the CE50 corresponds to an EPO concentration of 150 IU/L under physiological conditions, and this half-maximum response appears to be achieved with a roxadustat concentration of 10 µg/mL (CE50).
Based on pharmacodynamic considerations, the half-maximum concentration (CE50) represents the target concentration (CE50 = Ctarget). It is probably not by accident or simple chance, but by clinical experience, that the concentration producing the CE50, as stated in this review with a 10 µg/mL dose, corresponds closely to the target peak concentration established empirically with the usual 100 mg dose (1.5 mg/kg).
Because the molecular weight is stated at 352.34 g/mol for roxadustat, the concentration (CE50) of 10 µg/mL producing the half-maximum effect corresponds to a molar concentration of 28 µmol/L. From available studies, the corresponding half-maximum inhibitory concentration (CE50 equivalent IC50) was reported at 100 µmol/L for roxadustat, but is more similar to our result at 29 µmol/L for vadadustat [52]. In mice, the molar concentration producing the CE50 on EPO (CE50 equivalent EC50) was estimated to be much less but still comparable with 1.7 µmol/L for the HIF stabilizer PHI-1. Notably, the animal model also differed in the much higher PHI-1 dose of 30 mg/kg, but the sigmoidal Emax model was the same [66].
From the pharmacodynamic Hill equation and the Emax model, the time of effect duration (TED) can be derived. One special solution for the TED is the pharmacodynamic effect bisection time (TED50) that compares with the pharmacokinetic t½ [37]. For the two special conditions of H = 1.0 and Cpeak = CE50, the TED50 corresponds to the mean residence time used to describe compartment-free pharmacokinetics (TED50 = MRT).
Analogous to pharmacokinetics (Fig. 4), the TED50 can be found in published figures with the measured pharmacodynamic response. Graphical analysis yields a TED50 of 10 h (Fig. 3). The TED50 depends on dose and may increase up to 26 h in dialysis patients (Table 2). In patients with normal kidney function and NDD-CKD, the post-roxadustat response of endogenous EPO lasts for 24 h, contrasting to 48 h in dialysis-dependent patients [29, 56].

Roxadustat pharmacodynamics of the effect (E1) on EPO. By graphical analysis, a TED50 of 10 h can be seen from the published diagram [8]. The TED50 corresponds to a pharmacodynamic EPO t½ of 10 h. EPO erythropoetin, TED50 effect bisection time, BIW twice weekly, t½ elimination half-life
Given Cpeak and making instrumental the concentration at CE50 in the above equation for the TED50, a solution for the roxadustat Hill coefficient (H) can be found at H = 3.3, as calculated by numerical iteration (Table 2). This Hill coefficient (H = 3.3) concordantly compares with a coefficient estimate (H equivalent γ) of 2.8 in mice [66].
A threshold concentration has been postulated for the mouse model [66]. A Hill coefficient of > 2.0 and a threshold limit are compatible with time-dependent pharmacodynamics, contrasting to a Hill coefficient of < 2.0, which would indicate concentration-dependent pharmacodynamics [37].
From the available data, a threshold concentration (Cthreshold) can be predicted for roxadustat at 4.1 µg/mL (Cthreshold = CE50·19-1/H). Such a threshold concentration, and in consequence a ceiling concentration, has already been defined for the ESA darbepoetin [36, 47]. The time-dependence of effect (E1) would implicate that the drug concentration should not be less than the threshold concentration. This usually requires a short administration interval. The other HIF stabilizer vadadustat, with a dosing interval of 24 h, has an even shorter t½ of 5.8 h (Chavan [9]. Although the roxadustat t½ is approximately 12 h, the suggested administration interval is 48–72 h for a 100 mg dose, indicating that the target effect on ∆Hb more constantly and continuously follows downstream to the intermittent rise and decrease in EPO levels. When looking only at EPO, time-dependent kinetics would need a short administration interval of 24 h, but the dynamics of the ∆Hb response look different, allowing for less frequent dosing.
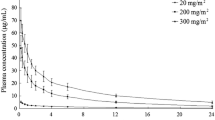
No clinically relevant accumulation will be seen with repetitively administered roxadustat. Using a graphical analysis tool, the roxadustat t½ after repetitive dosing can again be derived at 8.5 h (Fig. 4). The t½ average from published studies is 12 h (Table 1).

Roxadustat multiple-dose pharmacokinetics in a semi-logarithmic diagram. With repetitive dosing, no accumulation occurs. A t½ of 8.5 h can be estimated by graphical analysis. t½ elimination half-life
Upon integration of the Hill equation, one obtains the area under the effect-time curve (AUEC) after a single dose [39]. Again, a solution can be derived for the effect area (AUEC) of effect E1 on EPO even after repetitive dosing, with drug concentrations fluctuating between peaks and troughs [15].
The upstream effect area (AUEC) for roxadustat corresponds to the area under the EPO concentrations. The EPO effect area (AUEC1) represents the overall erythropoesis response to repetitive roxadustat administration (Table 2). With a 2.0 mg/kg three times weekly dose, the roxadustat peak is 12 µg/mL (Cpeak), the trough is 0.43 µg/mL (Ctrough), and the effect AUEC can be estimated at 1133 h·IU/L for normal kidney function but at 2906 h·IU/L for kidney failure. In Caucasians, the reported AUEC results were higher at 2630 and 4612 h·IU/L, respectively (Table 2); however, the pharmacodynamic model allows the prediction of these results, making our CE50 = 10 µg/mL and H = 3.33 estimates plausible.
With repetitive administration of 2.0 mg/kg, the trough concentration (Ctrough) was 0.43 µg/mL, much less than the predicted threshold concentration of 4.1 µg/mL (CE05 = CE50·19-1/H) that would be needed to produce a significant effect > 5% of Emax. The empirically chosen and clinically approved long administration interval indicates that the subsequent effect (E2) on hemoglobin (ΔHb) must be longer lasting than the 10-h bisection time of effect (E1) on EPO.
8.2 Irreversible Effect (E2) on Hemoglobin (∆Hb)
In contrast to the intermittently fluctuating and reversible EPO production, the effect on hemoglobin synthesis (∆Hb) might follow delayed pharmacodynamics, resembling an irreversible effect. Other than the intermittent effect on EPO levels, the effect on ∆Hb conveys a more continuous near-linear increase. Our hypothetical concept of an effect cascade is the interpretation of available clinical findings and observational data (ESM 1.2).
The effect (E1) on EPO exhibits oscillating fluctuations, as do the roxadustat concentrations (Figs. 2, 3, 4). In bone marrow, EPO binds to an erythroblast receptor. The cellular signaling cascade stimulates the division and differentiation of erythroid progenitor cells. The mechanism following this binding is no longer a molecular ligand-receptor interaction, but this binding results in the activation of a sustained genetic program of progenitor RBC production. The effect (E2) on hemoglobin exhibits a near linear, steady rise and sustained persistence (Fig. 5). A continuous and steady increase, not an instant rise, in hemoglobin occurs. This should be partially attributed to the fact that hemoglobin synthesis takes time. In addition, only a limited number of hematopoietic cells respond to EPO, and recruitment of EPO-responding cells may also take time [25].

Roxadustat effect on EPO and hemoglobin concentrations (ΔHb). With repetitive dosing, roxadustat concentrations rise and fall (Fig. 4), and, subsequently, EPO concentrations rise and fall (left). In contrast to the effect (E1) on EPO, the effect (E2) on hemoglobin (∆Hb) steadily increases (right). The fluctuating EPO effect (E1) is reversible and the sustained ΔHb effect (E2) might be interpreted as an irreversible effect. EPO erythropoetin, BIW twice weekly
The new RBCs contain hemoglobin that can be measured in blood. The duration or persistence of the roxadustat effect (E2) on hemoglobin synthesis depends on the erythrocyte cell lifespan and is not influenced by the intermittent fluctuations of the EPO concentrations. This effect is likely irreversible as long as the new RBCs survive.
Under normal conditions and without any drugs, the physiological EPO concentration is low at 12 IU/L [55]. Contrasting to the irreversible effect model for roxadustat proposed in this review, the hemoglobin response to high altitude followed a saturable and sigmoidal pattern: at 3454 m, the EPO concentrations rise from 14 to 32 U/L and a hemoglobin plateau is reached after 3 weeks, with a rapid decline within 2 weeks when at sea level again [65]. However, this capacity limited phenomenon might be due to altitude limits and not to hemoglobin limits. An EPO overshoot to > 1000 IU/L can be produced by high-dose roxadustat [71]. In agreement with irreversible pharmacodynamics, there is no ceiling of effect (E2) on hemoglobin, a hemoglobin overshoot can also be observed in patients with kidney diseases due to elevated erythropoietin levels [61].
The ∆Hb effect does not follow a steady and capacity limited function, but erythropoesis continuously increases and will ultimately suddenly be cut by an unsteady function, as predefined by the singular biological entity of the patient’s body. Contrasting to receptor-mediated, capacity limited reversible effects, the response to an irreversible effect is unsaturable, unless counter-regulatory mechanisms come into action.
In agreement with an irreversible effect, a sustained and stable hemoglobin level for 14 days and more, even after the end of roxadustat treatment, was observed [8]. The other observation in agreement with an irreversible effect is that the roxadustat dose can be decreased with the time that is needed to maintain the hemoglobin level in the high target range [12]. This decrease in weekly dose requirements from 5.2 to 3.3 mg/kg also pleads for a persistent non-fluctuating model to describe the roxadustat effect (E2) on hemoglobin. Different from the irreversible roxadustat E2 on hemoglobin, the reversible Emax model was needed to describe the capacity limited pharmacodynamics of the ESA darbepoetin on hemoglobin [36].
However, as opposed to a reversible effect, there is no possibility of deriving the duration of an irreversible effect (TED) from the pharmacodynamic parameters of the drug. The intensity of an irreversible effect depends on the pharmacokinetics (dose, volume of distribution, t½, target concentration, speed of absorption, or administration route) but not on the pharmacodynamic parameters. However, the duration or persistence of an irreversible effect does not depend on pharmacokinetic or pharmacodynamic parameters. This can also be seen for the roxadustat effect on hemoglobin.
The persistence of an irreversible effect could be derived from the generation time of new cells or from the survival time of the target cell. The irreversible effect can also depend on the intracellular persistence of an effector such as small-interfering RNA. For an irreversible effect, the effect duration time (TEDirrev) depends either on the regeneration period after cell death or on the lifespan of the new generated cell, or from the stability of an intracellular effector.
For the ∆Hb E2 of roxadustat on hemoglobin, the TED and measure of persistence is the RBC survival time. The normal lifespan of RBCs is 112 days or 16 weeks, corresponding to an erythrocyte t½ of 3 months or more [70]. The empiric-based prolonged dosing practice indicates that the target effect might not follow the reversible but long-lasting irreversible pharmacodynamics of roxadustat.
Through discrete pharmacokinetic alterations, kidney function may influence the pharmacodynamic response with regard to EPO exposure which is twofold stronger in CKD. However, the target effect on hemoglobin (ΔHb) will be 30% less compared with normal [29]. Although pharmacokinetic parameters such as AUC increase, the dose needed for an adequate hemoglobin response to roxadustat should be 1.5-fold higher in dialysis-dependent patients compared with controls [53, 54]. With comparable roxadustat dosing, the hemoglobin increase was 1.99 g/dL in NDD-CKD patients, but less in dialysis-dependent patients at only 0.52 g/dL [44]. Compared with CKD patients with preserved kidney function, dialysis-dependent patients received a 30% higher roxadustat dose but achieved only a 50% lower ΔHb of 0.9 g/dL [12]. Vadadustat, another HIF prolyl hydroxylase inhibitor, was likewise one-third less effective on hemoglobin in dialysis patients compared with CKD patients not yet in need of dialysis [49].
The pharmacodynamic response to EPO is the target effect, which is on hemoglobin (∆Hb). Loss of kidney function will likely be associated with a lower number of target cells where the HIF machinery on EPO production can work. Most likely, the myelopoetic proliferation of reticulocytes in response to EPO is also inhibited in uremia [44]. In addition, RBC lifespan (TEDirrev) may only be half the normal value in uremia [21].
9 Clinical Toxicities
In a meta-analysis of 30 studies comprising 13,146 patients [10], more adverse events were reported with roxadustat than with placebo, such as diarrhea (RR 1.21, 95% CI 1.00–1.47), nausea (RR 1.46, 95% CI 1.09–1.97), edema (RR 1.32, 95% CI 1.01–1.59), hyperkalemia (RR 1.27, 95% CI 1.05–1.54), and arterial hypertension (RR 1.34, 95% CI 1.02–1.76). With roxadustat, a rise in serum potassium and a decrease in blood bicarbonate have been reported [11]. Compared with exogenous ESA, roxadustat was associated with more frequent adverse events such as vomiting (RR 1.30, 95% CI 1.02–1.65), headache (RR 1.27, 95% CI 1.05–1.53), and thrombosis (RR 1.31, 95% CI 1.05–1.63) [10]. Roxadustat might unfavorably lead to the growth of renal cysts in autosomal dominant polycystic kidney disease (ADPKD) because the HIF pathway appears to be involved in cyst progression [43]. Roxadustat led to reduced thyroid-stimulating hormone levels in a multimorbid hemodialysis patient, but, to date, mechanism and clinical implications are unclear [33].
The frequency of treatment-emergent adverse events was high at 87% for both placebo and roxadustat [64]. In the largest study of CKD patients for up to 4 years, adverse events with roxadustat were reported to be comparable with placebo [23]. However, with roxadustat, the secondary efficacy endpoint of eGFR loss was steeper (−3.7 vs. −3.2 mL/min, p = 0.046). Numerically higher values were found for all-cause mortality (21% vs. 18%), urinary tract infections (13% vs. 8.0%), and treatment-related deaths (3.1% vs. 2.0%) compared with placebo in the intention-to-treat analysis (supplement to Fishbane [23]).
From the beginning of clinical HIF stabilizer development and during phases I–III, repeated concern was articulated that PHD inhibition might promote tumor growth [31]. Prolyl hydroxylase 3 is essential for the inhibition of fatty acid oxidation involved in anti-tumor immunity [58]. Furthermore, HIF activates the VEGF receptor with a proangiogenic response [42].
In patients new to dialysis, using pooled data from phase III trials, the risks of MACE were found to be significantly lower with roxadustat compared with epoetin-α [56]. In NDD-CKD patients, the 52-week trial with vadadustat was associated with a significantly increased MACE hazard ratio compared with darbepoetin [13]. Notably, vadadustat was administered once daily, which could be interpreted as an argument for longer dose intervals or even intermittent phases without HIF stabilizer therapy.
10 Conclusions and Expert Opinion
Roxadustat appears to be an effective therapy for anemia, with advantages particularly in NDD-CKD patients. One of its advantages is its oral availability combined with improved resorption and mobilization of iron. The hemoglobin response occurs after 4 weeks and the target hemoglobin concentration of 11 g/dL was observed after 12 weeks [6, 14]. However, long-term continuous treatment for more than 1 year may be associated with adverse events, which are still incompletely understood (Shutov [64]. Until further data are available, it might be reasonable to not target hemoglobin levels > 11 g/dL. The concept of an irreversible hemoglobin response (∆Hb) to roxadustat suggests that continuously maintained dosing may not be required.
Hypothesis 1: As a practical consequence from roxadustat pharmacokinetics/pharmacodynamics, one could suggest that a sequence of a reversible and irreversible roxadustat effect exists. This means that hemoglobin will still be produced for some time, even after stopping roxadustat administration.
As an advantage, it has been mentioned that roxadustat exerts an intermittent (not continuous) effect on HIF stabilization [24]. This particular advantage can be translated to a larger scale. Given the long-lasting response on RBCs, the limited use of roxadustat only in acute conditions, or the periodic dosing regimen with a cycling medication intermission, could be proposed. It might even be suggested that roxadustat only be used for transient treatment of acute states, but that the administration is limited to, for example, 3 months. Periodic roxadustat administration for 12 weeks until hemoglobin is > 10 g/dL could be followed by a medication interruption, and roxadustat could intermittently be discontinued. Such a regimen might maximize the therapeutic benefit and minimize potential harm, but would have to be studied in clinical trials.
Hypothesis 2: With regard to, as so far unclear, tumor risk, the fact that the roxadustat effect on hemoglobin is long-lasting could make a difference. The irreversible character of roxadustat dynamics might allow for only acute and temporary anemia treatment.
Such a discontinuous regimen might avoid a hemoglobin overshoot. Acute limited or cyclic roxadustat dosing periods to the target hemoglobin, with subsequent intermission of drug administration, could possibly reduce the suspected risks of tumor development. This dosing proposal might be worth investigating and to be proven or rejected by further studies.
References
Akizawa T, Iwasaki M, Otsuka T, Reusch M, Misumi T. Roxadustat Treatment of Chronic Kidney Disease-Associated Anemia in Japanese Patients Not on Dialysis: A Phase 2, Randomized, Double-Blind Placebo-Controlled Trial. Adv Ther. 2019;36(6):1438–54. https://doi.org/10.1007/s12325-019-00943-4.
Akizawa T, Otsuka T, Reusch M, Ueno M. Intermittent oral dosing of roxadustat in peritoneal dialysis chronic kidney disease patients with anemia: a randomized, Phase 3, Multicenter, Open-Label Study. Ther Apher Dial. 2020;24(2):115–25.
Akizawa T, Yamaguchi Y, Otsuka T, Reusch M. A Phase 3, Multicenter, Randomized, Two-Arm, Open-Label Study of Intermittent Oral Dosing of Roxadustat for the Treatment of Anemia in Japanese Erythropoiesis-Stimulating Agent-Naïve Chronic Kidney Disease Patients Not on Dialysis. Nephron. 2020;144(8):372–82. https://doi.org/10.1159/000508100.
Akizawa T, Nangaku M, Yonekawa T, Okuda N, Kawamatsu S, Onoue T, et al. Efficacy and Safety of Daprodustat Compared with Darbepoetin Alfa in Japanese Hemodialysis Patients with Anemia: a randomized, double-blind, Phase 3 Trial. Clin J Am Soc Nephrol. 2020;15(8):1155–65. https://doi.org/10.2215/CJN.16011219.
Akizawa T, Yamaguchi Y, Majikawa Y, Reusch M. Factors affecting the doses of roxadustat vs darbepoetin alfa for anemia treatment in hemodialysis patients. Ther Apher Dial. 2021;25(5):575–85. https://doi.org/10.1111/1744-9987.13609.
Bernhardt WM, Wiesener MS, Scigalla P, Chou J, Schmieder RE, Günzler V, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol. 2010;21(12):2151–6. https://doi.org/10.1681/ASN.2010010116.
Besarab A, Chernyavskaya E, Motylev I, Shutov E, Kumbar LM, Gurevich K, et al. Roxadustat (FG-4592): correction of anemia in incident dialysis patients. J Am Soc Nephrol. 2016;27(4):1225–33. https://doi.org/10.1681/ASN.2015030241.
Besarab A, Provenzano R, Hertel J, Zabaneh R, Klaus SJ, Lee T, et al. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transpl. 2015;30(10):1665–73.
Chavan A, Burke L, Sawant R, Navarro-Gonzales P, Vargo D, Paulson SK. Effect of moderate hepatic impairment on the pharmacokinetics of vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor. Clin Pharmacol Drug Dev. 2021;10(8):950–8. https://doi.org/10.1002/cpdd.927.
Chen H, Cheng Q, Wang J, Zhao X, Zhu S. Long-term efficacy and safety of hypoxia-inducible factor prolyl hydroxylase inhibitors in anaemia of chronic kidney disease: a meta-analysis including 13,146 patients. J Clin Pharm Ther. 2021;46(4):999–1009. https://doi.org/10.1111/jcpt.13385.
Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med. 2019;381(11):1011–22.
Chen N, Hao C, Peng X, Lin H, Yin A, Hao L, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med. 2019;381(11):1001–10.
Chertow GM, Pergola PE, Farag YMK, Agarwal R, Arnold S, Bako G, PRO2TECT Study Group, et al. Vadadustat in patients with anemia and non-dialysis-dependent CKD. N Engl J Med. 2021;384(17):1589–600. https://doi.org/10.1056/NEJMoa2035938.
Coyne DW, Roger SD, Shin SK, Kim SG, Cadena AA, Moustafa MA. Roxadustat for CKD-related anemia in non-dialysis patients. Kidney Int Rep. 2020;6(3):624–35. https://doi.org/10.1016/j.ekir.2020.11.034.
Czock D, Giehl M. Aminoglycoside pharmacokinetics and -dynamics: a nonlinear approach. Int J Clin Pharmacol Ther. 1995;33(10):537–9.
Del Balzo U, Signore PE, Walkinshaw G, Seeley TW, Brenner MC, Wang Q, et al. Nonclinical characterization of the hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat, a novel treatment of anemia of chronic kidney disease. J Pharmacol Exp Ther. 2020;374(2):342–53. https://doi.org/10.1124/jpet.120.265181.
Dhillon S. Roxadustat: first global approval. Drugs. 2019;79(5):563–72. https://doi.org/10.1007/s40265-019-01077-1.
Dias KA, Lawley JS, Gatterer H, Howden EJ, Sarma S, Cornwell WK 3rd, et al. Effect of acute and chronic xenon inhalation on erythropoietin, hematological parameters, and athletic performance. J Appl Physiol (1985). 2019;127(6):1503–10. https://doi.org/10.1152/japplphysiol.00289.2019.
Domínguez-Hüttinger E, Christodoulides P, Miyauchi K, Irvine AD, Okada-Hatakeyama M, Kubo M, et al. Mathematical modeling of atopic dermatitis reveals “double-switch” mechanisms underlying 4 common disease phenotypes. J Allergy Clin Immunol. 2017;139(6):1861-1872.e7.
Elnaem MH, Irwan NA, Abubakar U, Syed Sulaiman SA, Elrggal ME, Cheema E. Impact of medication regimen simplification on medication adherence and clinical outcomes in patients with long-term medical conditions. Patient Prefer Adherence. 2020;14:2135–45. https://doi.org/10.2147/PPA.S268499.
Erslev AJ, Besarab A. The rate and control of baseline red cell production in hematologically stable patients with uremia. J Lab Clin Med. 1995;126(3):283–6.
European Medicines Agency. Product information for Evrenzo. 24 Aug 2021. https://www.ema.europa.eu/en/documents/product-information/evrenzo-epar-product-information_en.pdf. Accessed 28 Oct 2021
Fishbane S, El-Shahawy MA, Pecoits-Filho R, Van BP, Houser MT, Frison L, et al. Roxadustat for treating anemia in patients with CKD Not on dialysis: results from a randomized phase 3 study. J Am Soc Nephrol. 2021;32(3):737–55. https://doi.org/10.1681/ASN.2020081150.
Fishbane S, El-Shahawy MA, Van BP, Little DJ. Authors’ reply. J Am Soc Nephrol. 2021;32(6):1537–8. https://doi.org/10.1681/ASN.2021030334.
Fogh J. Studies on the mechanism of the increased dose-response of erythropoietin after stimulation with erythropoietin. Blood. 1970;35(4):476–92.
Groenendaal-van de Meent D, Adel MD, Noukens J, Rijnders S, Krebs-Brown A, Mateva L, et al. Effect of Moderate Hepatic Impairment on the Pharmacokinetics and Pharmacodynamics of Roxadustat, an Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor. Clin Drug Investig. 2016;36(9):743–51. https://doi.org/10.1007/s40261-016-0422-y.
Groenendaal-van de Meent D, den Adel M, Rijnders S, Krebs-Brown A, Kerbusch V, Golor G, et al. The Hypoxia-inducible Factor Prolyl-Hydroxylase Inhibitor Roxadustat (FG-4592) and Warfarin in Healthy Volunteers: A Pharmacokinetic and Pharmacodynamic Drug-Drug Interaction Study. Clin Ther. 2016;38(4):918–28. https://doi.org/10.1016/j.clinthera.2016.02.010.
Groenendaal-van de Meent D, den Adel M, van Dijk J, Barroso-Fernandez B, El Galta R, Golor G, et al. Effect of Multiple Doses of Omeprazole on the Pharmacokinetics, Safety, and Tolerability of Roxadustat in Healthy Subjects. Eur J Drug Metab Pharmacokinet. 2018;43(6):685–92. https://doi.org/10.1007/s13318-018-0480-z.
Groenendaal-van de Meent D, Kerbusch V, Kaspera R, Barroso-Fernandez B, Galletti P, Klein GK, et al. Effect of Kidney Function and Dialysis on the Pharmacokinetics and Pharmacodynamics of Roxadustat, an Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor. Eur J Drug Metab Pharmacokinet. 2021;46(1):141–53. https://doi.org/10.1007/s13318-020-00658-w.
Groenendaal-van de Meent D, Kerbusch V, Barroso-Fernandez B, den Adel M, van Dijk J, Golor G, et al. Effect of the Phosphate Binders Sevelamer Carbonate and Calcium Acetate on the Pharmacokinetics of Roxadustat After Concomitant or Time-Separated Administration in Healthy Individuals. Clin Ther. 2021;43(6):1079–91. https://doi.org/10.1016/j.clinthera.2021.03.025.
Gupta N, Wish JB (2017) Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD (published erratum appears in: Am J Kidney Dis. 69(6):869. Am J Kidney Dis. 69(6):815-826. https://doi.org/10.1053/j.ajkd.2016.12.011.
Hann A, Nosalski E, Hermann PC, Egger J, Seufferlein T, Keller F. Chemotherapeutic agents eligible for prior dosing in pancreatic cancer patients requiring hemodialysis: a systematic review. Clin Nephrol. 2018;90(2):125–41. https://doi.org/10.5414/CN109327.
Ichii M, Mori K, Miyaoka D, Sonoda M, Tsujimoto Y, Nakatani S, et al. Suppression of thyrotropin secretion during roxadustat treatment for renal anemia in a patient undergoing hemodialysis. BMC Nephrol. 2021;22(1):104. https://doi.org/10.1186/s12882-021-02304-2.
Japanese Pharmaceuticals and Medical Devices Agency. Roxadustat - Report on the Deliberation Results. 2019. https://www.pmda.go.jp/files/000234811.pdf. Accessed 27 May 2021.
Jurado García JM, Torres Sánchez E, Olmos Hidalgo D, Alba CE. Erythropoietin pharmacology. Clin Transl Oncol. 2007;9(11):715–22. https://doi.org/10.1007/s12094-007-0128-y.
Keller F, Ludwig U, Czock D. Pharmacokinetic and pharmacodynamic considerations on the erythropoietin effect and adverse events of darbepoetin. Expert Opin Drug Metab Toxicol. 2015;11(1):139–47. https://doi.org/10.1517/17425255.2015.989832.
Keller F, with contributions from David Czock. Classical Pharmacokinetics and Concise Pharmacodynamics for Clinicians. Munich/Orlando: Dustri; 2020.
Kiss Z, Elliott S, Jedynasty K, Tesar V, Szegedi J. Discovery and basic pharmacology of erythropoiesis-stimulating agents (ESAs), including the hyperglycosylated ESA, darbepoetin alfa: an update of the rationale and clinical impact. Eur J Clin Pharmacol. 2010;66:331–40.
Krzyzanski W, Jusko WJ. Integrated functions for four basic models of indirect pharmacodynamic response. J Pharm Sci. 1998;87(1):67–72. https://doi.org/10.1021/js970168r.
Lappin KM, Mills KI, Lappin TR. Erythropoietin in bone homeostasis – implications for efficacious anemia therapy. Stem Cells Transl Med. 2021. https://doi.org/10.1002/sctm.20-0387.
Lewis R, Bewersdorf JP, Zeidan AM. Clinical management of anemia in patients with myelodysplastic syndromes: an update on emerging therapeutic options. Cancer Manag Res. 2021;13:645–57. https://doi.org/10.2147/CMAR.S240600.
Li N, Li Q, Bai J, Chen K, Yang H, Wang W, et al. The multiple organs insult and compensation mechanism in mice exposed to hypobaric hypoxia. Cell Stress Chaperones. 2020;25(5):779–91. https://doi.org/10.1007/s12192-020-01117-w.
Liu F, Wang J, Ye F, Fu H, Mao J. Roxadustat for renal anemia in ESRD from PKD patients: is it safe enough? J Am Soc Nephrol. 2021;32(4):1005.
Liu J, Zhang A, Hayden JC, Bhagavathula AS, Alshehhi F, et al. Roxadustat (FG-4592) treatment for anemia in dialysis-dependent (DD) and not dialysis-dependent (NDD) chronic kidney disease patients: a systematic review and meta-analysis. Pharmacol Res. 2020;155:104747. https://doi.org/10.1016/j.phrs.2020.104747.
Long G, Chen H, Wu M, Li Y, Gao L, Huang S, et al. Antianemia Drug Roxadustat (FG-4592) Protects Against Doxorubicin-Induced Cardiotoxicity by Targeting Antiapoptotic and Antioxidative Pathways. Front Pharmacol. 2020;11:1191.
Macdougall IC, Gray SJ, Elston O, Breen C, Jenkins B, Browne J, et al. Pharmacokinetics of novel erythropoiesis stimulating protein compared with epoetin alfa in dialysis patients. J Am Soc Nephrol. 1999;10(11):2392–5.
Macdougall IC. Optimizing the use of erythropoietic agents– pharmacokinetic and pharmacodynamic considerations. Nephrol Dial Transplant. 2002;17(Suppl 5):66–70. https://doi.org/10.1093/ndt/17.suppl_5.66.
Miró-Murillo M, Elorza A, Soro-Arnáiz I, Albacete-Albacete L, Ordoñez A, Balsa E, et al. Acute Vhl gene inactivation induces cardiac HIF-dependent erythropoietin gene expression. PLoS ONE. 2011;6(7):e22589. https://doi.org/10.1371/journal.pone.0022589.
Nangaku M, Farag YMK, deGoma E, Luo W, Vargo D, Khawaja Z. Vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, for treatment of anemia of chronic kidney disease: two randomized Phase 2 trials in Japanese patients. Nephrol Dial Transplant. 2021. https://doi.org/10.1093/ndt/gfaa060.
Ogawa C, Tsuchiya K, Tomosugi N, Maeda K. A Hypoxia-inducible factor stabilizer improves hematopoiesis and iron metabolism early after administration to treat anemia in hemodialysis patients. Int J Mol Sci. 2020;21(19):7153. https://doi.org/10.3390/ijms21197153.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. https://doi.org/10.1136/bmj.n71.
Portolés J, Martín L, Broseta JJ, Cases A. Anemia in chronic kidney disease: from pathophysiology and current treatments, to future agents. Front Med (lausanne). 2021;8:642296. https://doi.org/10.3389/fmed.2021.642296.
Provenzano R, Besarab A, Sun CH, Diamond SA, Durham JH, Cangiano JL, et al. Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat (FG-4592) for the Treatment of Anemia in Patients with CKD. Clin J Am Soc Nephrol. 2016;11(6):982–91. https://doi.org/10.2215/CJN.06890615.
Provenzano R, Besarab A, Wright S, Dua S, Zeig S, Nguyen P, et al. Roxadustat (FG-4592) Versus Epoetin Alfa for Anemia in Patients Receiving Maintenance Hemodialysis: A Phase 2, Randomized, 6- to 19-Week, Open-Label, Active-Comparator, Dose-Ranging, Safety and Exploratory Efficacy Study. Am J Kidney Dis. 2016;67(6):912–24.
Provenzano R, Tumlin J, Zabaneh R, Chou J, Hemmerich S, Neff TB, et al. Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat (FG-4592) for Treatment of Anemia in Chronic Kidney Disease: A Placebo-Controlled Study of Pharmacokinetic and Pharmacodynamic Profiles in Hemodialysis Patients. J Clin Pharmacol. 2020;60(11):1432–40. https://doi.org/10.1002/jcph.1648.
Provenzano R, Fishbane S, Szczech L, Leong R, Saikali KG, Zhong M, et al. Pooled analysis of roxadustat for anemia in patients with kidney failure incident to dialysis. Kidney Int Rep. 2020;6(3):613–23. https://doi.org/10.1016/j.ekir.2020.12.018.
Provenzano R, Shutov E, Eremeeva L, Korneyeva S, Poole L, Saha G, et al. Roxadustat for anemia in patients with end-stage renal disease incident to dialysis. Nephrol Dial Transplant. 2021;36(9):1717–30. https://doi.org/10.1093/ndt/gfab051.
Rathmell JC. Obesity, immunity, and cancer. N Engl J Med. 2021;384(12):1160–2. https://doi.org/10.1056/NEJMcibr2035081.
Rekić D, Kerbusch-Herben V, Någård M, Chou J, Huang J, Bradley C, et al. Pharmacokinetics of roxadustat: a population analysis of 2855 dialysis- and non-dialysis-dependent patients with chronic kidney disease. Clin Pharmacokinet. 2021;60(6):759–73. https://doi.org/10.1007/s40262-020-00974-z.
Schley G, Klanke B, Schödel J, Forstreuter F, Shukla D, Kurtz A, et al. Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol. 2011;22(11):2004–15. https://doi.org/10.1681/ASN.2010121249.
Shalhoub RJ, Rajan U, Kim VV, Goldwasser E, Kark JA, Antoniou LD. Erythrocytosis in patients on long-term hemodialysis. Ann Intern Med. 1982;97(5):686–90. https://doi.org/10.7326/0003-4819-97-5-686.
Shibata T, Nomura Y, Takada A, Aoki S, Katashima M, Murakami H. Evaluation of the effect of lanthanum carbonate hydrate on the pharmacokinetics of roxadustat in non-elderly healthy adult male subjects. J Clin Pharm Ther. 2018;43(5):633–9. https://doi.org/10.1111/jcpt.12729.
Shibata T, Nomura Y, Takada A, Ueno M, Katashima M, Yazawa R, et al. Evaluation of food and spherical carbon adsorbent effects on the pharmacokinetics of roxadustat in healthy nonelderly adult male Japanese subjects. Clin Pharmacol Drug Dev. 2019;8(3):304–13. https://doi.org/10.1002/cpdd.597.
Shutov E, Sułowicz W, Esposito C, Tataradze A, Andric B, Reusch M, et al. Roxadustat for the treatment of anemia in chronic kidney disease patients not on dialysis: a phase 3, randomized, double-blind, placebo-controlled study (ALPS). Nephrol Dial Transpl. 2021;26(9):1629–16397. https://doi.org/10.1093/ndt/gfab057.
Siebenmann C, Cathomen A, Hug M, Keiser S, Lundby AK, Hilty MP, et al. Hemoglobin mass and intravascular volume kinetics during and after exposure to 3,454-m altitude. J Appl Physiol (1985). 2015;119(10):1194–201. https://doi.org/10.1152/japplphysiol.01121.2014.
Singh I, Nagiec EE, Thompson JM, Krzyzanski W, Singh P. A systems pharmacology model of erythropoiesis in mice induced by small molecule inhibitor of prolyl hydroxylase enzymes. CPT Pharmacometrics Syst Pharmacol. 2015;4(2):e12. https://doi.org/10.1002/psp4.12.
Takada A, Shibata T, Shiga T, Groenendaal-van de Meent D, Komatsu K. Population pharmacokinetics of roxadustat in Japanese dialysis-dependent chronic kidney disease patients with anaemia. Br J Clin Pharmacol Epub. 2021. https://doi.org/10.1111/bcp.15023.
Terker AS, Sasaki K, Arroyo JP, Niu A, Wang S, Fan X, et al. Activation of hypoxia-sensing pathways promotes renal ischemic preconditioning following myocardial infarction. Am J Physiol Renal Physiol. 2021;320(4):F569–77. https://doi.org/10.1152/ajprenal.00476.2020.
Wilson JW, Shakir D, Batie M, Frost M, Rocha S. Oxygen-sensing mechanisms in cells. FEBS J. 2020;287:3888–906.
Ye L, Guo J, Jing LP, Peng GX, Zhou K, Li Y, et al. The life span of red blood cell in patients with severe/very severe aplastic anemia (in Chinese). Zhonghua Xue Ye Xue Za Zhi. 2018;39(2):137–42. https://doi.org/10.3760/cma.j.issn.0253-2727.2018.02.012.
Yu KH, Chou J, Klaus S, Schaddelee M, Kashiwa M, Takada A, et al. Comparable doses of FG-4592 have similar PK/PD in healthy Caucasian and Japanese subject. Nephrol Dial Transplant. 2013;28(Suppl 1):1368.
Zhang Y, Ren S, Xue H, Wang AY, Zou Y, Cai Y, et al. Roxadustat in treating anemia in dialysis patients (ROAD): protocol and rationale of a multicenter prospective observational cohort study. BMC Nephrol. 2021;22(1):28. https://doi.org/10.1186/s12882-021-02229-w.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
David Czock and Frieder Keller have no financial or intellectual conflicts of interest to declare.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Both authors consented to the publication of this article.
Availability of data and material code availability
Available from the corresponding author upon reasonable request.
Author contributions
FK conducted the primary literature search. DC analyzed the pharmacokinetic data and drafted the pharmacokinetic sections, and FK analyzed the pharmacodynamic data and drafted the pharmacodynamic sections. Both authors revised the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Czock, D., Keller, F. Clinical Pharmacokinetics and Pharmacodynamics of Roxadustat. Clin Pharmacokinet 61, 347–362 (2022). https://doi.org/10.1007/s40262-021-01095-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01095-x