
Abstract
Background and Objective
Little is known about acetaminophen (paracetamol) pharmacokinetics during pregnancy. The aim of this study was to develop a physiologically based pharmacokinetic (PBPK) model to predict acetaminophen pharmacokinetics throughout pregnancy.
Methods
PBPK models for acetaminophen and its metabolites were developed in non-pregnant and pregnant women. Physiological and enzymatic changes in pregnant women expected to impact acetaminophen pharmacokinetics were considered. Models were evaluated using goodness-of-fit plots and by comparing predicted pharmacokinetic profiles with in vivo pharmacokinetic data. Predictions were performed to illustrate the average concentration at steady state (Css,avg) values, used as an indicator for efficacy, of acetaminophen achieved following administration of 1000 mg every 6 h. Furthermore, as a measurement of potential hepatotoxicity, the molar dose fraction of acetaminophen converted to N-acetyl-p-benzoquinone imine (NAPQI) was estimated.
Results
PBPK models successfully predicted the pharmacokinetics of acetaminophen and its metabolites in non-pregnant and pregnant women. Predictions resulted in the lowest Css,avg in the third trimester (median [interquartile range]: 4.5 [3.8–5.1] mg/L), while Css,avg was 6.7 [5.9–7.4], 5.6 [4.7–6.3], and 4.9 [4.1–5.5] mg/L in non-pregnant, first trimester, and second trimester populations, respectively. Assuming a constant raised cytochrome P450 2E1 activity throughout pregnancy, the molar dose fraction of acetaminophen converted to NAPQI was highest during the first trimester (median [interquartile range]: 11.0% [9.1–13.4%]), followed by the second (9.0% [7.5–11.0%]) and third trimester (8.2% [6.8–10.1%]), compared with non-pregnant women (7.7% [6.4–9.4%]).
Conclusion
Acetaminophen exposure is lower in pregnant than in non-pregnant women, and is related to pregnancy duration. Despite these findings, higher dose adjustments cannot be advised yet as it is unknown whether pregnancy affects the toxicodynamics of NAPQI. Information on glutathione abundance during pregnancy and NAPQI in vivo data are required to further refine the presented model.
Similar content being viewed by others
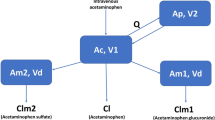

Simulations following administration of acetaminophen (paracetamol) 1000 mg every 6 h to both non-pregnant and pregnant women showed that the acetaminophen average concentration at steady state (Css,avg), was lowest in the third trimester (median: 4.5 mg/L), while Css,avg was 6.7 [5.9–7.4], 5.6, and 4.9 mg/L in non-pregnant, first trimester, and second trimester populations, respectively. |
Assuming a constant raised activity of cytochrome P450 2E1 throughout pregnancy, the median dose fraction of acetaminophen converted to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) was predicted to be 7.7%, 11.0%, 9.0%, and 8.2% in non-pregnant, first, second and third trimester populations, respectively. |
To better characterize the hepatotoxic risk of acetaminophen throughout these different stages of pregnancy, further investigations into NAPQI formation during different stages of pregnancy need to be combined with an assessment of potential changes in the detoxifying capacity (e.g., glutathione levels) of non-pregnant and pregnant women. |
1 Introduction
Pregnancy is associated with physiological changes that might affect the pharmacokinetics of many drugs [1]. If pregnant women are administered the same dose as non-pregnant women, potential pharmacokinetic changes may lead to either sub-therapeutic or toxic drug effects in the mother and/or fetus [2]. Since pregnant women are considerably underrepresented in clinical trials, information on optimal dosing during pregnancy is widely lacking [3]. As a result, because a quantitative and mechanistic understanding of pharmacokinetic alterations during pregnancy is often missing, determining of an appropriate dose is difficult [4]. Physiologically based pharmacokinetic (PBPK) modeling is considered a valuable tool for predicting pharmacokinetic changes throughout pregnancy and can be used to investigate pharmacokinetics and support dosing strategies [5]. PBPK models are multi-compartment models that mechanistically describe the processes of drug liberation, absorption, distribution, metabolism, and excretion through a set of differential equations [6].
Acetaminophen (paracetamol) is one of the most commonly used drugs during pregnancy [7]. Oral acetaminophen is used to treat mild to moderate pain and fever during pregnancy, while intravenous (IV) acetaminophen serves as an analgesic in the immediate postoperative period, e.g., after cesarean delivery [8]. Acetaminophen is eliminated through various metabolic pathways [9]. In non-pregnant healthy women, it is mainly metabolized to acetaminophen glucuronide via different uridine 5′-diphospho-glucuronosyltransferase (UGT) isoforms and to acetaminophen sulfate through sulfotransferases (SULT). In addition, it is metabolized by several cytochrome P450 (CYP) enzymes, primarily by CYP2E1, to the reactive (and therefore potentially toxic) metabolite N-acetyl-p-benzoquinone imine (NAPQI), and a small fraction is also excreted as unchanged drug in urine [10]. Under normal conditions, NAPQI is immediately detoxified by conjugation with glutathione. The glutathione conjugate is further metabolized to cysteine and mercapturate derivatives of acetaminophen [9]. At high doses, however, glutathione becomes depleted and NAPQI binds covalently to cellular proteins, thereby forming toxic protein adducts. This leads to mitochondrial dysfunction and early oxidative stress, which can ultimately result in hepatocellular necrosis [10, 11].
Due to limited data on acetaminophen pharmacokinetics in pregnant women, the main objective of this study was to develop a pregnancy PBPK model that can be used to (1) predict maternal pharmacokinetics of acetaminophen and its metabolites in pregnant women at delivery; and (2) extrapolate the pharmacokinetics of acetaminophen and its metabolites to earlier stages of pregnancy in order to compare the acetaminophen average concentration at steady state (Css,avg) and NAPQI formation, as a marker for efficacy and potential hepatotoxicity, respectively, to those in non-pregnant women.
2 Materials and Methods
2.1 Software
All PBPK models were built in PK-Sim® and MoBi® version 7.2.0 software, both part of the Open Systems Pharmacology Suite. All source codes and the herein developed models are publicly available on GitHub (accessible via http://www.open-systems-pharmacology.org). Reference PBPK models for adults were developed in PK-Sim®. The pregnancy PBPK models were built in MoBi® and subsequently exported to PK-Sim® for population simulations. WebPlotDigitizer (http://automeris.io/WebPlotDigitizer/) was used to extract data from published figures and digitize them. R (version 3.3.0) was used together with RStudio (version 1.1.453) for graphics creation and statistical analysis.
2.2 General Workflow
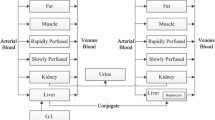
The workflow for constructing and translating a PBPK model to pregnant women (Fig. 1) has recently been described [12] and is summarized here, as it is relevant to this analysis. PBPK models for acetaminophen were developed for a non-pregnant population for IV administration and then, keeping distribution- and clearance-related parameters unchanged, for oral administration [13]. The demographic measures of the virtual subjects matched those of the in vivo study group, if the latter were reported. The non-pregnant PBPK model incorporated the PK-Sim® standard model structure comprising 18 compartments [14]. The model was evaluated by comparing the pharmacokinetic simulation with observed in vivo pharmacokinetic data taken from literature. If needed, drug-specific parameters were refined in the non-pregnant model by fitting the simulated plasma concentration–time curve to the observed data using Monte Carlo algorithm implemented in the software’s Parameter Identification toolbox. Thereafter, all drug-specific parameters, except the fraction unbound (fu), were fixed and the model was extrapolated to pregnant women by substituting the standard model structure with the pregnancy model structure, which includes nine additional compartments [2, 12]. Pharmacokinetic predictions were performed in a population of pregnant women that matched anthropometric measures of the in vivo study group of pregnant women. If not reported, the mean gestational age-specific demographic measures available in PK-Sim® were used. Finally, pharmacokinetic predictions were evaluated by comparing the results with in vivo pharmacokinetic data taken from literature.

Schematic workflow of pregnancy physiologically based pharmacokinetic model development and validation. Css,avg average concentration at steady state, IV intravenous, NAPQI N-acetyl-p-benzoquinone imine, PBPK physiologically based pharmacokinetic, phys-chem physicochemical, PK pharmacokinetic
2.3 Development of Physiologically Based Pharmacokinetic (PBPK) Models for Acetaminophen
A detailed overview of the development of PBPK models for acetaminophen is provided in the Electronic Supplementary Material (ESM_1). Input data used in the PBPK models are listed in Table 1. Briefly, acetaminophen was loaded from a template provided with the software that also includes a parameterization of the main metabolic pathways of both acetaminophen and its main metabolites. Of note, due to missing literature information, it was assumed in this template that the concentration of cysteine and mercapturate is equivalent to that of NAPQI [14]. Characteristics of comparison studies providing in vivo pharmacokinetic data are detailed in Table 2.
2.4 Enzymatic Changes During Pregnancy
The focus of this study was to predict maternal disposition of acetaminophen metabolized via UGT1A1, SULT1A1, and CYP2E1 and to quantify the fractional (molar acetaminophen) clearance via each of these pathways. Therefore, the literature was screened for quantitative information on pregnancy-related changes in the expression and activity of these enzymes.
2.4.1 Uridine 5′-Diphospho-Glucuronosyltransferase (UGT) 1A1
Literature data that allow informing the PBPK model with changes in UGT1A1 expression during human pregnancy are scarce. The approach used here relied on in vivo serum concentrations of bilirubin, an exclusive substrate of UGT1A1 [15]. In a cohort of 103 healthy pregnant women, the mean serum concentrations of free bilirubin were 25%, 43%, and 48% lower in the first, second, and third trimester, respectively, than the mean value in non-pregnant women [16]. From these figures, the fold increases in bilirubin clearance, a surrogate for UGT1A1 induction, were estimated to be 1.33, 1.75, and 1.92 in the first, second, and third trimester, respectively.
2.4.2 Sulfotransferase (SULT) 1A1
In human endometrial tissue, SULT1A1 activity in early pregnancy (10 weeks of gestational age), as assessed by the clearance of probe drugs, did not differ from that in non-pregnant subjects. Furthermore, expression of SULT1A1 was unaffected by progesterone [17]. In rat experiments, hepatic SULT1A1 expression was not substantially influenced by estradiol or progesterone [18]. Thus, no change in SULT1A1 expression throughout pregnancy was assumed.
2.4.3 Cytochrome P450 (CYP) 2E1
Due to conflicting data [19, 20] (see Sect. 4), it was not possible to quantitatively estimate pregnancy-induced changes in CYP2E1 with high certainty. Studying peripartum women 5 min after delivery, Kulo et al. [21] found a 1.8-fold higher clearance of acetaminophen to oxidative metabolites than in 12 weeks postpartum women; this finding was supported by Miners et al. [22]. As CYP2E1 is the major enzyme involved in acetaminophen oxidation, and since this study aims to predict exposure to NAPQI with as little uncertainty as possible, the value reported by Kulo et al. [21] was used here [23]. For earlier stages of pregnancy, the same increase was assumed. Although at earlier stages of pregnancy a lower value may be physiologically plausible, an 80% induction was used here because this value translates into the maximum NAPQI formation clearance that can probably be expected in vivo and thus represents the upper range of potential NAPQI formation during pregnancy (‘worst-case scenario’).
2.5 Statistical Analysis
The pharmacokinetics of acetaminophen were simulated in a population of 2000 virtual individuals following an oral administration dosing regimen of 1000 mg every 6 h for 48 h. The molar dose fractions of acetaminophen undergoing metabolism to its metabolites (acetaminophen glucuronide, acetaminophen sulfate, and NAPQI) were calculated. In addition, for each simulated or predicted concentration–time profile, the molar area under the plasma concentration–time curve (AUC) of acetaminophen, acetaminophen glucuronide, acetaminophen sulfate, and NAPQI was calculated. After this, the ratio of the AUC of the metabolite to that of acetaminophen (AUC ratio) was calculated. The molar dose fraction of acetaminophen converted to NAPQI as well as the molar AUC ratio of NAPQI/acetaminophen were used as surrogate markers for potential hepatotoxicity. Based on the AUC of acetaminophen, the Css,avg was calculated, in both non-pregnant and pregnant women, as Css,avg is a widely used measurement to assess acetaminophen efficacy [24].
2.6 Model Evaluation
Simulated and predicted pharmacokinetic profiles were compared with the observed in vivo data (Table 2). Additionally, ratios of predicted to observed mean pharmacokinetic parameters were calculated. Finally, pharmacokinetic simulations and predictions were evaluated with the use of goodness-of-fit plots, depicting observed versus simulated or predicted plasma concentrations. A sensitivity analysis was performed on CYP2E1 expression, ranging from 0 to 160% induction, to evaluate the impact of different CYP2E1 expression patterns on the pharmacokinetics of acetaminophen and NAPQI during the first trimester of pregnancy. A sensitivity analysis on CYP2E1 expression in the first trimester was performed as highest uncertainty was assumed in the model during this trimester due to an absolute lack of CYP2E1 expression patterns.
3 Results
3.1 PBPK Models for Acetaminophen
The plasma concentration–time profile of acetaminophen simulated in populations of non-pregnant women was in good agreement with in vivo pharmacokinetic data (Figs. 2a, 3a). All simulated mean plasma concentrations of acetaminophen after both oral and IV administration fell within a 2-fold error range (Fig. 4). In vivo urine data after IV administration in non-pregnant women were available for acetaminophen glucuronide, acetaminophen sulfate, and unchanged acetaminophen and are depicted in Fig. 3a. This resulted in adequate predictions of acetaminophen excreted unchanged in urine (Figs. 3a, 4). For the two major metabolites, glucuronide and sulfate, the model slightly underestimated urine concentrations (Figs. 3a, 4). Due to lack of NAPQI in vivo data, simulations of NAPQI plasma concentrations could not be evaluated. As indicated in Table 3, all simulated pharmacokinetic parameters in non-pregnant women fell well within a 1.5-fold error range. Ratios of simulated to observed pharmacokinetic parameters were not estimated for the metabolites in non-pregnant women after IV administration, as only one sample per patient was available. Overall, these PBPK models were deemed adequate and used for translation to pregnancy.

Median predicted (purple) and observed [33] (white dots) acetaminophen plasma concentration–time profiles in populations of a non-pregnant women and b first trimester pregnant women after administration of oral acetaminophen 650 mg. The shaded area denotes the predicted 95% confidence interval. The error bars denote the standard deviations of the observed data. NAPQI N-acetyl-p-benzoquinone imine

Median predicted and observed [31] acetaminophen plasma concentration–time profiles (left) and predicted and observed [31] acetaminophen inactive metabolites urine concentration–time profiles (right) in populations of a non-pregnant women after administration of intravenous acetaminophen 2000 mg and b third trimester pregnant women after administration of an intravenous acetaminophen 2000 mg loading dose followed by 1000 mg every 6 h. For the inactive metabolites, orange, pink, and green indicate acetaminophen glucuronide, acetaminophen sulfate, and unchanged acetaminophen in urine concentrations, respectively. The shaded area denotes the predicted 95% confidence interval. NAPQI N-acetyl-p-benzoquinone imine

Goodness-of-fit plot (predicted concentration versus observed concentration) for acetaminophen and its metabolites (acetaminophen unchanged, acetaminophen glucuronide, acetaminophen sulfate). For non-pregnant women, observed acetaminophen data were taken from Allegaert et al. [31], Mitchell et al. [34], and Beaulac-Baillargeon and Rocheleau [33]; for pregnant women, observed acetaminophen, acetaminophen glucuronide, and acetaminophen sulfate data were taken from Allegaert et al. [31] and Beaulac-Baillargeon and Rocheleau [33]. The solid line denotes the line of identity. The dotted lines denote the 2-fold error range. APAP acetaminophen, conc concentration, GLUC glucuronide, NP non-pregnant, P1 first trimester, P3 third trimester, SULF sulfate, U unchanged
After translation to pregnancy, the predicted plasma concentration–time profile of IV-administered acetaminophen in populations of pregnant women at delivery was in good agreement with the observed data (Fig. 3b). In vivo urine data at delivery were available for acetaminophen glucuronide, acetaminophen sulfate, and unchanged acetaminophen and are depicted in Fig. 3b. Concerning acetaminophen sulfate and unchanged acetaminophen in urine at delivery, one predicted mean plasma concentration was within the 2-fold error range, while the other one was outside. As for acetaminophen glucuronide, renal excretion appeared to be generally over predicted. Thereafter, the model was extrapolated to earlier stages of pregnancy. Figure 2b shows a good agreement between the predicted plasma concentration–time profile of acetaminophen in the first trimester of pregnancy after oral administration and the observed pharmacokinetic data. All simulated mean plasma concentrations of acetaminophen after both oral and IV administration fell within a 2-fold error range (Figs. 2, 3b, 4). Again due to a lack of NAPQI in vivo data, predictions of NAPQI plasma concentrations could not be evaluated. As indicated in Table 3, all predicted pharmacokinetic parameters of acetaminophen throughout pregnancy fell well within a 1.5-fold error range. The ratio of predicted to observed pharmacokinetic parameters was not estimated for the metabolites in pregnant women at term after IV administration, since only two samples per patient were available.
Regarding the plasma concentration–time profiles of oral acetaminophen 1000 mg in non-pregnant and pregnant populations (ESM_2A), maximum concentration (Cmax) values were higher in non-pregnant women and decreased with increasing duration of gestational week, while the AUC from time zero to 6 h (AUC0-6h) decreased with increasing duration of gestational age. Regarding the plasma concentration–time profiles of NAPQI after administration of oral acetaminophen 1000 mg in both the non-pregnant and pregnant women (ESM_2B), the highest Cmax and AUC0-6h were predicted during the first trimester of pregnancy.
The sensitivity analysis (ESM_3) revealed that changes in CYP2E1 expression substantially impact the simulated NAPQI plasma concentration, while the effect on the acetaminophen concentration is insignificant.
3.1.1 Acetaminophen Average Concentration at Steady State and Acetyl-p-Benzoquinone Imine (NAPQI) Exposure
Following oral administration of acetaminophen 1000 mg, the predicted Css,avg (median [interquartile range, IQR]) was 6.7 [5.9–7.4], 5.6 [4.6–6.3], 4.9 [4.1–5.5], and 4.5 [3.8–5.1] mg/L in non-pregnant women and in the first, second, and third trimester of pregnancy, respectively (Fig. 5a).

a Boxplot of the simulated average concentration at steady state (Css,avg; y-axis) values for a dose of acetaminophen 1000 mg administered orally to a non-pregnant and different pregnant populations discretized in trimesters (n = 2000). The box is the interquartile range (IQR) representing the range between the 25th and 75th percentile. The whiskers indicate 1.5 times the IQR. The line within the boxes indicates the generally accepted analgesic target concentration of 10 mg/L [24]. b Barplot of the predicted median and IQR fractions of metabolite formation to acetaminophen (expressed as percentage of molar acetaminophen dose) for non-pregnant and different pregnant populations discretized in trimesters (n = 2000). NAPQI N-acetyl-p-benzoquinone imine
Both the fraction (expressed as fraction of molar dose) of acetaminophen metabolized to NAPQI and the molar AUC ratio NAPQI/acetaminophen were used as measures for hepatotoxicity. The fractions and IQRs of acetaminophen converted to its metabolites, expressed on a molar basis, are illustrated in Fig. 5b. The dose fraction of acetaminophen metabolized to glucuronide was considerably increased in the second and third trimester (Table 4). On the basis of a ‘worse-case scenario’ (80% constant increase) pattern during pregnancy, the results suggest that the molar dose fraction of acetaminophen converted to NAPQI was highest during the first (median [IQR]: 11.0% [9.1–13.4%]) followed by the second (9.0% [7.5–11.0%]) and third trimester (8.2% [6.8–10.1%]), compared with non-pregnant women (7.7% [6.4–9.4%]), although a great inter-individual variability was observed, resulting in overlapping IQR between non-pregnant and pregnant women (Table 4).
Molar AUC ratios of acetaminophen metabolites (glucuronide, sulfate, and NAPQI) to acetaminophen are presented in Fig. 6. The median AUC ratio of acetaminophen sulfate to acetaminophen was lower in pregnancy than in non-pregnant women, with its nadir in the second trimester. The median AUC ratio of glucuronide to acetaminophen increased with advancing gestation. The median AUC ratio of NAPQI was highest during the first trimester followed by the second and third trimester. These findings suggest higher NAPQI formation in pregnant women than in non-pregnant women, especially during the first trimester.

Predicted area under the plasma concentration–time curve (AUC) from time zero to 6 h (AUC0-6h) metabolite to acetaminophen ratios of N-acetyl-p-benzoquinone imine (NAPQI) (a), acetaminophen sulfate (b), and acetaminophen glucuronide (c) in non-pregnant women and women at the first, second, and third trimester of pregnancy
4 Discussion
The many pregnancy PBPK models developed to assess the impact of pregnancy on pharmacokinetics have mainly focused on drugs that are eliminated via unchanged renal excretion or metabolism by phase I (CYP) pathways. Little attention has been paid to drugs metabolized by phase II enzymes. Although acetaminophen is one of the most frequently used drugs throughout pregnancy, little is known about the achieved Css,avg and the potential risk of hepatotoxicity at different stages of pregnancy at therapeutic dosages.
The developed pregnancy PBPK models could adequately predict the pharmacokinetics of acetaminophen throughout pregnancy. Based on these developed PBPK models, it was illustrated that following therapeutic dosage of 1000 mg of acetaminophen, the lowest Css,avg values were achieved in the third trimester (median [IQR]: 4.5 [3.8–5.1] mg/L), followed by the second trimester, first trimester, and non-pregnant state with Css,avg levels of 4.9 [4.1–5.5], 5.6 [4.6–6.3], and 6.7 [5.9–7.4] mg/L, respectively. Conversely, the molar dose fraction of acetaminophen converted to NAPQI was highest during the first (median [IQR]: 11.0% [9.1–13.4%]), followed by the second (9.0% [7.5–11.0%]) and third trimester (8.2% [6.8–10.1%]), compared with that simulated in non-pregnant women (7.7% [6.4–9.4%]). However, it has to be noted that a great inter-individual variability was observed, resulting in an overlapping IQR of NAPQI between non-pregnant and pregnant women (Table 4). The molar AUC ratio NAPQI/acetaminophen was highest during the first trimester and decreased in the second and third trimester. This decrease in NAPQI production can be explained by the fact that, as UGT1A1 activity increases, a higher molar fraction of acetaminophen is being glucuronidated, leaving less substrate available for oxidation via CYP2E1 (assuming a ‘worst-case scenario’ with an 80% constant increase). Our findings suggest that acetaminophen exposure is likely to be lower in pregnant than in non-pregnant women, and is related to pregnancy duration. Despite these findings, dose adjustments cannot be advised yet, as toxicity related to more NAPQI production during pregnancy compared with non-pregnant women may occur. Information on glutathione abundance during pregnancy and NAPQI in vivo data are required to further refine the presented model. Quantitative information on gestation-specific changes in enzyme activity available in literature was incorporated in the model. The main caveat of such a knowledge-driven approach is the available amount and quality of the information in the scientific literature. If these are insufficient, biologically plausible assumptions must be made. Previously, well-validated PBPK models have been developed of acetaminophen in other special populations [25,26,27]. Although these models could be used to further validate the predictions of both acetaminophen and its metabolites throughout different stages of pregnancy once adequate clinical data are available, those models were not used here as, for example, it was unknown whether pregnancy itself changes the levels of the co-factors for acetaminophen glucuronidation and sulfation. Sparse information was available on pregnancy-induced changes in UGT1A1 activity, which hampers an accurate and continuous description of these changes in the pregnancy PBPK model. Therefore, these changes in the presented pregnancy PBPK model were informed by reported reductions in the mean serum concentrations of free bilirubin in the first, second, and third trimester of pregnancy [16]. While bilirubin is an exclusive substrate of UGT1A1 [15], the mean serum concentration may not accurately represent underlying changes in clearance. For example, it is unknown if bilirubin production during pregnancy is different. Therefore, it has not been taken into account. Interestingly, UGT1A1-mediated clearance was slightly overestimated in the presented PBPK models, suggesting that factors other than an increase in clearance, at least partly, may contribute to the observed reductions in free bilirubin serum concentrations. One potential factor is the metabolism of acetaminophen through UGTs other than only UGT1A1 (as implemented in this PBPK model). However, limited evidence is available on which other UGTs contribute to acetaminophen metabolism and the fractions metabolized via different UGTs. While UGT1A6 and UGT1A9 have been suggested to play an important role in acetaminophen metabolism [28, 29], no quantitative information can currently be generated from available data. Moreover, high inter-individual variability in acetaminophen glucuronidation, partly caused by polymorphism, may hamper the prediction of metabolism in humans. Finally, discrete changes do not capture the full dynamics of changes in biological systems (such as the increase in gene expression) and a continuous description of UGT1A1 activity changes during pregnancy may be more accurate. Yet, a clear lack of data prevented a finer resolution of UGT1A1 activity changes. Additionally, it should also be assessed whether upregulation of UGT1A1 is tissue specific and how that translates into in vivo pharmacokinetics. Further in vitro or in vivo studies addressing this issue are clearly needed.
Although a very small dose fraction of acetaminophen is eliminated by CYP2E1, accurate characterization of this pathway is crucial since it is involved in hepatotoxicity. However, conflicting and limited data are available on changes in CYP2E1 enzyme expression throughout human pregnancy. Lee et al. [19] reported that CYP2E1 gene expression in female hepatocytes is induced by human placental lactogen (hPL; human chorionic somatomammotropin). Plasma concentrations of hPL increase progressively during pregnancy, with Cmax values of 5–7 mg/L being reached near term. The CYP2E1 induction by hPL was concentration dependent, yet the hPL concentration exhibiting 50% of the maximal induction could not be estimated because the effect did not reach a plateau [19]. To determine whether the increased CYP2E1 messenger RNA (mRNA) levels led to a corresponding increase in protein expression in human hepatocytes, enzymatic activity protein and metabolic activity of CYP2E1 were analyzed by using western blot and the CYP2E1 probe drug chlorzoxazone, respectively. The formation of 6-hydroxychlorzoxazone was increased significantly in the majority of hepatocytes pretreated with hPL. Taken together, these results indicate that, at physiological concentrations reached during pregnancy, hPL induces CYP2E1 expression [19]. In another study, medroxyprogesterone acetate increased CYP2E1 activity [20]. The activity increase was concentration dependent in the range of 0–100 µmol/L; the highest level amounted to 170.8% of the control level observed at 50 µmol/L [20]. While it is difficult to extrapolate these in vitro findings to a quantitative CYP2E1 activity change in vivo, the 80% increase in CYP2E1 activity reported in term pregnant women was used in the herein presented pregnancy PBPK models [21, 22]. Furthermore, although a lower value may be plausible at earlier stages of pregnancy, an 80% induction was used because this value translates into the maximum of NAPQI exposure that can probably be expected in vivo, if a monotonous increase in CYP2E1 activity during pregnancy is assumed, thereby constituting a ‘worst-case scenario’. Additional scenarios of different CYP2E1 induction patterns in the first trimester have been assessed in a sensitivity analysis given in the Electronic Supplementary Material. As the objective of the present study was to address maternal exposure to acetaminophen and its metabolites, fetal NAPQI exposure was not modeled here, which is clearly of great interest. Although technically feasible, such consideration is out of the scope and should be further investigated.
Due to missing literature information on the glutathione-S-transferase enzyme, it was assumed that glutathione tissue concentrations in pregnant women are similar to those in non-pregnant subjects. However, if glutathione-S-transferase activities are lower in pregnant women, NAPQI concentrations can be expected to be even higher. If this is not explicitly taken into consideration, the model presented here would predict a too low increase in the generated NAPQI amount, bearing the potential of falsely inferring that there is no substantially increased risk of hepatotoxicity. While the incorporation of explicit NAPQI detoxification kinetics and changes in glutathione-S-transferase expression are beyond the scope of this study, they should be carefully examined before any dose recommendations are inferred from the model predictions.
The pregnancy PBPK models were used to investigate acetaminophen pharmacokinetics throughout pregnancy after a therapeutic dosage (1000 mg). The Css,avg of acetaminophen and NAPQI exposure, was a marker for efficacy and potential hepatotoxicity, respectively, to non-pregnant women. In this study, Css,avg was chosen as it is a widely used measure to express the efficacy of acetaminophen [24]. It should be noted that the Css,avg (i.e., a pharmacokinetic target) might not be completely the same as effective analgesia (pharmacodynamic). It is unknown if the pharmacodynamic target changes throughout pregnancy. Using the pharmacokinetic target as a meaningful measure, the results revealed that throughout all stages, for both non-pregnant as well as pregnant women, Css,avg is not reached, with the lowest value during the third trimester. The question arises if this is the correct target concentration for non-pregnant adults, as in older people (whose volume of distribution and clearance are lower than those in young healthy adults) a target concentration of 10 mg/L is reached [30]. Further research should therefore focus on which Css,avg should be achieved for effective pain relief throughout different stages of pregnancy, taking safety into account. As emphasized before, investigations into NAPQI formation during different stages of pregnancy need to be combined with an assessment of the changes of the detoxifying capacity (e.g., glutathione levels) of non-pregnant and pregnant women to determine the optimal dose of acetaminophen throughout these different stages of pregnancy. Future research should not only focus on in vitro and in vivo studies related to UGT and CYP2E1 expression throughout pregnancy, but also on qualification of these model predictions in relation to NAPQI/oxidative metabolite concentrations.
5 Conclusion
The developed pregnancy PBPK models successfully predicted pharmacokinetics of paracetamol and its metabolites at different stages of pregnancy. Predictions resulted in the lowest Css,avg in the third trimester, followed by increased number of weeks of gestational age and non-pregnant state. Conversely, the molar dose fraction of acetaminophen metabolized to NAPQI was highest during the first trimester, followed by the second trimester, third trimester, and non-pregnant women. AUC ratio NAPQI/acetaminophen was highest during the first, followed by the second and third trimester. Although it has to be noted that a great inter-individual variability was observed, resulting in an overlapping IQR of NAPQI between non-pregnant and pregnant women, these findings suggest that acetaminophen exposure is, on average, likely to be lower in pregnant than in non-pregnant women and that this reduction may be related to pregnancy duration. Despite these findings, dose adjustments cannot be advised yet, as toxicity related to more NAPQI production during pregnancy than in non-pregnant women may occur. Information on glutathione abundance during pregnancy and NAPQI in vivo data are required to further refine the presented model.
References
Pariente G, Leibson T, Carls A, Adams-Webber T, Ito S, Koren G. Pregnancy-associated changes in pharmacokinetics: a systematic review. PLoS Med. 2016;13(11):e1002160. https://doi.org/10.1371/journal.pmed.1002160.
Dallmann A, Ince I, Solodenko J, Meyer M, Willmann S, Eissing T, et al. Physiologically based pharmacokinetic modeling of renally cleared drugs in pregnant women. Clin Pharmacokinet. 2017;56(12):1525–41. https://doi.org/10.1007/s40262-017-0538-0.
Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br J Clin Pharmacol. 2014;77(3):554–70. https://doi.org/10.1111/bcp.12207.
Jogiraju VK, Avvari S, Gollen R, Taft DR. Application of physiologically based pharmacokinetic modeling to predict drug disposition in pregnant populations. Biopharm Drug Dispos. 2017;38(7):426–38. https://doi.org/10.1002/bdd.2081.
Dallmann A, Pfister M, van den Anker J, Eissing T. Physiologically based pharmacokinetic modeling in pregnancy: a systematic review of published models. Clin Pharmacol Ther. 2018;104(6):1110–24. https://doi.org/10.1002/cpt.1084.
Gerlowski LE, Jain RK. Physiologically based pharmacokinetic modeling: principles and applications. J Pharm Sci. 1983;72(10):1103–27.
Allegaert K, van den Anker JN. Perinatal and neonatal use of paracetamol for pain relief. Semin Fetal Neonatal Med. 2017;22(5):308–13. https://doi.org/10.1016/j.siny.2017.07.006.
Kulo A, Van Calsteren K, Van De Velde M, Mulabegovic N, Verbesselt R, De Hoon JN, et al. Weight, pregnancy and oral contraceptives affect intravenous paracetamol clearance in young women. Eur Rev Med Pharmacol Sci. 2014;18(5):599–604.
Flint RB, Mian P, van der Nagel B, Slijkhuis N, Koch BC. Quantification of acetaminophen and its metabolites in plasma using UPLC-MS: doors open to therapeutic drug monitoring in special patient populations. Ther Drug Monit. 2017;39(2):164–71. https://doi.org/10.1097/ftd.0000000000000379.
Prescott LF. Kinetics and metabolism of paracetamol and phenacetin. Br J Clin Pharmacol. 1980;10(Suppl 2):291S–8S.
Rumack BH. Acetaminophen hepatotoxicity: the first 35 years. J Toxicol Clin Toxicol. 2002;40(1):3–20.
Dallmann A, Solodenko J, Ince I, Eissing T. Applied concepts in PBPK modeling: how to extend an open systems pharmacology model to the special population of pregnant women. CPT Pharmacomet Syst Pharmacol. 2018;7(7):419–31. https://doi.org/10.1002/psp4.12300.
Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacomet Syst Pharmacol. 2016;5(10):516–31. https://doi.org/10.1002/psp4.12134.
Krauss M, Schaller S, Borchers S, Findeisen R, Lippert J, Kuepfer L. Integrating cellular metabolism into a multiscale whole-body model. PLoS Comput Biol. 2012;8(10):e1002750. https://doi.org/10.1371/journal.pcbi.1002750.
Bosma PJ, Seppen J, Goldhoorn B, Bakker C, Oude Elferink RP, Chowdhury JR, et al. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem. 1994;269(27):17960–4.
Bacq Y, Zarka O, Brechot JF, Mariotte N, Vol S, Tichet J, et al. Liver function tests in normal pregnancy: a prospective study of 103 pregnant women and 103 matched controls. Hepatology. 1996;23(5):1030–4. https://doi.org/10.1002/hep.510230514.
Rubin GL, Harrold AJ, Mills JA, Falany CN, Coughtrie MW. Regulation of sulphotransferase expression in the endometrium during the menstrual cycle, by oral contraceptives and during early pregnancy. Mol Hum Reprod. 1999;5(11):995–1002.
Klaassen CD, Liu L, Dunn RT 2nd. Regulation of sulfotransferase mRNA expression in male and female rats of various ages. Chem Biol Interact. 1998;109(1–3):299–313.
Lee JK, Chung HJ, Fischer L, Fischer J, Gonzalez FJ, Jeong H. Human placental lactogen induces CYP2E1 expression via PI 3-kinase pathway in female human hepatocytes. Drug Metab Dispos. 2014;42(4):492–9. https://doi.org/10.1124/dmd.113.055384.
Zhang JW, Liu Y, Li W, Hao DC, Yang L. Inhibitory effect of medroxyprogesterone acetate on human liver cytochrome P450 enzymes. Eur J Clin Pharmacol. 2006;62(7):497–502. https://doi.org/10.1007/s00228-006-0128-9.
Kulo A, Peeters MY, Allegaert K, Smits A, de Hoon J, Verbesselt R, et al. Pharmacokinetics of paracetamol and its metabolites in women at delivery and post-partum. Br J Clin Pharmacol. 2013;75(3):850–60. https://doi.org/10.1111/j.1365-2125.2012.04402.x.
Miners JO, Robson RA, Birkett DJ. Paracetamol metabolism in pregnancy. Br J Clin Pharmacol. 1986;22(3):359–62.
Dallmann A, Ince I, Coboeken K, Eissing T, Hempel G. A physiologically based pharmacokinetic model for pregnant women to predict the pharmacokinetics of drugs metabolized via several enzymatic pathways. Clin Pharmacokinet. 2018;57(6):749–68. https://doi.org/10.1007/s40262-017-0594-5.
Gibb IA, Anderson BJ. Paracetamol (acetaminophen) pharmacodynamics: interpreting the plasma concentration. Arch Dis Child. 2008;93(3):241–7. https://doi.org/10.1136/adc.2007.126896.
Zurlinden TJ, Reisfeld B. Physiologically based modeling of the pharmacokinetics of acetaminophen and its major metabolites in humans using a Bayesian population approach. Eur J Drug Metab Pharmacokinet. 2016;41(3):267–80. https://doi.org/10.1007/s13318-015-0253-x.
Howell BA, Yang Y, Kumar R, Woodhead JL, Harrill AH, Clewell HJ 3rd, et al. In vitro to in vivo extrapolation and species response comparisons for drug-induced liver injury (DILI) using DILIsym: a mechanistic, mathematical model of DILI. J Pharmacokinet Pharmacodyn. 2012;39(5):527–41. https://doi.org/10.1007/s10928-012-9266-0.
Zurlinden TJ, Heard K, Reisfeld B. A novel approach for estimating ingested dose associated with paracetamol overdose. Br J Clin Pharmacol. 2016;81(4):634–45. https://doi.org/10.1111/bcp.12796.
Court MH, Freytsis M, Wang X, Peter I, Guillemette C, Hazarika S, et al. The UDP-glucuronosyltransferase (UGT) 1A polymorphism c.2042C>G (rs8330) is associated with increased human liver acetaminophen glucuronidation, increased UGT1A exon 5a/5b splice variant mRNA ratio, and decreased risk of unintentional acetaminophen-induced acute liver failure. J Pharmacol Exp Ther. 2013;345(2):297–307.
Court MH, Duan SX, von Moltke LL, Greenblatt DJ, Patten CJ, Miners JO, et al. Interindividual variability in acetaminophen glucuronidation by human liver microsomes: identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J Pharmacol Exp Ther. 2001;299(3):998–1006.
Mian P, van Esdonk MJ, Olkkola KT, de Winter BCM, Liukas A, Spriet I, et al. Population pharmacokinetic modelling of intravenous paracetamol in fit older people displays extensive unexplained variability. Br J Clin Pharmacol. 2019;85(1):126–35. https://doi.org/10.1111/bcp.13770.
Allegaert K, Peeters MY, Beleyn B, Smits A, Kulo A, van Calsteren K, et al. Paracetamol pharmacokinetics and metabolism in young women. BMC Anesthesiol. 2015;15(1):163. https://doi.org/10.1186/s12871-015-0144-3.
Rayburn W, Shukla U, Stetson P, Piehl E. Acetaminophen pharmacokinetics: comparison between pregnant and nonpregnant women. Am J Obstet Gynecol. 1986;155(6):1353–6.
Beaulac-Baillargeon L, Rocheleau S. Paracetamol pharmacokinetics during the first trimester. Eur J Clin Pharmacol. 1994;46(5):451–4.
Mitchell MC, Hanew T, Meredith CG, Schenker S. Effects of oral contraceptive steroids on acetaminophen metabolism and elimination. Clin Pharmacol Ther. 1983;34(1):48–53.
Shinoda S, Aoyama T, Aoyama Y, Tomioka S, Matsumoto Y, Ohe Y. Pharmacokinetics/pharmacodynamics of acetaminophen analgesia in Japanese patients with chronic pain. Biol Pharm Bull. 2007;30(1):157–61. https://doi.org/10.1248/bpb.30.157.
Liukas A, Kuusniemi K, Aantaa R, Virolainen P, Niemi M, Neuvonen PJ, et al. Pharmacokinetics of intravenous paracetamol in elderly patients. Clin Pharmacokinet. 2011;50(2):121–9.
Prescott LF, Speirs GC, Critchley JAJH, Temple RM, Winney RJ. Paracetamol disposition and metabolite kinetics in patients with chronic renal failure. Eur J Clin Pharmacol. 1989;36(3):291–7. https://doi.org/10.1007/bf00558162.
Beaulac-Baillargeon L, Rocheleau S. Paracetamol pharmacokinetics in pregnancy. Drug Investig. 1993;6(3):176–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Paola Mian is supported by the Sophia Stichting Wetenschappelijk Onderzoek (SSWO; S16-08), received a Short Term Minor (STM-2017) Grant from the Stichting Sophia Kinderziekenhuis Fonds to conduct this research, and received a travel grant from Erasmus Trustfonds and the Dr. Catharine van Tussenbroek, Mevr. Speleers Pharmacy Grant.
Conflict of interest
Marc Pfister is a part-time consultant for Certara; this consulting work is not related to this study. Paola Mian, John N. van den Anker, Kristel van Calsteren, Pieter Annaert, Dick Tibboel, Karel Allegaert have no conflicts of interest to declare. André Dallmann is an employee of Bayer AG, a company which is part of the Open Systems Pharmacology (OSP) member team and hence involved in OSP software development.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mian, P., van den Anker, J.N., van Calsteren, K. et al. Physiologically Based Pharmacokinetic Modeling to Characterize Acetaminophen Pharmacokinetics and N-Acetyl-p-Benzoquinone Imine (NAPQI) Formation in Non-Pregnant and Pregnant Women. Clin Pharmacokinet 59, 97–110 (2020). https://doi.org/10.1007/s40262-019-00799-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00799-5