
Abstract
Background
Lorlatinib demonstrated efficacy (including intracranial activity) in patients with anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) in a phase I/II study (NCT01970865).
Background and Objective
This analysis describes the pharmacokinetics (PK) of lorlatinib following single and multiple dosing.
Methods
This ongoing, multicenter, open-label, single-arm, phase I/II trial enrolled patients with ALK-positive or c-ros oncogene 1 (ROS1)-positive advanced NSCLC. In phase I, patients received escalating doses of lorlatinib (10–200 mg orally once daily) and twice-daily doses of 35, 75, and 100 mg in continuous 21-day cycles. In phase II, lorlatinib was administered at a starting dose of 100 mg once daily in continuous 21-day cycles. Parameters investigated included the potential for lorlatinib to inhibit/induce cytochrome P450 (CYP) 3A; the absorption/metabolism of lorlatinib and its major metabolite PF-06895751; and differences in these parameters between Asian and non-Asian patients.
Results
Data were available for 54 patients from phase I and 275 patients from phase II. Lorlatinib plasma exposure increased dose proportionally after single doses of 10–200 mg, and slightly less than dose proportionally after multiple doses. Lorlatinib clearance increased following multiple dosing compared with single dosing, indicating autoinduction. The area under the concentration-time curve from time zero to time τ (the dosing interval; AUCτ) of PF-06895751 was approximately 80% higher than that of lorlatinib after multiple dosing. Lorlatinib exhibited brain penetration. Furthermore, no overt differences in single- and multiple-dose PK parameters between the Asian and non-Asian patients were observed.
Conclusions
Lorlatinib is highly brain penetrant and exhibits autoinduction after multiple dosing. There appears to be no inherent differences in lorlatinib PK between healthy subjects and cancer patients, or between Asian and non-Asian patients.
ClinicalTrials.gov NCT01970865.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Multiple daily dosing of lorlatinib induced cytochrome P450 (CYP) 3A4 and resulted in autoinduction of lorlatinib metabolism. |
Continued dosing for up to 20 cycles showed no evidence for additional changes in lorlatinib exposure. |
Brain penetration was demonstrated, with cerebral spinal fluid concentrations that were 70% of lorlatinib free plasma concentrations. |
There were no clinically significant differences in single- and multiple-dose pharmacokinetic parameters between the Asian and non-Asian patients. |
1 Introduction
Lorlatinib is a potent, third-generation inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) tyrosine kinases that is currently approved in the US, European Union, Japan, and many other countries for the treatment of patients with ALK-positive metastatic non-small cell lung cancer (NSCLC) [1,2,3]. Lorlatinib was specifically developed to penetrate the blood–brain barrier and have broad potency against common ALK-resistance mutations [4, 5].
Two absorption, metabolism, and excretion studies conducted in healthy participants have shown that lorlatinib is primarily eliminated via metabolism, with little renal excretion (< 5% of administered dose recovered in urine) [6]. The most abundant circulating human metabolite of lorlatinib is PF-06895751, which is pharmacologically inactive and is generated by the oxidative cleavage of the lorlatinib amide and aromatic ether bonds [6]. Lorlatinib is metabolized primarily by cytochrome P450 (CYP) 3A and UGT1A4, and to a lesser extent by CYP2C8, CYP2C19, CYP3A5, and UGT1A3 [2]. In addition, in vitro studies indicated that lorlatinib is a time-dependent inhibitor as well as an inducer of CYP3A and that it activates the pregnane X receptor (PXR) [2]. Hence, lorlatinib has the potential of influencing its own metabolism.
The safety and efficacy data from the phase I/II B7461001 study (NCT01970865) have been previously reported [7,8,9]. That study established the recommended clinical dose of lorlatinib 100 mg once daily and demonstrated systemic and intracranial activity in patients with advanced ALK-positive or ROS1-positive NSCLC, including patients who had progressed after treatment with crizotinib or second-generation tyrosine kinase inhibitors (TKIs) [7,8,9,10]. Adverse events (AEs) reported with lorlatinib were generally mild or moderate and managed with dosing modifications and supportive care [9]. The most common treatment-related AEs with lorlatinib were hypercholesterolemia and hypertriglyceridemia.
The B7461001 study comprised two parts (phase I and phase II), plus a midazolam substudy and a Japanese lead-in cohort (LIC). In that study, lorlatinib pharmacokinetics (PK) were evaluated at single dose and steady state (after 15 days of continuous dosing) as a secondary objective of the phase I and phase II portions. The parameters investigated included the absorption and metabolism of lorlatinib and the major human circulating lorlatinib metabolite PF-06895751, both blood and urinary concentrations, and differences in these parameters between Asian and non-Asian patients, including a subset of Japanese patients. Since lorlatinib showed the potential to simultaneously inhibit and induce CYP3A in vitro, the midazolam substudy assessed the net clinical effect of lorlatinib on the CYP3A enzyme via the probe substrate, midazolam.
2 Methods
2.1 Trial Design and Patients
Details of the B7461001 study (ClinicalTrials.gov identifier: NCT01970865) have been previously reported [7,8,9]. Briefly, this ongoing, multicenter, open-label, single-arm, phase I/II trial enrolled patients with ALK-positive or ROS1-positive advanced NSCLC with or without central nervous system (CNS) metastases. Patients using strong or moderate CYP3A4 inhibitors or strong CYP3A4 inducers were not eligible for inclusion [7,8,9].
The phase I portion of the trial evaluated escalating doses of lorlatinib, administered orally, from 10 to 200 mg once daily, as well as twice-daily doses of 35, 75, and 100 mg in continuous 21-day cycles, with no days off in between. For most phase I patients, a Day −7 lead-in dose of lorlatinib was administered to characterize single-dose PK. A phase I substudy, comprising the same patients from the main study who were administered the 25 mg once-daily and 150 mg once-daily lorlatinib doses, was conducted to investigate the potential for lorlatinib to inhibit or induce CYP3A using midazolam as a probe CYP3A substrate. Patients received a single 2 mg oral dose of midazolam on Day −7, then received another single 2 mg oral dose of midazolam concurrently with lorlatinib on Cycle 1 Day 15.
The recommended phase II dose was selected to be 100 mg once daily [8]. In the phase II portion of the trial, lorlatinib was administered orally at a starting dose of 100 mg once daily in continuous 21-day cycles. Patients were enrolled into six different expansion cohorts based on their ALK or ROS1 status and previous therapy [9]. The cohorts were defined as EXP-1, ALK treatment-naïve; EXP-2, prior crizotinib only; EXP-3, prior crizotinib or other TKI and one or two prior regimens of chemotherapy; EXP-4, two prior TKIs; EXP-5, three prior TKIs; and EXP-6, ROS1 and any prior therapy. Dose modifications were permitted to manage toxicities at the investigator’s discretion. For a subset of phase II patients participating in serial PK profiling, a single dose of lorlatinib 100 mg once daily was administered on Day −7 to characterize lorlatinib single-dose PK. In this subset, there was an attempt to enrol approximately three Japanese patients in order to evaluate lorlatinib single-dose PK in Japanese patients. In addition to these phase II Japanese patients, a separate LIC enrolled only Japanese patients who were treated with lorlatinib 100 mg once daily.
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Council for Harmonization Good Clinical Practice Guidelines, and all local regulatory requirements were followed. Each patient provided written informed consent before participation.
2.2 Pharmacokinetic (PK) Assessments
In both phase I and phase II, plasma PK parameters, including the maximum plasma concentration (Cmax), time to Cmax (Tmax), and area under the plasma concentration versus time curve (AUC) for lorlatinib and the metabolite PF-06895751, were determined for both single and multiple doses of lorlatinib. The specific bioanalytical methods used have been previously published [11, 12]. Blood samples were collected for serial PK profiling of lorlatinib up to 120 h postdose on Day −7 and up to 24 h postdose on Cycle 1 Day 15, for all phase I patients and a subset of phase II patients. Additionally, sparse PK samples were collected on Days 1 and 8 of Cycle 1, on Day 1 of Cycles 2–5 for both phase I and phase II, and on Day 1 of Cycles 6, 8, and 10 for phase II.
For patients participating in the midazolam substudy, 24-h serial blood samples for lorlatinib PK were collected postdose on Cycle 1 Days 1 and 15, and 24-h serial blood samples for midazolam PK were collected after administration of a single 2 mg oral dose of midazolam on Day −7 and on Cycle 1 Day 15 (concurrently with lorlatinib). Urine samples for the measurement of lorlatinib were also collected for patients in the midazolam substudy.
To evaluate the potential differences in PK in Japanese patients, blood samples were collected during phase II for serial PK profiling of lorlatinib and its metabolites in the Japanese patients (up to 120 h postdose on Day −7 and up to 24 h postdose on Cycle 1 Day 15). Sparse PK samples including predose samples were collected on Cycle 1 Day 8 (only from patients who underwent serial PK sampling), Day 1 of Cycles 2–6, and Day 1 of each other cycle thereafter. The separate Japan LIC patients underwent serial PK sampling up to 24 h postdose on Cycle 1 Days 1 and 15 and sparse PK sampling on Day 1 of Cycles 2–6, 8, and 10.
In both phase I and II, cerebral spinal fluid (CSF) was collected with time-matched plasma samples from clinically appropriate patients who were to undergo a lumbar puncture.
PK parameters for lorlatinib, PF-06895751, and midazolam were calculated for each patient and each treatment, as applicable, employing standard noncompartmental analysis using an internally validated software system (eNCA, version 2.2.4; Pfizer, Groton, CT, USA). The linear-log trapezoidal method was used for AUC estimation. Plasma samples with concentrations below the lower limit of quantification were set to 0 for summary statistics and PK analysis. Actual PK sampling times were used in the derivation of PK parameters. Nominal time was assumed for PK parameter calculations if the actual time was missing.
2.3 Additional Assessment of Cytochrome P450 (CYP) 3A4/5 Inhibition/Induction
The effect of lorlatinib on CYP3A4/5 inhibition/induction was also evaluated by measurement of the 4ß-hydroxycholesterol/cholesterol ratio in blood samples and urinary 6ß-hydroxycortisol/cortisol ratio over the time course of measurement during phase I [13, 14].
2.4 Statistical Analysis
The PK concentration analysis set of lorlatinib was defined as all patients treated (including Day −7 dose) who had at least one concentration of lorlatinib. The PK concentration analysis set for midazolam was defined as all patients treated with midazolam (including Day −7 dose) who had at least one concentration of midazolam, while the PK parameter analysis population was defined as all enrolled patients who received at least one dose of study medication (including Day −7 dose, not including midazolam) and had sufficient information to estimate at least one of the PK parameters of interest (Cmax or AUC) for lorlatinib. The midazolam analysis set included patients who had received at least one dose of midazolam and for which at least one midazolam PK parameter of interest (Cmax or AUC) was available.
All reported PK parameters were summarized descriptively using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA) and no additional statistical tests were performed. The PK parameters AUC from time zero to the time of the last quantifiable concentration (AUClast), AUC from time zero extrapolated to infinite time (AUC∞), area under the concentration-time curve from time zero to time τ (the dosing interval; AUCτ), Cmax, trough concentration (Ctrough), apparent oral clearance (CL/F), and apparent volume of distribution (V/F) were summarized using the summary statistics numbers, arithmetic mean, median, percentage coefficient of variation (%CV), standard deviation (SD), minimum, maximum, geometric mean, and geometric %CV. The PK parameter Tmax was summarized using the summary statistics number, median, minimum, and maximum, and the PK parameters terminal elimination half-life (t½), observed accumulation ratio (Rac), and steady-state accumulation ratio (Rss) were summarized using the summary statistics number, arithmetic mean, median, %CV, SD, minimum, and maximum.
3 Results
3.1 Patients
In phase I, a total of 54 patients were treated with lorlatinib: three patients each in the 10, 25, 50, and 150, and 200 mg once-daily cohorts; 12 patients in the 75 mg once-daily cohort; 17 patients in the 100 mg once-daily cohort; three patients in the 35 and 75 mg twice-daily dosing cohorts; and four patients in the 100 mg twice-daily cohort. All treated patients were evaluable and were included in the PK analysis (N = 54). Of these patients, 22 were male and 32 were female; 37 patients were White, 3 were Black, 7 were Asian, 1 was of other ethnicity, and 6 were of unspecified race (Table 1). The mean (SD) age was 51.9 years (12.8), height was 169.0 cm (11.5), and weight was 71.1 kg (18.0). Six patients, three from the 25 mg once-daily cohort and three from the 150 mg once-daily cohort, were enrolled in the midazolam substudy.
In phase II, a total of 275 patients were treated with lorlatinib 100 mg once daily—30, 27, 60, 65, 46, and 47 patients in cohorts EXP-1, EXP-2, EXP-3, EXP-4, EXP-5, and EXP-6, respectively. In addition, three patients were enrolled in the Japan LIC. All treated patients (phase II and the Japan LIC) except one were evaluable for PK assessments and were included in the phase II PK analysis (N = 277). Of these patients, 119 were male and 158 were female; 132 patients were White, 3 were Black, 105 were Asian, 12 were other ethnicities, and 25 were of unspecified race (Table 1). The mean (SD) age was 53.4 years (12.0), height was 166.0 cm (10.5), and weight was 67.6 kg (17.1). Of the 277 patients, 19 had full PK sampling, which allowed for at least one single-dose lorlatinib PK parameter estimation, and 22 had samples allowing for at least one multiple-dose lorlatinib PK parameter estimation.
3.2 Single-Dose Lorlatinib PK Results
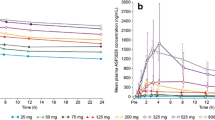
Median lorlatinib plasma concentration-time profiles following single oral doses of lorlatinib are shown in Fig. 1a and b. Following single doses of 10–200 mg, lorlatinib was absorbed rapidly, with median Tmax values of 1.09–2.00 h, and showed biphasic decline, with a mean plasma t½ of 17.2–27.2 h across all doses. Lorlatinib PK parameter values are summarized descriptively in electronic supplementary Table S1. Lorlatinib dose-normalized exposures did not appear to change across the 10–200 mg dose range (electronic supplementary Fig. S1).

Median plasma lorlatinib concentration-time profiles following a single oral doses (Day −7) linear scale; b single oral doses (Day −7) semi-logarithmic scale; c multiple oral doses (Cycle 1 Day 15) linear scale; and d multiple oral doses (Cycle 1 Day 15) semi-logarithmic scale. BID twice daily, QD once daily
In phase II, lorlatinib was absorbed rapidly, with a median value of 1.15 h following a single dose of 100 mg on Day −7 (Table 2). Following attainment of Cmax, the disposition of lorlatinib declined, with a mean t½ of 23.6 h.
3.3 Multiple-Dose Lorlatinib PK Results
Median plasma lorlatinib concentration-time profiles following multiple oral doses in phase I are shown in Fig. 1c and d. Plasma PK parameter values following multiple-dose administration are summarized descriptively in Table 3. On Cycle 1 Day 15 of multiple-dose administration, lorlatinib was absorbed rapidly, with median Tmax values of ≤ 2 h across the entire range of doses, from 10 mg to 200 mg on either once-daily or twice-daily dosing schedules.
Urinary recovery of unchanged lorlatinib following multiple doses was low, with < 0.5% (n = 3) of the dose recovered unchanged in urine for the 100 mg once-daily dosing regimen. Geometric mean renal clearance was 61.3 mL/h. Linear plots of individual and geometric mean dose-normalized AUCτ and Cmax by dose for the once- and twice-daily regimens are shown in Fig. 2.

Linear plot of lorlatinib dose-normalized PK parameters versus dose following multiple oral doses: a AUCτ with QD dosing; b Cmax with QD dosing; c AUCτ with BID dosing; and d Cmax with BID dosing. Circles represent individual values and stars represent the geometric mean. AUCτ area under the concentration-time profile from time zero to time τ, the dosing interval, where τ = 24 h, BID twice daily, Cmax maximum observed plasma concentration, PK pharmacokinetic, QD once daily
In general, plasma lorlatinib dose-normalized AUCτ and Cmax slightly decreased across the 10–200 mg once-daily dose range based on visual comparison of individual and geometric mean Cmax and AUCτ values by dose. Arithmetic mean values for the observed Rac, comparing AUCτ for multiple-dose administration with that for single-dose administration, ranged from 1.0 to 1.5 for the once-daily dosing regimen and 1.2 to 2.1 for the twice-daily dosing regimen.
Arithmetic mean values for the Rss, comparing AUCτ for multiple-dose administration with AUC∞ for single-dose administration, were consistently < 1 (ranging from 0.54 to 0.99), suggesting net autoinduction of lorlatinib following multiple oral dosing.
In phase II, on Cycle 1 Day 15 of multiple-dose administration (100 mg once daily), lorlatinib was absorbed rapidly, with a median Tmax value of 1.96 h (Table 2). Lorlatinib plasma concentrations appeared to reach steady state by 15 days with repeated 100 mg once-daily dosing. The Ctrough of lorlatinib across the phase II cohorts and the Japan LIC were fairly consistent, with median values of approximately 100 ng/mL and geometric means (in groups with n ≥3) ranging from 46.4 to 138.5 ng/mL over the period between Cycle 2 Day 1 and Cycle 20 Day 1 (electronic supplementary Table S2). After 100 mg once-daily dosing of lorlatinib, the arithmetic mean value for Rac was 1.08; the arithmetic mean Rss value was 0.66.
3.4 PF-06895751 PK
An evaluation of the steady-state plasma PK of the most abundant human circulating lorlatinib metabolite, PF-06895751, was performed for 10 patients following repeated 100 mg once-daily administration of lorlatinib. On Cycle 1 Day 15, the PF-06895751 geometric mean AUCτ was 4127 ng·h/mL and the geometric mean Cmax was 193.7 ng/mL, with a median Tmax of 8.1 h. The geometric mean molar ratio for AUCτ of PF-06895751 to lorlatinib was 1.799.
3.5 Effect of Lorlatinib on Midazolam PK
Median midazolam plasma concentrations were substantially lowered in the presence of multiple oral doses of lorlatinib (25 mg once daily [n = 3] and 150 mg once daily [n = 3]) compared with those observed when midazolam (2 mg) was administered alone (Fig. 3 and electronic supplementary Table S3). For the 150 mg once-daily cohort, evaluable midazolam PK data were only available for two patients. Midazolam median Tmax was 0.5 h with or without lorlatinib (25 mg once daily and 150 mg once daily). Following attainment of Cmax, the decline in midazolam plasma concentrations was faster when coadministered with lorlatinib; the mean estimate of the midazolam elimination t½ decreased from 5 to 3 h following coadministration with lorlatinib (25 mg once daily). Following the coadministration of midazolam with lorlatinib (25 mg once daily or 150 mg once daily), the midazolam AUC∞ and Cmax values were lower compared with when midazolam was administered alone. The CL/F of midazolam increased from 36.7 and 45.1 L/h when administered alone, to 93.9 and 124.2 L/h when coadministered with lorlatinib 25 mg once daily and 150 mg once daily, respectively. Midazolam AUClast geometric mean values (%CV) decreased from 51.3 (47%) to 20.4 (18%) ng·h/mL and from 36.5 (20%) to 14.4 (25%) ng·h/mL, respectively, with 25 mg once-daily and 150 mg once-daily lorlatinib dosing. Likewise, midazolam Cmax geometric mean values (%CV) decreased from 16.1 (42%) to 9.7 (40%) ng/mL and from 11.6 (48%) to 5.73 (43%) ng/mL, respectively, with 25 mg once-daily and 150 mg once-daily lorlatinib dosing.

Median plasma midazolam concentration-time profiles following a single 2 mg dose alone and in the presence of a 25 mg QD lorlatinib and b 150 mg QD lorlatinib. MDZ midazolam, QD once daily
3.6 Lorlatinib PK Based on Ethnicity
Twelve non-Asian and seven Asian patients (of whom four were Japanese) had single-dose lorlatinib concentration-time data evaluable for PK analysis, and 11 non-Asian and 11 Asian patients (of whom seven were Japanese) had multiple-dose lorlatinib concentration-time data evaluable for PK analysis. Median lorlatinib plasma concentration-time profiles for Asian versus non-Asian patients after single and multiple 100 mg lorlatinib dosing are shown in Fig. 4.

Median plasma lorlatinib concentration-time profiles following a single oral doses at lead-in Day −7, and b multiple oral doses of lorlatinib at Cycle 1 Day 15 in Asian versus non-Asian patients
After multiple dosing, on Cycle 1 Day 15, the median peak concentrations of lorlatinib in Asian patients were slightly higher than those in non-Asian patients (644.8 vs. 515.5 ng/mL). Following single-dose lorlatinib administration, a 52% higher lorlatinib Cmax was noted in Asian patients, but changes in AUC were minimal (Table 4). The ratios of the adjusted geometric means (expressed as percentages) for lorlatinib AUC∞ and Cmax (90% confidence interval [CI]) were 110.0% (80.5–150.4%) and 152.4% (116.2–199.9%), respectively, for the Asian population compared with the non-Asian population.
Following multiple dosing, lorlatinib plasma exposure (AUCτ) was similar in Asian and non-Asian patients (Table 4). The ratios of the adjusted geometric means for lorlatinib AUCτ and Cmax (90% CI) Cycle 1 Day 15 were 110.7% (83.7–146.5%) and 125.1% (93.7–166.9%), respectively, for the Asian population compared with the non-Asian population. Similar trends for lorlatinib PK following single and multiple 100 mg once-daily doses were observed in Japanese patients compared with non-Asian patients (data not shown).
The PK parameters of the metabolite PF-06895751 following multiple-dose administration of lorlatinib 100 mg once daily, by ethnicity, are shown in Table 5. The molar PF-06895751 to lorlatinib ratios for AUCτ were similar in the Asian and Japanese populations (1.60 vs. 1.63).
3.7 Cerebral Spinal Fluid Results in Phases I and II
Over the course of the study, CSF concentrations and time-matched plasma concentrations of lorlatinib were available for four patients from phase I and one patient from phase II. The CSF concentrations ranged from 2.64 to 125 ng/mL (electronic supplementary Table S3). The mean CSF/free plasma ratio data from phase I was published previously and was reported to be 0.75 [8]. An additional CSF/free plasma ratio measurement from one patient from the phase II portion is reported here and was 0.68.
3.8 Lorlatinib Effect on CYP3A as Measured by 4ß-Hydroxycholesterol/Cholesterol and 6ß-Hydroxycortisol/Cortisol Ratios
At the 100 mg once-daily dose of lorlatinib, both 4ß-hydroxycholesterol/cholesterol and 6ß-hydroxycortisol/cortisol ratios reached maximal values by Day 8 of multiple dosing (electronic supplementary Fig. S2).
4 Discussion
In vitro studies indicated that lorlatinib is a time-dependent inhibitor, as well as an inducer, of CYP3A via PXR activation [6]; thus, it was unclear if the net effect on CYP3A would be induction or inhibition. In the phase I midazolam substudy, lorlatinib was demonstrated to be a net moderate inducer of CYP3A. In this substudy, coadministration of midazolam 2 mg with repeated 25 mg and 150 mg once-daily lorlatinib dosing decreased the oral AUC∞ and Cmax of midazolam. The increase in lorlatinib clearance after multiple dosing across all dose levels further demonstrates that lorlatinib is a net inducer of its own metabolism. Both the urinary 6β-hydroxycortisol/cortisol ratio and blood 4β-hydroxycholesterol/cholesterol ratio indicated that maximum induction of CYP3A was reached by Day 8 of multiple dosing with lorlatinib 100 mg once daily. Urinary recovery of unchanged lorlatinib following multiple doses was low (< 0.5%). Although it is possible that with lorlatinib induction, steady state is reached before day 15, as evidenced by the stable plasma cholesterol and urine cortisol ratios achieved on day 8, it is clear that by day 15 of continuous dosing, steady state is achieved. The urine cortisol ratios additionally confirm what the plasma cholesterol ratios show, i.e. lorlatinib induction reaches an eventual steady state.
In the phase I portion of this study, lorlatinib exposure increased in a dose-proportional manner after single doses of 10–200 mg, and in a slightly less than dose-proportional manner after multiple doses of 10–200 mg once daily. Lorlatinib was absorbed rapidly and showed biphasic decline across all doses.
In phase II, lorlatinib steady state was reached by 15 days following multiple oral doses of 100 mg once daily. At the 100 mg once-daily dose of lorlatinib, the mean Rac was 1.08, compared with a predicted accumulation ratio of 2.0 for a drug with a terminal plasma t½ of approximately 24 h that is administered once daily. The mean Rss was 0.66, whereas an Rss of approximately 1.0 is to be expected with linear PK. This further demonstrates a net autoinduction effect with multiple dosing of lorlatinib.
Although it is clear that lorlatinib steady state was observed by Cycle 1 Day 15, one limitation of the study is that a limited number of samples were collected between Cycle 1 Day 1 and Cycle 1 Day 15. This indicates a need for PK sampling prior to Cycle 1 Day 15 in future studies for better characterization of lorlatinib time-to-completion of autoinduction. Despite the limited number of patients in the midazolam substudy, the results clearly show that while lorlatinib has both the potential to inhibit and induce CYP3A in vitro, the net clinical effect after multiple dosing is induction Similarly, fewer patients were included in the analysis of ethnicity, which was why no statistical calculations were performed. However, the data should provide confidence when prescribing lorlatinib for patients of different ethnicities. Note that for two concentrations, nominal time was used for PK parameter calculations because the actual times were missing. This was for two different participants, one at 9 h and one at 48 h postdose. Thus, this is unlikely to have caused substantial bias.
There was no evidence of additional changes in lorlatinib exposure with long-term dosing of 100 mg once daily (up to 20 cycles) after steady state (Cycle 1 Day 15) was reached. The AUCτ of PF-06895751, the major human circulating metabolite, was about 80%, higher than that of lorlatinib, after multiple 100 mg once-daily dosing. With PF-06895751 being pharmacologically inactive, there are no clinical implications of these findings.
No overt differences in multiple-dose PK parameters between the Asian and non-Asian patients were seen, Japanese patients accounted for about 60% of the Asian patients in this study and demonstrated similar trends in both lorlatinib and PF-06895751 PK compared with non-Asian patients.
The measured CSF/free plasma ratios of lorlatinib from the additional patient in the phase II portion of this study further support the prior published evidence from the phase I portion [8] that lorlatinib is a brain-penetrating drug. Lorlatinib mean CSF concentration reached over 70% of lorlatinib free-plasma concentrations. The systemic effective concentrations for lorlatinib inhibition of the wild-type ALK gene arrangement, L1196M resistance mutation, and G1202R resistance mutation are reported to be 7.6, 62, and 150 ng/mL, respectively [8]. Given that lorlatinib is 66% protein bound (fraction unbound of 0.34) [2], the CNS unbound effective concentrations for lorlatinib inhibition of the wild-type ALK gene arrangement, L1196M resistance mutation, and G1202R resistance mutation can be estimated to be 2.6, 21, and 51 ng/mL, respectively. Of the five patients who had CSF samples drawn (electronic supplementary Table S4), all exceeded the effective concentration target for the wild-type ALK gene rearrangement. Additionally, of the five patients with available CSF data, four patients exceeded the target concentrations for the L1196M mutation and three exceeded that for the G120R mutation.
Lorlatinib AUC and Cmax values in patients with NSCLC in this trial were comparable with those that have been observed in healthy subjects, indicating no inherent differences in PK between healthy subjects and cancer patients [11, 12].
5 Conclusion
Lorlatinib PK were fully characterized in this study in patients with NSCLC, including the evaluation of single- and multiple-dose exposures, dose proportionality, accumulation with multiple dosing, effect of lorlatinib on midazolam PK, and evaluation of urinary recovery of the drug. Multiple oral dosing of lorlatinib 100 mg daily induced CYP3A4 and resulted in autoinduction of lorlatinib metabolism, stabilizing by Day 15. Continued dosing for up to 20 cycles showed no evidence of additional changes in lorlatinib exposure. Brain penetration was conclusively demonstrated, as measured mean CSF concentrations were 70% of lorlatinib free plasma concentrations.
References
Syed YY. Lorlatinib: First Global Approval. Drugs. 2019;79(1):93–8.
Pfizer Inc. LORBRENA® (lorlatinib): Prescribing information. 2021. http://labeling.pfizer.com/ShowLabeling.aspx?id=11140. Accessed 17 Jan 2021.
European Medicines Agency. Lorviqua (lorlatinib). 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/lorviqua. Accessed 17 Jan 2021.
Johnson TW, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57(11):4720–44.
Basit S, et al. First macrocyclic 3rd-generation ALK inhibitor for treatment of ALK/ROS1 cancer: clinical and designing strategy update of lorlatinib. Eur J Med Chem. 2017;134:348–56.
Stypinski D, et al. Metabolism, excretion, and pharmacokinetics of lorlatinib (PF-06463922) and evaluation of the impact of radiolabel position and other factors on comparability of data across 2 ADME studies. J Clin Pharmacol. 2020;60(9):1254–67.
Shaw AT, et al. Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: a multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2019;20(12):1691–701.
Shaw AT, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18(12):1590–9.
Solomon BJ, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. 2018;19(12):1654–67.
Rodig SJ, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15(161):5216–23.
Patel M, et al. The effect of itraconazole on the pharmacokinetics of lorlatinib: results of a phase I, open-label, crossover study in healthy participants. Invest New Drugs. 2020;38(1):131–9.
Chen J, et al. The effect of rifampin on the pharmacokinetics and safety of lorlatinib: results of a phase one, open-label, crossover study in healthy participants. Adv Ther. 2020;37(2):745–58.
Diczfalusy U, et al. 4β-hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br J Clin Pharmacol. 2011;71(2):183–9.
Lutz U, et al. Quantification of cortisol and 6 beta-hydroxycortisol in human urine by LC-MS/MS, and gender-specific evaluation of the metabolic ratio as biomarker of CYP3A activity. J Chromatogr B Anal Technol Biomed Life Sci. 2010;878(1):97–101.
Acknowledgements
The authors would like to thank the study participants and study site personnel. Additionally, they would also like to thank Kimberly Lee, Global Product Development, Pfizer Inc., for her contributions to the design, conduct, and data collection of the study. Medical writing support was provided by Paul O’Neill, PhD, and Meredith Rogers, MS, CMPP, of CMC AFFINITY, McCann Health Medical Communications, and was funded by Pfizer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Pfizer Inc.
Conflict of interest
Joseph Chen, Yazdi K. Pithavala, and Melissa T. O’Gorman are employees of Pfizer Inc. and may own stock or stock options in Pfizer. Lee P. James, Karen J. Klamerus, and Ganesh Mugundu are former employees of Pfizer Inc. and may own stock or stock options in Pfizer.
Data availability
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the US and/or EU, or (2) in programs that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Ethics approval
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Council for Harmonization Good Clinical Practice Guidelines, and all local regulatory requirements were followed.
Consent to Participate
Each patient provided written informed consent before participation. Each investigator received prospective approval of the study protocol, protocol amendments, informed consent documents, and all other relevant documents from their individual institution’s review board or ethics committee.
Author contributions
JC, LPJ, KK, GM, YKP designed and performed the research. MTO analyzed the data. JC and YKP interpreted the data. All authors drafted the manuscript or revised it critically for important intellectual content and approved the version to be submitted. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Chen, J., O’Gorman, M.T., James, L.P. et al. Pharmacokinetics of Lorlatinib After Single and Multiple Dosing in Patients with Anaplastic Lymphoma Kinase (ALK)-Positive Non-Small Cell Lung Cancer: Results from a Global Phase I/II Study. Clin Pharmacokinet 60, 1313–1324 (2021). https://doi.org/10.1007/s40262-021-01015-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01015-z