
Abstract
Introduction
Neglected tropical diseases (NTDs) affect more than one billion people, mainly living in developing countries. For most of these NTDs, treatment is suboptimal. To optimize treatment regimens, clinical pharmacokinetic studies are required where they have not been previously conducted to enable the use of pharmacometric modeling and simulation techniques in their application, which can provide substantial advantages.
Objectives
Our aim was to provide a systematic overview and summary of all clinical pharmacokinetic studies in NTDs and to assess the use of pharmacometrics in these studies, as well as to identify which of the NTDs or which treatments have not been sufficiently studied.
Methods
PubMed was systematically searched for all clinical trials and case reports until the end of 2015 that described the pharmacokinetics of a drug in the context of treating any of the NTDs in patients or healthy volunteers.
Results
Eighty-two pharmacokinetic studies were identified. Most studies included small patient numbers (only five studies included >50 subjects) and only nine (11 %) studies included pediatric patients. A large part of the studies was not very recent; 56 % of studies were published before 2000. Most studies applied non-compartmental analysis methods for pharmacokinetic analysis (62 %). Twelve studies used population-based compartmental analysis (15 %) and eight (10 %) additionally performed simulations or extrapolation. For ten out of the 17 NTDs, none or only very few pharmacokinetic studies could be identified.
Conclusions
For most NTDs, adequate pharmacokinetic studies are lacking and population-based modeling and simulation techniques have not generally been applied. Pharmacokinetic clinical trials that enable population pharmacokinetic modeling are needed to make better use of the available data. Simulation-based studies should be employed to enable the design of improved dosing regimens and more optimally use the limited resources to effectively provide therapy in this neglected area.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Neglected tropical diseases affect a major part of the global population, but treatments have generally not been optimized. |
We provide a comprehensive systematic overview of performed pharmacokinetic studies in all 17 neglected tropical diseases, advantages and drawbacks of different methodologies, and gaps in pharmacokinetic research through which neglected tropical diseases therapeutics can be further improved. |
For most neglected tropical diseases, adequate pharmacokinetic studies were found lacking or completely absent, pediatric patients have largely been ignored, and population-based modeling and simulation techniques have not generally been applied. |
To more optimally use the limited available resources in this neglected area, more emphasis should be given to simulation-based pharmacokinetic studies enabling the design of improved dosing regimens. |
1 Introduction
Neglected tropical diseases (NTDs) represent a wide range of infectious afflictions, which are prevalent mostly in tropical and subtropical countries and have one common characteristic: they all affect people living in deep poverty. All NTDs are heavily debilitating, causing life-long disability, which can be directly fatal if left untreated. At the moment, over 1.4 billion people are affected by at least one NTD, and they are the cause of death for over 500,000 people annually [1, 2]. There are currently 17 NTDs as defined by the World Health Organization (WHO), which include protozoal, bacterial, helminth, and viral infections [1]. An overview of their transmission, geography, and burden of disease is provided in Table 1. Collectively, the NTDs belong to the most devastating of communicable diseases, not only in terms of global health burden (26.1 million disability-adjusted life-years) [3, 4], but also in terms of impact on development and overall economic productivity in low- and middle-income countries [3, 5].
The currently available treatments for NTDs are an outdated arsenal generally considered to be insufficient for NTD control and elimination [5]. Many of the currently available drugs were developed over 50 years ago and many of them exhibit high toxicity [5]. For example, the only available drug to treat late-stage human African trypanosomiasis (or sleeping sickness) caused by T. b. rhodesiense is melarsoprol, an arsenic compound, developed in the 1940s, which is itself lethal to 5 % of treated patients owing to post-treatment reactive encephalopathy [6]. In many regions, pentavalent antimony-containing compounds are still the treatment of choice for visceral leishmaniasis (VL) and cutaneous leishmaniasis, which have been in use since the 1930s. Therapeutic failure is generally thought to result from sub-therapeutic dosing and shortened treatment durations [7]. As a consequence, clinical antimonial drug resistance in Leishmania has yielded the drug useless in various geographical regions. At the same time, the upper limit of dosing of antimonials is limited by severe toxicities, such as pancreatitis and cardiotoxicity [7, 8]. Examples like these emphasize the role of dose optimization and pharmacokinetic (PK) studies for treatments against NTDs, where there is often only a small therapeutic window between treatment failure, engendering drug resistance, and drug toxicity.
Despite the urgent need for new, safer, and more efficacious treatments for NTDs, there is insufficient interest from the pharmaceutical industry to invest in drug development for these diseases because of the limited financial incentive. This paradigm has led to a fatal imbalance in drug development: although NTDs account for 12 % of the global disease burden, only 1 % of all approved drugs during the past decade was developed for these diseases. None of these approved drugs were a new chemical entity, and just 0.5 % of all clinical trials in the past decade were dedicated to NTDs [9].
Owing to the lack of innovation as a result of the absence of financial incentives and the continued use of drugs developed many decades ago, dose-optimization studies or studies in specific patient populations particularly affected by NTDs (e.g., pediatric or HIV co-infected patients) have rarely been reported. While a comprehensive and quantitative overview is currently lacking, only a few clinical trials on NTDs appear to have included studies on the pharmacokinetics of the therapeutic compounds that were under clinical investigation. Rational drug therapy is based on the assumption of a causal relationship between exposure and response. Therefore, characterizing the pharmacokinetics of a drug is of utmost importance. Conventionally, non-compartmental analysis (NCA) methods were used for PK analysis, but these are less powerful and informative for typical NTD PK studies, which are sparse and heterogeneous in nature. NCA has a low power to identify true covariate effects and does not allow for simulations of alternative dosing regimens. Population-based modeling and simulation techniques are therefore more appropriate to describe and predict the relationship between exposure (pharmacokinetics), response (pharmacodynamics), individual patient characteristics, and other covariates of interest (e.g., body weight, sex, and concomitant medication). These pharmacometric methods have become standard in drug development worldwide, and have been recommended by the US Food and Drug Administration and the European Medicines Agency for PK–pharmacodynamic (PD) data analysis and clinical trial design, particularly in pediatric and small-sized patient populations [10–12]. Nevertheless, these methodologies appear to be systematically underused to address NTDs, likely because their advent occurred much later than the time when many of these drugs were developed.
To better understand to what extent clinical PK studies have contributed to optimization of treatment regimens for NTDs, we performed a systematic review of published clinical PK studies on NTD therapeutics. We hypothesize that for many of the NTD therapeutics, proper PK studies and thus a rationale for their dosing are plainly missing, and that only a few of these studies use modeling and simulation tools. By providing a comprehensive overview of performed PK studies, we illustrate the advantages and drawbacks of different PK methodologies and we identity the gaps in PK research for particular NTDs to indicate the areas where NTD therapeutics can be further improved.
2 Methods
2.1 Study Identification
We performed a systematic literature review following applicable criteria of the most current PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines [13], the PRISMA Checklist is in Appendix 1. The MEDLINE database was systematically searched through PubMed for all human clinical PK studies until September 2015 that described the clinical pharmacokinetics of a drug in the treatment of any of the NTDs. For instance, the search term used for studies for Chagas disease was: ((Chagas disease[Title/Abstract] OR American trypanosomiasis[Title/Abstract])) AND (pharmacokinetics[Title/Abstract] OR pharmacokinetic[Title/Abstract]). Reviews were excluded from the search, as well as preclinical research and research concerning animals other than humans. The search was limited to publications in English. A full list of all the search terms used is shown in Supplemental Table 1.
Secondary literature was identified using the bibliographies of the primary identified literature and by specifically querying PubMed using the drug name in combination with the disease. Because we were particularly interested in the application of population PK approaches in NTDs, the abstracts of the Population Approach Group Europe conference [14] were also searched using the same search terms. No specific protocol was developed for this systematic review.
2.2 Study Selection
Records were initially screened to identify relevant publications based on title and abstract. If the abstract lacked sufficient detail, the full publication was assessed. The aim of this study was the identification of clinical PK studies in the context of the treatment of NTDs, and therefore studies were excluded if the study’s subjects were not healthy subjects (phase I studies) or patients diagnosed with one of the NTDs; or if the drug of interest was symptomatic treatment (e.g., suppression of fever) or for treatment of concomitant diseases instead of the NTD itself (primary criteria). Articles with only pharmacodynamic results or only reporting a bioanalytical method were also excluded.
2.3 Assessment of Pharmacokinetic Data Analysis Methods
The methods used to analyze the PK data were extracted from the identified records and qualitatively categorized as follows, in order of level of complexity: (I) comparison of average trough/steady-state concentrations, (II) NCA, (III) individual-based compartmental analysis, (IV) population-based compartmental analysis, and (V) the use of simulations and/or extrapolations. In category I, studies were included that basically compared a drug concentration at a single time point between different formulations or different patient groups. In category II, we included studies that described concentration-time profiles or PK parameters by using NCA techniques [15]. Analyses in category III used non-linear equations to describe individual concentration-time curves, by using theoretical compartments and inter-compartmental transfer rates, deriving individual PK parameters that can be averaged. In population-based analysis (category IV), similar techniques are being used, but with simultaneous estimation of both inter- and intra-individual variability (nonlinear mixed-effects models). The derived model is descriptive for the entire population and can subsequently be used for predictions and simulations, and potentially for extrapolation to for instance other populations (additional category V).
2.4 Extraction and Analysis of Data
Besides the PK data analysis method, other data that were extracted from the identified study reports were: administered compound, measured analytes (parent compound and/or metabolites), route of administration, PK sample matrix, the type and number of subjects, and particularly whether pediatric patients were included in the study. Additionally, the main conclusions were extracted from all studies in a qualitative way, focused on the study recommendations in regard to dose adjustments or other treatment optimizations. The risk of bias in these recommendations, for instance when used analysis methods were insufficient to support these treatment recommendations, was gauged and reported if detected. Given the nature of extracted data, only a simple descriptive analysis was conducted, summarizing individual studies.
3 Results
3.1 Study Characteristics
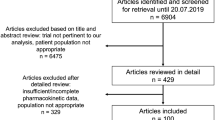
The primary literature search identified 431 unique publications. After screening, 341 publications were excluded based on the primary criteria. Combined with additional articles through secondary sources, 82 publications were eventually included in this systematic review (Fig. 1). No full texts were available for six studies; however, the abstracts of these publications contained all the information to be extracted and they did not need to be excluded. The search and inclusion results stratified per NTD are shown in Supplemental Table 1. A summary of all identified PK studies together with their main characteristics is shown in Table 2.

Study flow diagram. NTDs neglected tropical diseases
For four out of the 17 (24 %) NTDs, not a single PK study could be identified, these were yaws, dracunculiasis, dengue/chikungunya/zika and soil-transmitted helminthiases. For six (41 %) other NTDs, fewer than five PK studies had been reported. Most studies had included small patient numbers, only five studies (6.1 %) had included >50 subjects (Table 2). Pediatric patients were included in nine (11 %) studies. The majority of these studies were not very recent; 56 % of studies were published before 2000; the frequency of studies per year is depicted in Fig. 2. Concerning the used analysis methods, some studies employed multiple analysis methods, e.g., both comparison of steady-state concentrations and NCA (Table 2). When looking at the most complicated method employed in the study, most studies used NCA methods for PK analysis (62 %). Twelve studies (15 %) used population-based compartmental analysis, of which eight (10 %) additionally performed simulations or extrapolation. Regarding the aim of the studies, 38 studies (46 %) focused on describing the pharmacokinetics of a compound without further interpretations. Only five studies (6 %) evaluated exposure-response relationships. Although some of these studies reported side effects [16–18], none of these attempted to relate drug exposure to observed toxicity. However, relatively many studies (28 %) evaluated drug–drug and food interactions. This is owing to the frequent use of combination therapies for the treatment of NTDs, and the implementation of overlapping prophylactic mass drug administrations, e.g., onchocerciasis, lymphatic filariasis, and schistosomiasis.

Number of identified clinical pharmacokinetic publications on neglected tropical diseases stratified per year
3.2 Pharmacokinetic Studies per Neglected Tropical Disease
Based on the cause of the infection, NTDs can be divided into four groups: diseases caused by protozoal parasites, bacteria, helminthes, and viruses (an extensive overview is provided in Table 1). The protozoal NTDs are all caused by kinetoplastid parasites: Chagas disease, human African trypanosomiasis, and leishmaniasis. Bacteria, a large and diverse group of prokaryotic microorganisms, cause Buruli ulcer, leprosy (both caused by Mycobacteria), trachoma, and yaws. Helminthes, commonly known as parasitic worms, are large multicellular organisms. The helminth NTDs are cysticercosis/taeniasis, dracunculiasis, echinococcosis, food-borne trematodiases, lymphatic filariasis, onchocerciasis, schistosomiasis, and the soil-transmitted helminthiases. Viral NTDs include the arboviral disease dengue (plus chikungunya and zika) and rabies. A general overview of medicines that are currently in use for NTDs is listed in Table 3 [1, 19].
We discuss the most salient identified PK studies for NTD therapies, focusing on studies that played a role in treatment optimization. A full overview of identified studies can be found in Table 2.
3.2.1 Chagas Disease
Around 5.7 million people worldwide are affected by Chagas disease (also known as American trypanosomiasis), which is caused by the Trypanosoma cruzi parasite [20]. The acute phase of the disease is asymptomatic in most patients. During the chronic phase, patients can experience cardiac, digestive, or neurological symptoms, which complications lead in many patients to fatality in the late chronic stage mostly decades after the start of infection. However, Chagas disease can be cured when treatment is initiated at the acute or early chronic stage. Currently, the only two drugs with proven efficacy in Chagas disease are nifurtumox and benznidazole (Table 3). Clinical PK studies were found for three drugs: allopurinol riboside [21, 22], nifurtimox [23], and benznidazole [24–26] (Table 2).
Allopurinol was not further evaluated for the treatment of Chagas disease after demonstrating suboptimal exposure [21], which could not be sufficiently increased by probenecid co-administration decreasing the drug’s renal excretion [22]. A population PK modeling and simulation approach was used to estimate the exposure of infants to nifurtimox via breastmilk of patients [23]. Transfer of nifurtimox into breastmilk appeared limited and unlikely to lead to significant exposure in infants, yielding nifurtimox safe to use for breastfeeding patients. The first PK study on benznidazole was published in 1980 [24]. Very recently, population-based analyses were performed in children [25] and in adults [26]. Model-based simulations in these studies suggested that the adult daily dose intervals in chronic Chagas patients could be prolonged, while benznidazole concentrations were kept within the target range, potentially simplifying the treatment regimen.
3.2.2 Human African Trypanosomiasis
Human African trypanosomiasis, also known as sleeping sickness, is transmitted by the tsetse fly and caused by T. b. rhodesiense, resulting in an acute infection, and T. b. gambiense, leading to a more chronic infection (Table 1). Without treatment, the infection of the central nervous system is ultimately fatal [27]. There are currently four treatments in use for the two different stages of human African trypanosomiasis (Table 3), all of which exhibit substantial toxicities: pentamidine, suramin, melarsoprol, and nifurtimox plus eflornithine. Clinical PK studies were identified for three of these treatments: pentamidine [28, 29], melarsoprol [30–33], and eflornithine [34–36]. Additionally, PK studies were found for fexinidazole, a drug currently still in late-phase clinical development [37, 38].
Pharmacokinetics played an important role in the optimization of eflornithine therapy. Based on cerebrospinal fluid (CSF) and plasma PK data from late-stage T. b. gambiense trypanosomiasis, a new dosing regimen was proposed for eflornithine, including different infusion intervals, and increased doses in children, based on body surface area instead of body weight [34]. Later, it was shown that the current dosing of oral eflornithine did not result in adequate therapeutic plasma and CSF concentrations in adult patients [35]. Recently, a population-based PK–PD model for the different stereoisomers of eflornithine was developed reanalyzing previous PK data and showed the importance of stereoselective exposure, which provided an explanation why oral eflornithine had failed so far for late-stage human African trypanosomiasis patients [36].
Melarsoprol pharmacokinetics in plasma and CSF was assessed using compartmental methods in 19 trypanosomiasis patients, after which the typical exposure for safer alternative dose regimens could be simulated [31]. However the PK–PD relationships for melarsoprol remain unclear: melarsoprol PK parameters and CSF/plasma exposure were not significantly different in refractory compared with cured patients [32] and arsenic urinary excretion was not predictive of either toxicity or efficacy of melarsoprol [30].
Fexinidazole, a nitroimidazole-compound currently in clinical development for human African trypanosomiasis, and its active metabolites were studied in healthy volunteers. The study showed the need for concomitant food intake, which increases the bioavailability of this compound substantially, and identified a target dose for the first in-patient studies [37, 38].
3.2.3 Leishmaniasis
Leishmaniasis is caused by various species of Leishmania parasites that are transmitted by sandflies, with different and widespread geographical regions of distribution, leading to distinctly different clinical presentations. Cutaneous leishmaniasis is most prevalent and has the potential to progress into mucocutaneous leishmaniasis. Visceral leishmaniasis is the most severe clinical form and is inevitably fatal if left untreated. Treatment of leishmaniasis depends on the type of disease, parasite species, and on the availability of treatment depending on the geographical location. Local chemotherapeutic treatment with intralesional pentavalent antimonials or paromomycin cream can be an option for cutaneous leishmaniasis, although some species or severe/diffuse disease are rather treated systemically with either parenteral antimonials, liposomal amphotericin B, pentamidine or oral miltefosine, ketoconazole, and fluconazole [39]. Recommended treatments for VL are, depending on species and geographical location, either parenteral (liposomal) amphotericin B, the antimonial sodium stibogluconate, paromomycin, oral miltefosine, or combinations of sodium stibogluconate with paromomycin (East Africa) or liposomal amphotericin B plus paromomycin/miltefosine (India). Several clinical PK studies were conducted in leishmaniasis, and have helped most notably to optimize dose regimens for miltefosine for VL [40–43], for antimonials for cutaneous leishmaniasis [44–46] and for VL [47, 48], to quantify exposure to paromomycin in VL [49], and to assess systemic penetration of topical paromomycin formulations [50]. Few studies have been performed in the context of leishmaniasis on allopurinol [21, 22] and sitamaquine [51] of which both are not in clinical use at the moment.
Comparing the two pentavalent antimonial compounds in use for leishmaniasis, meglumine antimoniate, and sodium stibogluconate, equivalent systemic exposure was shown for the active component pentavalent antimony, possibly indicating that they can be used interchangeably [48]. In cutaneous leishmaniasis, PK studies on parenteral sodium stibogluconate demonstrated wide variability in drug exposure [44], but also penetration of the active component antimony in the skin, with no differences between normal skin and lesions [52]. The first pediatric study of meglumine antimoniate showed that drug exposure is significantly lower in children than in adults treated with the same linear weight-adjusted (mg/kg) regimen, possibly indicating that children are currently undertreated [53]. Only a descriptive analysis of the pharmacokinetics was performed, which did not suggest or evaluate alternative dose regimens for children.
Systemic penetration of paromomycin and gentamycin after application of two different topical formulations in cutaneous leishmaniasis patients was assessed using compartmental methods [50]. While gentamycin remained largely undetectable in plasma, paromomycin accumulated to 5–9 % of typical trough concentrations achieved after a standard intramuscular administration of 15 mg/kg paromomycin, indicating little concern for systemic drug toxicity of the topical formulations.
Most PK studies in leishmaniasis were conducted on the oral drug miltefosine. In 2008, the first population PK model for this drug was developed on data from Dutch military personnel who contracted L. major cutaneous leishmaniasis in Afghanistan [40]. This analysis showed that miltefosine is eliminated at a much slower rate than expected, which has potential implications for emerging drug resistance and the required contraception period owing to the teratogenicity of miltefosine. A subsequent simulation study focused on the translation of the reproductive safety limit in animal studies to Indian female VL patients. New recommendations for the duration of contraceptive cover after miltefosine treatment were provided based on these findings [41]. In a model-based study, miltefosine exposure appeared to be lower in children than in adults treated with the same mg/kg dose, possibly explaining increased failure rates observed in pediatric VL patients. A new dosing algorithm based on allometric scaling was proposed and was evaluated by Monte Carlo simulations [42]. Recently, a PK–PD model of miltefosine in Nepalese VL patients indeed identified a PK–PD relationship between miltefosine exposure and long-term treatment relapse [43]. The confirmed underexposure in children, reinforces the need for implementing the earlier proposed allometric miltefosine dosing regimen for VL [42, 43].
3.2.4 Buruli Ulcer
Buruli ulcer is an ulcerating infection caused by Mycobacterium ulcerans, leading to long-term functional disability, loss of productivity, and stigmatization. Antimicrobial treatment is particularly effective in small lesions and at an early stage of infection, it reduces healing time, recurrence rate, and the need for surgical intervention [54]. Different combinations of antimicrobials are used, depending on available resources and the stage of the disease. The most widely accepted combination is oral rifampicin with intramuscular streptomycin, the oral combination of rifampicin with clarithromycin is still under clinical evaluation. Only a single PK study could be identified for Buruli ulcer, which studied systemic pharmacokinetics of rifampicin and clarithromycin in patients using population compartmental methods [55].
In this study, the counteracting interaction effects (both cytochrome P450 3A4 and P-glycoprotein) of clarithromycin and rifampicin on each other’s pharmacokinetics were investigated. Eventually, it was suggested that a doubled dose of clarithromycin should be evaluated in future clinical studies to ensure an increased time above the minimum inhibitory concentration [55].
3.2.5 Leprosy
Leprosy can be divided into paucibacillary and multibacillary disease. If not treated in an early phase, it results in lifelong neuropathy and disability. A combination of drugs is needed because of the emergence of drug resistance. In 1995, the WHO supplied free multi-drug therapy to leprosy patients in all endemic countries, which led to a dramatic decrease in prevalence. For paucibacillary treatment, the recommended all oral treatment combination is rifampicin plus dapsone, for multibacillary treatment; this combination should be extended with clofazimine (Table 3). Various PK studies have been conducted on dapsone [56–59], clofazimine [60], and specific drug–drug interaction studies focusing on the interactions between dapsone, clofazimine, and rifampicine using various formulations [61–64]. A few studies focused on thalidomide pharmacokinetics [65, 66], which is currently largely considered obsolete because of its teratogenicity. PK studies for leprosy were mainly performed in the 1980/90s and generally using NCA methods (Table 2).
A study on dapsone and its main active metabolite monoacetyldapsone in leprosy patients concluded that the standard 100-mg/day dose was sufficient to maintain therapeutic plasma concentrations in relation to in vitro susceptibility values [58]. Nevertheless, dose adjustments might be needed for obese patients treated with this regimen [59]. Various drug–drug interaction studies did not reveal clinical significant interactions, although little is known about the required minimal effective exposure in leprosy [61–64].
Pharmacokinetics of clofazimine has been analyzed using compartmental methods after various fed and fasting conditions to determine food effects and the relative bioavailability [60]. A high-fat meal increased bioavailability significantly and was therefore considered preferable, although exposure–effect relationships for clofazimine in leprosy have not been properly established.
3.2.6 Trachoma
Trachoma is the leading infectious cause of blindness worldwide. The infection of the eye by Chlamydia trachomatis can be divided into two clinical stages: initial active trachoma (inflammation) and cicatricial disease (eyelid scarring). Active trachoma is mostly seen in young children and cicatricial disease and eventual blindness are typically seen in adults. Treatment and prevention of trachoma consists of surgery and mass drug administration of antibiotic treatment. The WHO recommends either single-dose oral azithromycin or topical tetracycline. Because trachoma commonly geographically overlaps with other NTDs such as onchocerciasis and lymphatic filariasis, regional elimination initiatives for these diseases in terms of mass drug administrations are often aimed to be combined. Pharmacokinetic studies have therefore focused on drug–drug interactions between azithromycin and drugs used in mass drug administration for these other NTDs (ivermectin and albendazole) [67, 68].
Ivermectin exposure appeared to be increased in healthy volunteers in combination with azithromycin and the authors recommended subsequent modeling and simulation to predict and evaluate an optimal dosing regimen for this drug combination [67]. A subsequent population PK analysis of the same data showed the benefit of modeling and simulation by pinpointing that the mechanism of this interaction was an increase in bioavailability and demonstrating that maximum expected ivermectin exposures after concomitant administration of azithromycin were still within a well-tolerated range, meaning that combining these drugs in mass drug administrations should be feasible [68].
3.2.7 Endemic Treponematoses
Endemic treponematoses are a group of chronic bacterial infections, related to venereal syphilis, caused by treponemes that mainly affect the bones and/or skin causing localized lesions. The spectrum of diseases includes yaws, endemic syphilis (bejel), and pinta. Yaws is the most prevalent form of non-venereal treponematosis, and while rarely fatal, it can lead to chronic disfigurement and disability. Treatment consists of a single dose of long-acting penicillin or oral azithromycin. No PK studies could be identified for drugs used to treat endemic treponematoses.
3.2.8 Cysticercosis/Taeniasis
Cysticercosis and taeniasis are both caused by species of the Taenia tapeworm. Taeniasis is the intestinal infection with adult tapeworms. This mild disease is an important cause for transmission of cysticercosis, an infection with the larval stage of the pork tapeworm Taenia solium that can cause life-threatening clinical manifestations. The most severe form is neurocysticercosis in which the larval cysts are located in the central nervous system and cause severe neurological symptoms. The treatment of (neuro-)cysticercosis is not fully established. Besides symptomatic treatment (antiepileptics), it remains debated whether, and if so in which cases, antiparasitic and concomitant anti-inflammatory treatment to reduce inflammation associated with the dying organism are indicated. The main antiparasitic agents used in cysticercosis are albendazole and praziquantel, while the supportive anti-inflammatory therapy can be corticosteroids or methotrexate. Pharmacokinetic studies are available for both albendazole [69–71, 75] and praziquantel [72–74], and have focused on drug–drug interactions [71–74].
Albendazole sulfoxide, the main metabolite of albendazole, has been studied in several clinical trials on neurocysticercosis. Despite the absence of an established PK–PD relationship, these studies suggested based on the area under the concentration-time curve and steady-state trough concentrations that albendazole administration could be changed from the current clinical practice of three times daily, to twice daily [70, 75]. Conversely, a small descriptive study in children advised an opposite dose adjustment, given the increased clearance in children [69]. Drug–drug interaction studies indicated that there were no interactions with antiepileptic drugs and that dexamethasone even decreased the elimination rate of albendazole [71]. Co-administration of the antiparasitic praziquantel increased albendazole sulfoxide exposure possibly synergizing the efficacy of both drugs when administered together [74].
Drug–drug interaction studies with praziquantel demonstrated that exposure was decreased in combination with dexamethasone and anti-epileptic drugs possibly related to induction of cytochrome P450-mediated hepatic metabolism [72]. Conversely, co-administration of the histamine H2-receptor antagonist cimetidine was demonstrated to prolong exposure of praziquantel, suggesting the possibility for further improvement of efficacy of this single-day therapy [73].
3.2.9 Dracunculiasis
Dracunculiasis is also known as guinea-worm disease. The infection is transmitted by drinking unfiltered water containing the larvae of Dracunculus medinensis. Treatment consists of slow extraction of the worm combined with wound care and pain management. There is no specific chemotherapy indicated to treat dracunculiasis and no PK studies were found.
3.2.10 Echinococcosis
There are four species of Echinococcus tapeworms that can cause infection in humans. Humans are an incidental host; with transmission through for example, contaminated environmental water. The two main types of disease are cystic echinococcosis and alveolar echinococcosis, both characterized by the slow growth of cyst-like larvae, usually in the liver. Development of active disease can take multiple years. Oral albendazole is the chemotherapy of choice for both disease types, sometimes combined with surgery or percutaneous drainage of the cysts. Albendazole is poorly absorbed from the gastrointestinal tract and most PK studies have focused on improving the bioavailability of the compound [76–78].
The pharmacokinetics of albendazole and its main metabolite, albendazole sulfoxide, have been studied in patients with echinococcosis [76]. It was shown that extrahepatic cholestasis, a common symptom of echinococcosis, delayed the absorption and decreased the elimination rate of albendazole. Another study looked at bioequivalence between a novel emulsified formulation of albendazole compared with a standard oral tablet formulation [77]. To improve the low bioavailability of albendazole, co-administration with cimetidine was studied [78]. The high inter-individual variability in drug exposure and the various possible contradictory effects of cimetidine on both absorption (increased) by decreasing the gastrointestinal pH and metabolism by cytochrome P450 enzyme inhibition for both albendazole and its sulfoxide metabolite complicated the descriptive interpretation of the results from this study [78].
3.2.11 Food-Borne Trematodiases
Food-borne trematodiases are zoonotic infections caused by parasitic flatworms, so-called ‘liver flukes’, which can result in clonorchiasis, opisthorchiasis, fascioliasis, and paragonimiasis. Transmission cycles differ widely, but generally involve ingestion of food contaminated with the parasite larvae. The worms are mainly located in the liver and gall bladder or in the lung (paragonimiasis). Different anti-helminthic compounds are used, depending on the infecting worm (Table 3), but praziquantel and triclabendazole are two of the main drugs in use for this group of diseases. Pharmacokinetic studies were found for both these drugs [79–82], although no studies were found in the context of paragonimiasis (lung fluke). Given the liver damage caused by the flukes, many PK studies focused on a disease effect on cytochrome P450 enzyme-mediated metabolism of the compounds, which appeared most prominent for the cytochrome P450 3A4 substrate praziquantel [79].
The bioavailability of triclabendazole, the drug of choice for fascioliasis, and total exposure to active metabolites were shown to be greatly increased by concomitant food intake [81]. Descriptive PK parameters were not different between fascioliasis patients and healthy subjects, indicating the absence of a disease effect on triclabendazole metabolism despite obvious liver damage [82].
Praziquantel, the anti-helminth drug of choice for both opisthorchiasis and clonorchiasis, appeared to have a reduced clearance rate in advanced opisthorchiasis infection, compared with early-stage disease and post-recovery, presumably owing to liver impairment [79]. In clonorchiasis patients, a sustained-release formulation was tested to allow for a single-dose treatment with praziquantel. Despite a similar area under the concentration-time curve, the sustained-release formulation with lower maximal concentration and peak time showed unsatisfactory efficacy compared with single-dose normal-release praziquantel [80].
3.2.12 Lymphatic Filariasis
Lymphatic filariasis, also known as elephantiasis, is a disfiguring disease caused by filarial nematodes (roundworms) and is a major cause of disability and social stigma in endemic areas. The filarial worms are transmitted by mosquitoes and cause an infection of the lymphatic system and skin, leading to massive edema formation in, for example, extremities and genitalia. Current treatment is generally through mass drug administration with the aim to stop transmission of the disease by killing the microfilarial stage of the parasite, using albendazole plus either ivermectin in regions with onchocerciasis (i.e., African countries) or albendazole plus diethylcarbamazine in all other regions. Pharmacokinetic studies in the context of lymphatic filariasis were found for both these combinations [83–85]. Doxycycline has been proposed as a treatment to kill also adult worms, but no PK studies could be identified for this drug in this context.
Co-administration of diethylcarbamazine and albendazole was investigated in healthy volunteers from areas where lymphatic filariasis is endemic [83]. Whereas large inter-individual variability in exposure of all drugs was observed, no significant interaction was detected. To assess the safety of albendazole mass drug administration during breast feeding, the pharmacokinetics of albendazole and metabolites was studied in the breast milk of treated women. Albendazole and albendazole sulfoxide achieved low penetration into breast milk and was not considered to be harmful for breastfed infants [85].
3.2.13 Onchocerciasis
Onchocerciasis, also known as river blindness, is caused by the filarial nematode Onchocerca volvulus, which is transmitted through the bites of blackfly that breed near rivers. It results in various clinical manifestations, such as pruritus, subcutaneous nodules, onchocercal skin disease, and blindness. The therapeutic targets are the young microfilariae located, for example, in the skin, as well as the adult worms (macrofilariae) located generally in the subcutaneous nodules. The clinical approach to treatment is mainly focused on interrupting transmission through mass drug administration programs with ivermectin (focused on killing the microfilariae) at 6- to 12-monthly intervals, sometimes in combination with albendazole owing to an overlap with lymphatic filariasis co-infection. For individual treatment, doxycycline is used in combination with ivermectin. Various studies have investigated the pharmacokinetics of albendazole and ivermectin in onchocerciasis patients, focusing on dose-finding, food-effect, compliance, disease-effect, tissue distribution, and drug–drug interactions [86–90]. No PK studies were found for doxycycline. Other less established treatments include: suramin (too toxic and costly [91]), moxidectin (under development [92]), and amocarzine (insufficient efficacy [93, 94]).
Combining albendazole and ivermectin appeared to be safe and not to result in any PK interactions; albendazole co-administration offered no advantage over ivermectin alone in terms of efficacy against onchocerciasis [86, 87]. A fatty meal increased the bioavailability of albendazole fourfold and concomitant food intake should thus be recommended [86]. However, ivermectin pharmacokinetics was shown to be not affected by either food or alcohol intake [90].
Ivermectin PK parameters were similar between healthy volunteers and onchocerciasis patients and the drug was shown to penetrate in fat, skin, infected nodules, and even isolated parasites from these patients [89]. Compliance to non-observed ivermectin therapy in mass drug administration programs could be assessed through plasma concentration monitoring [88].
Pharmacokinetics of moxidectin, a veterinary anti-parasitic, was studied in healthy lactating women, including excretion into breast milk [92]. Moxidectin exposure in infants via breast milk was estimated to be 8.37 % of the maternal dose, but PK information from young children is necessary to fully understand the implications of this indirect exposure.
The bioavailability of amocarzine, an experimental drug for onchocerciasis, appeared to be poor in fasting conditions. Additionally, the dosing interval was suggested to be shortened to twice-daily administration to increase exposure [93]. A subsequent study showed improved bioavailability of amocarzine and exposure to its N-oxide metabolite with food intake [94].
3.2.14 Schistosomiasis
Schistosomiasis is caused by Schistosoma blood flukes, whose life cycle is dependent on fresh water snails. Humans are infected through skin contact with contaminated water. The localization of the infection can vary depending on the infective Schistosoma species and can develop in the intestines, liver, spleen, lungs, bladder, or urinary tract. The acute phase is characterized by a transient hypersensitivity reaction associated with tissue migration of the larvae. Chronic infection can result in many different clinical manifestations such as hematuria (urogenital) or blood in the stool (intestinal), depending on the infected organs. Control of schistosomiasis is based on large-scale treatment mainly using praziquantel, which was the topic of most PK studies in schistosomiasis [16, 95–97]. The hepatic and renal dysfunction associated with chronic infection have been the focus of various praziquantel PK studies, which is hepatically metabolized and renally cleared. We also found descriptive PK studies for the experimental drugs metrifonate [98] and oxamniquine [99].
In one schistosomiasis case with chronic kidney failure, praziquantel plasma pharmacokinetics seemed not to be affected. This could indicate that advanced schistosomiasis can be treated with the regular praziquantel dose [95]. In patients infected by Schistosoma mansoni with various degrees of hepatic dysfunction, both the time to maximal concentrations as well as the area under the concentration-time curve increased proportionally with the degree of hepatic insufficiency [16]. Despite these PK differences, efficacy appeared not to be affected and dose adjustments based on hepatic function were not advised [16]. Pharmacokinetic parameters of two formulations of praziquantel were compared, to investigate a slower release formulation [96].
3.2.15 Soil-Transmitted Helminthiases
Soil-transmitted helminthiases are a diverse set of diseases caused by intestinal worms and often affect the most poor and rural communities. The main species that infect people through contact with contaminated soil are the roundworm (Ascaris), the whipworm (Trichuris), and the hookworms (Necator and Ancylostoma). Treatment for these infections is mainly through administration of antihelminths such as albendazole and mebendazole. Preventive treatment to reduce transmission to endemic populations is also widely used. No PK studies were identified.
3.2.16 Dengue, Chikungunya, and Zika
Dengue is the most prevalent arthropod-borne viral disease. The Flavivirus infection can cause a wide range of clinical manifestations of which severe hemorrhagic dengue is potentially fatal. Chikungunya and zika, two other flaviviruses that are also transmitted by mosquitos, both cause acute febrile polyarthralgia and arthritis. There are no specific therapeutic treatments available for dengue, chikungunya, or zika, although a few vaccines are currently in development. However, no PK studies were identified for these viral infections.
3.2.17 Rabies
Rabies is caused by a range of lyssaviruses and usually starts with non-specific symptoms during the prodromal phase, but once a patient is symptomatic the infection usually leads to progressive encephalopathy and is virtually always fatal. There are no established antiviral treatment regimens for rabies, although various post-exposure prophylaxis schedules based on vaccine therapy with or without rabies-specific immunoglobulins are being used to prevent development of symptomatic disease. Some PK studies have been performed on the kinetics of administered anti-rabies antibodies [17, 18] and administered human interferon to support an early immune response [100].
Pharmacokinetics of human leukocyte interferon in CSF was compared between systemic and local intraventricular direct administration [100]. This study demonstrated that interferon levels in the CSF could be maintained at potentially therapeutically active levels, also by systemic administration. Two other studies looked at immunoglobulin antibody administrations to increase rabies antibody titers. In one study, two sources of equine rabies immunoglobulin were compared in terms of antigen-binding fragments, which showed similar time profiles, but no bioequivalence [17]. A phase I study with a recombinant human IgG1 anti-rabies monoclonal antibody determined the required dose to use in future post-exposure prophylaxis studies based on antibody pharmacokinetics [18].
4 Discussion
This is the first comprehensive and systematic review of clinical PK studies undertaken in the field of NTDs. Our study highlights the paucity of PK data available for the treatments used against NTDs and the lack of application of modeling and simulation techniques in this particular clinical area. For various NTDs (endemic treponematoses, dracunculiasis, dengue/chikungunya, and soil-transmitted helminthiasis), no PK studies could be identified at all, while for others only very few studies (<5) were found (Buruli ulcer, trachoma, echinococcosis, lymphatic filariasis, food-borne trematodiases, and rabies). For diseases such as soil-transmitted helminthiases and rabies, this lack of PK studies is in stark contrast to their massive global burden of disease (2700 and 26,400 deaths per year and 5.19 and 1.46 million disability-adjusted life-years, respectively). Whereas for some of these diseases dedicated chemotherapeutic options have never been available (e.g., dracunculiasis and dengue), for other NTDs, multiple drugs have been in clinical use for decades as part of established treatment guidelines, but information on PK studies is lacking. Owing to the consistent lack of research and development funding for treatment of NTDs, very little innovation has been witnessed in the past century for the management of NTDs. For example, suramin, pentamidine, and melarsoprol, which were discovered in 1920, 1940, and 1949, respectively, are still being used for human African trypanosomiasis and leishmaniasis management. For all of these drugs, no or very little PK studies have been performed since their introduction (we identified one, two, and five studies, respectively), while their live-threatening toxicities and the continuous threat of emerging drug resistance requires continued optimization of these dose regimens. The gap of knowledge on pharmacokinetics and PK–PD relationships limiting treatment optimization and adaptation has been highlighted before, e.g., for praziquantel [101] and schistosomiasis [102], but has largely been neglected for other NTDs previously.
4.1 Limitations
Because our systematic review focused on clinical PK studies, the term ‘pharmacokinetics’ was the most central search term in our analysis. However, this term itself was only been introduced in the field of pharmacology in 1953 by Dost and has been popularized in the two decades afterwards [103]. Given that several current drugs against NTDs have been in use for a few decades already, it could be that various older publications for these particular NTD drugs were not identified because they potentially did not make mention of the term ‘pharmacokinetics’. This might also explain that our two oldest identified studies date from 1980 [24, 98], which might thus be a biased observation. While drug development activities in the field of NTDs have substantially increased during the past 15 years, mainly through increased political awareness and novel innovation mechanisms such as the product development partnership Drugs for Neglected Diseases initiative (http://www.dndi.org), this appeared not to be reflected in the number of PK studies conducted. There is no particular increasing trend in the number of clinical PK reports for NTDs since 2000 (Fig. 2). On the contrary, more than half of all identified PK studies were published prior to 2000, with a peak of publications in the mid-1990s (seven studies in 1993). This might indicate that, despite innovative breakthroughs and increased clinical trial activities in the field of NTDs, PK studies are still being neglected; an observation corroborated by a recent review that found only 4/382 active clinical trials on NTDs directed at PK studies [9]. Regarding the type of PK analysis, there was an increasing trend of using a population approach to analyze the PK data over the past decade (Fig. 2).
A limitation in our search strategy was the English language restriction, potentially missing papers, e.g. in French from African journals or Chinese from Asian journals. Additionally, (national) journals from countries/regions where NTDs are endemic are not particularly well covered by PubMed/MEDLINE. Theoretically, this might have precluded our access to some literature, but given the topic of our literature analysis this potential bias is probably in reality very small or even non-existent and more relevant for clinical publications than PK publications.
While the list of NTDs used can be variable, the word ‘neglected’ in that term generally refers to the lack of interest from the pharmaceutical industry and the overall lack of funding and innovation in terms of therapeutic research and development for these diseases, but also to neglect by health ministries in countries where infected people live, by the World Bank, or relative to human immunodeficiency virus/acquired immune deficiency syndrome, tuberculosis, and malaria. Typically NTDs are infectious diseases closely interrelated to poverty and socioecological systems promoting close contact between affected populations, vectors, and animal reservoirs. Malaria, tuberculosis, and human immunodeficiency virus/acquired immune deficiency syndrome are not considered as NTDs. The list of NTDs has since the turn of the century often been expanded to more than 30 diseases and disease complexes [104]. We adhered to the list of NTDs put forward by the WHO [1], which contains 17 items of which various are disease ‘complexes’, such as ‘soil-transmitted helminthiases’, comprising multiple clinical infectious disorders.
4.2 Challenges in Clinical Pharmacokinetic Studies in Patients with Neglected Tropical Diseases
In the rural settings in which NTD clinical trials take place, collecting samples, maintaining cold chains, and generally performing large clinical trials are logistically challenging. Obtaining useful blood samples from patients can be practically difficult owing to a lack of laboratory infrastructure and restrictive clinical characteristics such as anemia. NTDs typically affect the poorest of the poor and disadvantaged populations, inherently constituting ethical difficulties, while language barriers and illiteracy make it difficult to acquire informed consent from patients. Additionally, following up patients after their treatment, e.g., to sample the elimination phase of a drug or identify long-term outcome, is often practically impossible, e.g., in nomadic populations. Moreover, there is limited research and development funding available for the clinical development of drugs for NTDs [9]. For all these reasons, clinical trials on NTDs therefore typically result in small and heterogeneous datasets. This is illustrated by our systematic review, as 94 % of the identified studies included small patient numbers (n < 50).
Patients in PK studies on NTDs are highly heterogeneous owing to variability in clinical characteristics such as degree of liver impairment, malnourishment, or concomitant underlying infections, which subsequently can lead to large inter-individual variability in PK parameters. Many studies in this review, e.g. almost all studies on cysticercosis and taeniasis [69, 70, 72–75], reported large inter-individual variabilities in PK profiles and parameters. That large unexplained inter-individual variability can limit conclusions of trials is also exemplified in our review. For instance, in one study on Buruli ulcer, no significant differences in PK parameters could be found between two treatment groups because of the small population size and high degree of inter-patient variability [55]. In another study on albendazole in echinococcosis, a dose-dependent increase of the active metabolite’s maximum plasma concentration could not be identified because of high intra- and inter-individual variability in bioavailability [78]. Furthermore, in the PK studies, some attention has been paid to drug–drug and food interactions (28 % of studies), but only a few studies (11 %) made suggestions for optimizing dosing. Furthermore, little attention has been paid to exposure-response relationships (6 % of studies), which is of high importance to make proper decisions regarding dosing schemes.
4.3 Advantages of Pharmacometric Techniques
To optimize treatment regimens and to design efficient and cost-effective clinical trials, the use of population-based analyses can provide substantial advantages. The US Food and Drug Administration and the European Medicines Agency recommend the use of pharmacometrics in data analysis and clinical trial designs, especially in pediatrics or small patient groups [10–12]. Modeling and simulation techniques are pivotal in designing and simulating dosing regimens and trials and are a useful tool to extrapolate proposed regimens, e.g., from healthy to diseased populations. Particularly for pediatrics, the application of quantitative pharmacometric methods has been considered essential to increase the success rates of clinical trials [105]. Not only in a priori pediatric trial design, but particularly also in the a posteriori analysis of collected PK (and PD) data, pharmacometric methods are needed to deal with the typical pediatric challenges of small study populations and a low number of measurements [106]. For many NTDs, e.g., human African trypanosomiasis, leishmaniasis, soil-transmitted helminthiases, schistosomiasis, and dengue, more than 50 % of the burden of disease is occurring in children [4].
The use of population-based approaches can aid in understanding and explaining the heterogeneity in pharmacokinetics and dealing with sparse sampling and smaller sample sizes, which are all so typical for studies in NTDs (see above). Physiological covariates could be identified to understand factors that contribute to variability among patients, even with a smaller sample size, unstructured sampling time points, and sparse data per individual [107]. More information could be gained from studies with high intra- and inter-individual variability when part of the variability can be explained. Therefore, more emphasis on identification of appropriate covariate and error models could be particularly helpful, which requires the application of pharmacometrics [108].
An additional complication with infectious diseases is the need for combination therapy because of the fear of emerging (or already existing) drug resistance of the causative pathogen, and also because of the presence of co-infections. Identifying and understanding drug–drug interactions is therefore important for NTDs, that often appear in conjunction with malaria, human immunodeficiency virus, or tuberculosis, and also here the use of pharmacometrics has been recommended [108–110].
4.4 Extrapolations and Simulation-Based Studies
Only 11 % of the PK studies in our review included pediatric populations, which is in dire contrast to the actual burden of disease of most NTDs, as mentioned above. Again, population PK studies offer many advantages in this context. Pharmacokinetic models derived from the adult population can be extrapolated to pediatric populations based on appropriately identified physiological covariate models [111]. Nevertheless, no studies were identified in this review that actually employed such extrapolation. Similar applications could be useful to extrapolate between ethnic groups or between patients from different geographical regions, all of which are relevant owing to the wide geographical distribution of many NTDs (Table 1). Models could also be used to bridge PK results between different diseases, as some NTD drugs are (primarily) used for other diseases. For example, rifampicin is used for tuberculosis, with various published population PK models available, which could be extrapolated to assess PK–PD relationships for its use in leprosy and Buruli ulcer patients.
Once PK and PK–PD models are developed and exposure-response relationships are established, optimal dosing schemes can be designed to reach the desired drug effect in all patients. This can be achieved in a cost-effective way by PK or PK–PD simulations of various dose regimens, taking into account the actual variability in patient characteristics in the target population. In our review, we identified only four studies that actually made recommendations for alternative dose regimens. These four studies performed population PK analyses and subsequent simulations with the developed models to define optimal doses [25, 26, 38, 42]. Other PK studies, mainly employing non-compartmental or individual compartmental analyses (together 78 %), typically made simple non-specific suggestions to lower or increase the dose, or conclude that more research was needed to suggest alternative doses. Additionally, for at least four studies we identified, a re-analysis performed using population PK methods provided more details about the PK–PD relationship or optimal dose regimens, compared with previous reports of the same PK datasets using conventional analysis methods [26, 36, 38, 42].
Simulation-based studies can also help to limit and assess potential toxicity of dose regimens. For example, one study investigated the pharmacokinetics of benznidazole in Chagas disease patients [26], and simulations revealed that the studied dose might lead to overexposure in most patients. In another study, simulations showed that the maximum ivermectin exposure used in combination with azithromycin was in accordance with the safe exposure that was observed previously [68]. Furthermore, we identified two studies that assessed the toxicity of a drug to infants exposed by breast feeding [23, 41]. In the first study, the drug exposure in breast milk was simulated by only using data from literature, and consequently the exposure of the drug to the infant was predicted. This gave insight into the potential exposure and toxicity in infants without exposing actual infants to the drug. Another example was the use of population-based simulations and data from animal reproductive studies to define human contraceptive cover periods for the potentially teratogenic drug miltefosine for leishmaniasis [41]. Often there is a lack of women included in clinical trials, but still insight can be gained in drug exposure to women by the use of simulation and extrapolation techniques. All these examples highlight the opportunities of applying pharmacometric methods over more conventional analysis methods in assessing therapeutics for NTDs.
5 Conclusion
This review provides an overview of the current status and gaps in PK research for NTDs, as well as the lack of population-based modeling in the performed clinical trials. Simulation and extrapolation tools have been minimally applied. For many NTDs, no registered therapy is available, and more clinical PK trials are needed to establish evidence-based treatments and define PK–PD relationships. To make future clinical trials feasible and affordable, population PK modeling should be used to optimally analyze the often sparse data available and simulation-based studies should be used to inform trial design. This would minimize risks and maximize success rates of clinical trials, and optimally use the limited funding available in this neglected clinical area.
References
World Health Organization. Neglected tropical diseases. 2015. http://www.who.int/neglected_diseases/diseases/en/. Accessed 14 Jan 2016.
The END fund; Ending Neglected Diseases. NTD overview. 2015. http://www.end.org/whatwedo/ntdoverview. Accessed 14 Jan 2016.
Hotez PJ, Alvarado M, Basáñez M-G, et al. The Global Burden of Disease Study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl Trop Dis. 2014;8:e2865.
Murray CJL, Vos T, Lozano R, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–223.
Hotez P, Ottesen E, Fenwick A, Molyneux D. The neglected tropical diseases: the ancient afflictions of stigma and poverty and the prospects for their control and elimination. Adv Exp Med Biol. 2006;582:23–33.
Kennedy PG. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013;12:186–94.
Amato VS, Tuon FF, Bacha HA, Neto VA, Nicodemo AC. Mucosal leishmaniasis: current scenario and prospects for treatment. Acta Trop. 2008;105:1–9.
Sundar S, Sinha PR, Agrawal NK, et al. A cluster of cases of severe cardiotoxicity among kala-azar patients treated with a high-osmolarity lot of sodium antimony gluconate. Am J Trop Med Hyg. 1998;59:139–43.
Pedrique B, Strub-Wourgaft N, Some C, et al. The drug and vaccine landscape for neglected diseases (2000–11): a systematic assessment. Lancet Glob Health. 2013;1:e371–9.
European Medicines Agency. Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population. 2006. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000087.jsp. Accessed 14 Jan 2016.
Food and Drug Administration. Guidance for industry: population pharmacokinetics. 1999. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133184.pdf. Accessed 14 Jan 2016.
Food and Drug Administration. Guidance for industry: exposure-response relationships: study design, data analysis and regulatory applications. 2003. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072109.pdf. Accessed 14 Jan 2016.
Moher D, Shamseer L, Clarke M, et al. Preferred Reporting Items for Systematic Review and Meta-analysis Protocols (PRISMA-P) 2015 statement. Syst Rev. 2015;4:1–9.
Population Approach Group Europe. PAGE-meeting. 2015. http://www.page-meeting.org/default.asp?keuze=search&orderby=&keywordNumber=&keywordAuthor=&keywordTitle=&keywordContent=&meeting=. Accessed 28 Sep 2016.
Gabrielsson J, Weiner D, editors. Pharmacokinetic and pharmacodynamic data analysis: concepts and applications. 4th ed. Stockholm: Taylor & Francis; 2007.
El Guiniady MA, el Touny MA, Abdel-Bary MA, et al. Clinical and pharmacokinetic study of praziquantel in Egyptian schistosomiasis patients with and without liver cell failure. Am J Trop Med Hyg. 1994;51:809–18.
Lang J, Attanath P, Quiambao B, et al. Evaluation of the safety, immunogenicity, and pharmacokinetic profile of a new, highly purified, heat-treated equine rabies immunoglobulin, administered either alone or in association with a purified. Vero-cell rabies vaccine. Acta Trop. 1998;70:317–33.
Gogtay N, Thatte U, Kshirsagar N, et al. Safety and pharmacokinetics of a human monoclonal antibody to rabies virus: a randomized, dose-escalation phase 1 study in adults. Vaccine. 2012;30:7315–20.
Post T, editor. UpToDate. Waltham: UpToDate; 2015.
Bern C. Chagas’ disease. N Engl J Med. 2015;373:456–66.
Shapiro TA, Were JB, Danso K, et al. Pharmacokinetics and metabolism of allopurinol riboside. Clin Pharmacol Ther. 1991;49:506–14.
Were JBO, Shapiro TA. Effects of probenecid on the pharmacokinetics of allopurinol riboside. Antimicrob Agents Chemother. 1993;37:1193–6.
Garcia-Bournissen F, Altcheh J, Panchaud A, Ito S. Is use of nifurtimox for the treatment of Chagas disease compatible with breast feeding? A population pharmacokinetics analysis. Arch Dis Child. 2010;95:224–8.
Richle RW, Raaflaub J. Difference of effective antitrypanosomal dosages of benznidazole in mice and man: chemotherapeutic and pharmacokinetic results. Acta Trop. 1980;37:257–61.
Altcheh J, Moscatelli G, Mastrantonio G, et al. Population pharmacokinetic study of benznidazole in pediatric Chagas disease suggests efficacy despite lower plasma concentrations than in adults. PLoS Negl Trop Dis. 2014;8:e2907.
Soy D, Aldasoro E, Guerrero L, et al. Population Pharmacokinetics of benznidazole in adult patients with Chagas disease. Antimicrob Agents Chemother. 2015;59:3342–9.
World Health Organization. Human African trypanosomiasis. 2015. http://www.who.int/trypanosomiasis_african/en/. Accessed 14 Jan 2016.
Bronner U, Doua F, Ericsson O, et al. Pentamidine concentrations in plasma, whole blood and cerebrospinal fluid during treatment of Trypanosoma gambiense infection in Côte d’Ivoire. Trans R Soc Trop Med Hyg. 1991;85:608–11.
Bronner U, Gustafsson LL, Doua F, et al. Pharmacokinetics and adverse reactions after a single dose of pentamidine in patients with Trypanosoma gambiense sleeping sickness. Br J Clin Pharmacol. 1995;39:289–95.
Harrison SM, Harris RW, Bales JD Jr. Attempt to correlate urine arsenic excretion with clinical course during melarsoprol therapy of patients with Rhodesian trypanosomiasis. Am J Trop Med Hyg. 1997;56:632–6.
Burri C, Baltz T, Giroud C, et al. Pharmacokinetic properties of the trypanocidal drug melarsoprol. Chemotherapy. 1993;39:225–34.
Burri C, Keiser J. Pharmacokinetic investigations in patients from northern Angola refractory to melarsoprol treatment. Trop Med Int Health. 2001;6:412–20.
Bronner U, Brun R, Doua F, et al. Discrepancy in plasma melarsoprol concentrations between HPLC and bioassay methods in patients with T. gambiense sleeping sickness indicates that melarsoprol is metabolized. Trop Med Int Health. 1998;3:913–7.
Milord F, Loko L, Ethier L, et al. Eflornithine concentrations in serum and cerebrospinal fluid of 63 patients treated for Trypanosoma brucei gambiense sleeping sickness. Trans R Soc Trop Med Hyg. 1993;87:473–7.
Na-Bangchang K, Doua F, Konsil J, et al. The pharmacokinetics of eflornithine (alpha-difluoromethylornithine) in patients with late-stage T. b. gambiense sleeping sickness. Eur J Clin Pharmacol. 2004;60:269–78.
Jansson-Löfmark R, Na-Bangchang K, Björkman S, et al. Enantiospecific reassessment of the pharmacokinetics and pharmacodynamics of oral eflornithine against late-stage Trypanosoma brucei gambiense sleeping sickness. Antimicrob Agents Chemother. 2015;59:1299–307.
Tarral A, Blesson S, Mordt OV, et al. Determination of an optimal dosing regimen for fexinidazole, a novel oral drug for the treatment of human African trypanosomiasis: first-in-human studies. Clin Pharmacokinet. 2014;53:565–80.
Gualano V, Felices M, Evene E, et al. Dose regimen assessement for oral fexinidazole, a new antimicrobial agent of the treatment of HAT: population pharmacokinetic analysis of fexinidazole and 2 metabolites. Presented at the 21st Population Approach Group in Europe Meeting, Venice, 5–8 Jun 2012.
Blum J, Buffet P, Visser L, et al. LeishMan recommendations for treatment of cutaneous and mucosal leishmaniasis in travelers. J Travel Med. 2014;21:116–29.
Dorlo TP, van Thiel PP, Huitema AD, et al. Pharmacokinetics of miltefosine in old world cutaneous leishmaniasis patients. Antimicrob Agents Chemother. 2008;52:2855–60.
Dorlo TPC, Balasegaram M, Lima MA, et al. Translational pharmacokinetic modelling and simulation for the assessment of duration of contraceptive use after treatment with miltefosine. J Antimicrob Chemother. 2012;67:1996–2004.
Dorlo TPC, Huitema ADR, Beijnen JH, De Vries PJ. Optimal dosing of miltefosine in children and adults with visceral leishmaniasis. Antimicrob Agents Chemother. 2012;56:3864–72.
Dorlo TPC, Rijal S, Ostyn B, et al. Failure of miltefosine in visceral leishmaniasis is associated with low drug exposure. J Infect Dis. 2014;210:146–53.
Al Jaser M, El-Yazigi A, Croft SL. Pharmacokinetics of antimony in patients treated with sodium stibogluconate for cutaneous leishmaniasis. Pharm Res. 1995;12:113–6.
Vásquez L, Scorza Dagert JV, Scorza JV, et al. Pharmacokinetics of experimental pentavalent antimony after intramuscular administration in adult volunteers. Curr Ther Res Clin Exp. 2006;67:193–203.
Zaghloul IY, Radwan MA, Al Jaser MH. Clinical efficacy and pharmacokinetics of antimony in cutaneous leishmaniasis patients treated with sodium stibogluconate. J Clin Pharmacol. 2010;50:1230–7.
Reymond J-M, Desmeules J. Sodium stibogluconate (pentostan) overdose in a patient with acquired immunodeficiency syndrome. Ther Drug Monit. 1998;20:714–6.
Chulay JD, Fleckenstein L, Smith DH. Pharmacokinetics of antimony during treatment of visceral leishmaniasis with sodium stibogluconate or meglumine antimoniate. Trans R Soc Trop Med Hyg. 1988;82:69–72.
Musa AM, Younis B, Fadlalla A, et al. Paromomycin for the treatment of visceral leishmaniasis in Sudan: a randomized, open-label, dose-finding study. PLoS Negl Trop Dis. 2010;4:e855.
Ravis WR, Llanos-Cuentas A, Sosa N, et al. Pharmacokinetics and absorption of paromomycin and gentamicin from topical creams used to treat cutaneous leishmaniasis. Antimicrob Agents Chemother. 2013;57:4809–15.
Sundar S, Sinha PK, Dixon SA, et al. Pharmacokinetics of oral sitamaquine taken with or without food and safety and efficacy for treatment of visceral leishmaniais: a randomized study in Bihar. India. Am J Trop Med Hyg. 2011;84:892–900.
Al Jaser M, El-Yazigi A, Kojan M, Croft SL. Skin uptake, distribution, and elimination of antimony following administration of sodium stibogluconate to patients with cutaneous leishmaniasis. Microbiology. 1995;39:516–9.
Cruz A, Rainey PM, Herwaldt BL, et al. Pharmacokinetics of antimony in children treated for leishmaniasis with meglumine antimoniate. J Infect Dis. 2007;195:602–8.
Nienhuis WA, Stienstra Y, Thompson WA, et al. Antimicrobial treatment for early, limited Mycobacterium ulcerans infection: a randomised controlled trial. Lancet. 2010;375:664–72.
Alffenaar JWC, Nienhuis WA, De Velde F, et al. Pharmacokinetics of rifampin and clarithromycin in patients treated for Mycobacterium ulcerans infection. Antimicrob Agents Chemother. 2010;54:3878–83.
Pieters FA, Zuidema J. Intra-adipose administration of monoacetyldapsone to healthy volunteers. Int J Lepr Other Mycobact Dis. 1986;54:510–6.
Pieters FA, Zuidema J. The absolute oral bioavailability of dapsone in dogs and humans. Int J Clin Pharmacol Ther Toxicol. 1987;25:396–400.
Garg SK, Kumar B, Bakaya V, et al. Plasma dapsone and its metabolite monoacetyldapsone levels in leprotic patients. Int J Clin Pharmacol Ther Toxicol. 1988;26:552–4.
Moura FM, Dias RM, Araujo EC, et al. Dapsone and body mass index in subjects with multibacillary leprosy. Ther Drug Monit. 2013;36:261–3.
Nix DE, Adam RD, Auclair B, et al. Pharmacokinetics and relative bioavailability of clofazimine in relation to food, orange juice and antacid. Tuberculosis. 2004;84:365–73.
Venkatesan K, Mathur A, Girdhar BK, Bharadwaj VP. The effect of clofazimine on the pharmacokinetics of rifampicin and dapsone in leprosy. J Antimicrob Chemother. 1986;18:715–8.
Venkatesan K, Chauhan SL, Girdhar A, Girdhar BK. Bioavailability of dapsone on oral administration of dapsomine: a comparative evaluation. Indian J Lepr. 1993;65:157–61.
Mehta J, Gandhi I, Sane S, Wamburkar M. Effect of clofazimine and dapsone on rifampicin (Lositril) pharmacokinetics in multibacillary and paucibacillary leprosy cases. Lepr Rev. 1986;57:67–76.
Pieters FA, Woonink F, Zuidema J. Influence of once-monthly rifampicin and daily clofazimine on the pharmacokinetics of dapsone in leprosy patients in Nigeria. Eur J Clin Pharmacol. 1988;34:73–6.
Teo SK, Scheffler MR, Kook KA, et al. Thalidomide dose proportionality assessment following single doses to healthy subjects. J Clin Pharmacol. 2001;41:662–7.
Teo SK, Colburn WA, Thomas SD. Single-dose oral pharmacokinetics of three formulations of thalidomide in healthy male volunteers. J Clin Pharmacol. 1999;39:1162–8.
Amsden GW, Gregory TB, Michalak CA, et al. Pharmacokinetics of azithromycin and the combination of ivermectin and albendazole when administered alone and concurrently in healthy volunteers. Am J Trop Med Hyg. 2007;76:1153–7.
El-Tahtawy A, Glue P, Andrews EN, et al. The effect of azithromycin on ivermectin pharmacokinetics: a population pharmacokinetic model analysis. PLoS Negl Trop Dis. 2008;2:e236.
Jung H, Sanchez M, Gonzalez-Astiazaran A, et al. Clinical pharmacokinetics of albendazole in children with neurocysticercosis. Am J Ther. 1997;4:23–6.
Sanchez M, Suastegui R, Gonzalez-Esquivel D, et al. Pharmacokinetic comparison of two albendazole dosage regimens in patients with neurocysticercosis. Clin Neuropharmacol. 1993;16:77–82.
Takayanagui OM, Lanchote VL, Marques MP, Bonato PS. Therapy for neurocysticercosis: pharmacokinetic interaction of albendazole sulfoxide with dexamethasone. Ther Drug Monit. 1997;19:51–5.
Na-Bangchang K, Vanijanonta S, Karbwang J. Plasma concentrations of praziquantel during the therapy of neurocysticerosis with praziquantel, in the presence of antiepileptics and dexamethasone. Southeast Asian J Trop Med Public Health. 1995;26:120–3.
Jung H, Medina R, Castro N, et al. Pharmacokinetic study of praziquantel administered alone and in combination with cimetidine in a single-day therapeutic regimen. Antimicrob Agents Chemother. 1997;41:1256–9.
Garcia HH, Lescano AG, Lanchote VL, et al. Pharmacokinetics of combined treatment with praziquantel and albendazole in neurocysticercosis. Br J Clin Pharmacol. 2011;72:77–84.
Jung H, Hurtado M, Sanchez M, et al. Clinical pharmacokinetics of albendazole in patients with brain cysticercosis. J Clin Pharmacol. 1992;32:28–31.
Cotting J, Zeugin T, Steiger U, Reichen J. Albendazole kinetics in patients with echinococcosis: delayed absorption and impaired elimination in cholestasis. Eur J Clin Pharmacol. 1990;38:605–8.
Mingjie W, Shuhua X, Junjie C, et al. Albendazole-soybean oil emulsion for the treatment of human cystic echinococcosis: evaluation of bioavailability and bioequivalence. Acta Trop. 2002;83:177–81.
Schipper HG, Koopmans RP, Nagy J, et al. Effect of dose increase or cimetidine co-administration on albendazole bioavailability. Am J Trop Med Hyg. 2000;63:270–3.
Na Bangchang K, Karbwang J, Pungpak S, et al. Pharmacokinetics of praziquantel in patients with opisthorchiasis. Southeast Asian J Trop Med Public Health. 1993;24:717–23.
Choi M-H, Chang B-C, Lee S-J, et al. Therapeutic evaluation of sustained-releasing praziquantel (SRP) for clonorchiasis: phase 1 and 2 clinical studies. Korean J Parasitol. 2006;44:361.
Lecaillon JB, Godbillon J, Campestrini J, et al. Effect of food on the bioavailability of triclabendazole in patients with fascioliasis. Br J Clin Pharmacol. 1998;45:601–4.
El-Tantawy WH, Salem HF, Mohammed Safwat NA. Effect of fascioliasis on the pharmacokinetic parameters of triclabendazole in human subjects. Pharm World Sci. 2007;29:190–8.
Shenoy RK, Suma TK, John A, et al. The pharmacokinetics, safety and tolerability of the co-administration of diethylcarbamazine and albendazole. Ann Trop Med Parasitol. 2002;96:603–14.
Sarin R, Dash AP, Dua VK. Albendazole sulphoxide concentrations in plasma of endemic normals from a lymphatic filariasis endemic region using liquid chromatography. J Chromatogr B Anal Technol Biomed Life Sci. 2004;799:233–8.
Abdel-Tawab AM, Bradley M, Ghazaly EA, et al. Albendazole and its metabolites in the breast milk of lactating women following a single oral dose of albendazole. Br J Clin Pharmacol. 2009;68:737–42.
Awadzi K, Hero M, Opoku NO, et al. The chemotherapy of onchocerciasis XVII: a clinical evaluation of albendazole in patients with onchocerciasis; effects of food and pretreatment with ivermectin on drug response and pharmacokinetics. Trop Med Parasitol. 1994;45:203–8.
Awadzi K, Edwards G, Duke BOL, et al. The co-administration of ivermectin and albendazole: safety, pharmacokinetics and efficacy against Onchocerca volvulus. Ann Trop Med Parasitol. 2003;97:165–78.
Okonkwo PO, Ogbuokiri JE, Ofoegbu E, Klotz U. Protein binding and ivermectin estimations in patients with onchocerciasis. Clin Pharmacol Ther. 1993;53:426–30.
Baraka OZ, Mahmoud BM, Marschke CK, et al. Ivermectin distribution in the plasma and tissues of patients infected with Onchocerca volvulus. Eur J Clin Pharmacol. 1996;50:407–10.
Homeida MM, Malcolm SB, ElTayeb AZ. The lack of influence of food and local alcoholic brew on the blood level of Mectizan® (ivermectin). Acta Trop. 2013;127:97–100.
Chijioke CP, Umeh RE, Mbah AU, et al. Clinical pharmacokinetics of suramin in patients with onchocerciasis. Eur J Clin Pharmacol. 1988;54:249–51.
Korth-Bradley JM, Parks V, Chalon S, et al. Excretion of moxidectin into breast milk and pharmacokinetics in healthy lactating women. Antimicrob Agents Chemother. 2011;55:5200–4.
Lecaillon JB, Dubois JP, Awadzi K, et al. Pharmacokinetics of CGP 6140 (amocarzine) after oral administration of single 100–1600 mg doses to patients with onchocerciasis. Br J Clin Pharmacol. 1990;30:625–8.
Lecaillon JB, Poltera AA, Zea-Flores G, et al. Influence of food related to dose on the pharmacokinetics of amocarzine and of its N-oxide metabolite, CGP 13 231, after oral administration to 20 onchocerciasis male patients from Guatemala. Trop Med Parasitol. 1991;42:286–90.
Pehrson PO, Bengtsson E, Diekmann HW, Groll E. Treatment with praziquantel in a patient with schistosomiasis and chronic renal failure. Trans R Soc Trop Med Hyg. 1983;77:687–8.
Mandour ME, El Turabi H, Homeida MMA, et al. Pharmacokinetics of praziquantel in healthy volunteers and patients with schistosomiasis. Trans R Soc. 1990;84:389–93.
Valencia CI, Catto BA, Webster LT, et al. Concentration time course of praziquantel in Filipinos with mild Schistosoma japonicum infection. Southeast Asian J Trop Med Public Health. 1994;25:409–14.
Nordgren I, Holmstedt BO, Bengtsson E, Finkel A. Plasma levels of metrofonate and dichlorvos during treatment of schistosomiasis with Bilarcil®. Am J Trop Med Hyg. 1980;29:426–30.
Daneshmend TK, Homeida MA. Oxamniquine pharmacokinetics in hepatosplenic schistosomiasis in the Sudan. J Antimicrob Chemother. 1987;19:87–93.
Merigan TC, Baer GM, Winkler WG, et al. Human leukocyte interferon administration to patients with symptomatic and suspected rabies. Ann Neurol. 1984;16:82–7.
Olliaro P, Delgado-Romero P, Keiser J. The little we know about the pharmacokinetics and pharmacodynamics of praziquantel (racemate and R-enantiomer). J Antimicrob Chemother. 2014;69:863–70.
Wilby KJ, Gilchrist SE, Ensom MHH. A review of the pharmacokinetic implications of schistosomiasis. Clin Pharmacokinet. 2013;52:647–56.
Wagner JG. History of pharmacokinetics. Pharmacol Ther. 1981;12:537–62.
Hotez PJ, Yamey G. The evolving scope of PLoS neglected tropical diseases. PLoS Negl Trop Dis. 2009;3:e379.
Gobburu JVS. Pharmacometrics 2020. J Clin Pharmacol. 2010;50:151S–7S.
Jadhav PR, Kern SE. The need for modeling and simulation to design clinical investigations in children. J Clin Pharmacol. 2010;50:121S–9S.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development. Part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst. Pharmacol. 2013;2:e38.
Pillai G, Davies G, Denti P, et al. Pharmacometrics: opportunity for reducing disease burden in the developing world: the case of Africa. CPT Pharmacometrics Syst Pharmacol. 2013;2:e69.
Davies GR, Hope W, Khoo S. Opinion: the pharmacometrics of infectious disease. CPT Pharmacometrics Syst Pharmacol. 2013;2:e70.
Svensson EM, Acharya C, Clauson B, et al. Pharmacokinetic interactions for drugs with a long half-life: evidence for the need of model-based analysis. AAPS J. 2016;18:171–9.
European Medicines Agency (EMA). Reflection paper on extrapolation of efficacy and safety in paediatric medicine development. 2016. http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2016/04/WC500204187.pdf. Accessed 4 Oct 2016.
Acknowledgments
We thank Dr. Steven Kern from the Bill and Melinda Gates Foundation (Seattle, USA) for his critical review of the manuscript and helpful comments.
Author contributions
LV performed the literature survey, carried out the data extraction, and drafted an initial version of the report. TD conceived of the study and wrote subsequent drafts and the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No specific funding was received for the conduct of this systematic review.
Conflict of interest
TD is a consultant for the non-profit organization Drugs for Neglected Diseases initiative, Geneva, Switzerland.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Verrest, L., Dorlo, T.P.C. Lack of Clinical Pharmacokinetic Studies to Optimize the Treatment of Neglected Tropical Diseases: A Systematic Review. Clin Pharmacokinet 56, 583–606 (2017). https://doi.org/10.1007/s40262-016-0467-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0467-3