
Abstract
Background and Objective
Cytomegalovirus (CMV) is a common opportunistic infection after allogenic hematopoietic stem cell transplantation (allo-HSCT). Letermovir, an inhibitor of CMV DNA terminase, is approved for CMV prophylaxis in allo-HSCT patients. We report the final results of post-marketing surveillance of letermovir in Japan.
Methods
The case report forms were drafted in part by the Japanese Data Center for Hematopoietic Cell Transplantation using data elements in the Transplant Registry Unified Management Program and sent to individual HSCT centers to decrease the burden of reporting. Hematopoietic stem cell transplantation patients who received letermovir between May 2018 and May 2022 were registered. Data collected included physician-assessed adverse events/adverse drug reactions and clinical effectiveness (development of CMV disease, CMV antigen status, and use of preemptive therapy).
Results
A total of 821 HSCT patients were included in the safety analyses. Adverse drug reactions occurred in 11.33% of patients, with serious adverse drug reactions in 3.05%. The five most common adverse drug reactions were nausea (1.58%), renal impairment (1.46%), and acute graft versus host disease, CMV test positive, and hepatic function abnormal (0.61% each). A total of 670 patients were eligible for effectiveness analyses. Among these patients, 16.57% and 28.66% required preemptive therapy through week 14 and week 48, respectively. In addition, relatively few patients developed CMV disease throughout the follow-up period (1.34% at week 14 and 3.85% at week 48).
Conclusions
This final analysis of post-marketing surveillance with up to 48 weeks follow-up period in Japan provides further evidence supporting the safety profile and effectiveness of letermovir for CMV prophylaxis in patients undergoing allo-HSCT in real-world settings.
Plain Language Summary
Cytomegalovirus (CMV) infection is common after allogenic hematopoietic stem cell transplantation and causes both directly and indirectly a serious disease that frequently results in the death or severe outcomes for the affected patient. Letermovir is a drug that inhibits CMV replication and infection and can be administered to prevent CMV infection in at-risk patients undergoing allogenic hematopoietic stem cell transplantation. After it was approved in Japan, a post-marketing surveillance was started in order to confirm the safety profile and effectiveness of letermovir in clinical practice in Japan. The data collected included the adverse drug reactions during treatment and the effectiveness of letermovir. In this article, we describe the final results of this survey. The most common adverse drug reactions were nausea (1.58% of patients), renal impairment (1.46%), and acute graft versus host disease, CMV test positive, and hepatic function abnormal (0.61% each). There were few cases of myelosuppression, which is frequently seen in patients treated with ganciclovir/valganciclovir, and blood cells recovered steadily over time. Cytomegalovirus antigens were detected in 38.36% of patients through 48 weeks. Preemptive therapy was initiated to 28.66% of patients for up to 48 weeks. Cytomegalovirus disease was infrequent, occurring in 3.85% of patients. Overall, these findings are in alignment with the currently approved product label and provide further evidence supporting the consistent safety profile and effectiveness of letermovir for CMV prophylaxis in patients in Japan undergoing allogenic hematopoietic stem cell transplantation in clinical practice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
We report the final results of a preemptive therapy to assess the safety profile and effectiveness of letermovir for cytomegalovirus (CMV) prophylaxis for up to 48 weeks in patients undergoing allogenic hematopoietic stem cell transplantation in Japan. |
Adverse drug reactions and serious adverse drug reactions occurred in 11.33% and 3.05% of the patients, respectively. At weeks 14 and 48, the rates of CMV antigen positivity were 21.34% and 38.36%, 16.57%, and 28.66% of patients required preemptive therapy, and 1.44% and 3.85% developed CMV disease, respectively. |
Overall, the results of this large-scale preemptive therapy provide further evidence to support the safety profile and effectiveness of letermovir for CMV prophylaxis in real-world settings in Japan. |
1 Introduction
Cytomegalovirus (CMV) is one of the most common opportunistic infections in patients undergoing hematopoietic stem cell transplantation (HSCT) [1] that usually occurs as a result of reactivation in patients with seropositivity for CMV. Notably, CMV was reported to reactivate in up to 70% of HSCT recipients without prophylaxis [2,3,4]. Cytomegalovirus reactivation is associated with a poor prognosis in HSCT patients by increasing the risks of complications and subsequently mortality [2, 5, 6]. A recent study revealed that CMV reactivation significantly increased the risks of both overall mortality and non-relapse mortality after allogenic-HSCT (allo-HSCT), with hazard ratios (HRs) of 1.46 and 1.41, respectively [7].
Although ganciclovir prophylaxis can effectively prevent CMV reactivation following HSCT, it was not reported to improve survival, which has been explained by increased infection in neutropenic settings, and the utility of ganciclovir prophylaxis is limited by its toxicity [8, 9]. Preemptive therapy (PET) involving early administration of antiviral drugs based on frequent CMV monitoring can also reduce the risk of developing CMV disease [1, 10,11,12]. However, this strategy is not completely a satisfiable solution to manage CMV infection in allo-HSCT patients because it was reported that CMV reactivation requiring PET was associated with increased risks of overall mortality and non-relapse mortality in allo-HSCT recipients [7]. Considering the significant impact of CMV reactivation on prognosis and the limited therapeutic options, alternative strategies were needed to help prevent CMV reactivation and reduce the excess morbidity and mortality associated with this complication in allo-HSCT patients.
Letermovir inhibits CMV DNA terminase, responsible for producing the terminal complex, and thus disrupts viral replication [13]. A large phase III clinical trial [14, 15] confirmed that letermovir prophylaxis reduces clinically significant CMV infection (CMV disease or CMV viremia leading to PET) among allo-HSCT recipients, a finding supported by several real-world studies [16,17,18,19,20,21]. Following the phase III trial, letermovir was approved in the USA in 2017 and in Japan and the European Union in 2018 [22, 23]. Letermovir was indicated in Japan upon initial approval for the prevention of CMV disease in allo-HSCT patients regardless of CMV serostatus. In consideration of its effectiveness, letermovir is now widely recommended in clinical guidelines, including in Japan, for preventing CMV infection in patients undergoing allo-HSCT [12, 24, 25].
At the time of approval, the clinical experience of letermovir in Japan was limited because the phase III trial enrolled only 36 Japanese patients [14]. Although several small real-world studies have been reported in recent years [26,27,28,29], there was limited information on the use of letermovir in clinical practice. As part of the approval conditions for letermovir in Japan, retrospective post-marketing surveillance (PMS) was initiated to record its safety profile and effectiveness in clinical use. Interim results of the PMS were published in 2021 for 461 patients treated with letermovir as of March 2021, out of a total of 932 registered patients [30]. Here, we report the final results of the PMS for 838 registered patients for whom the case report forms (CRFs) had been collected by 20 May, 2022.
2 Methods
2.1 PMS Design and Patients
In Japan, HSCT centers are required to report outcome data to the Japanese Society for Transplantation and Cellular Therapy and the Japanese Data Center for Hematopoietic Cell Transplantation using the Transplant Registry Unified Management Program (TRUMP) [31,32,33]. The TRUMP dataset is fixed annually and is used to generate annual report/summary slides for HSCT activities and outcomes and for other research purposes [31, 32]. For this PMS, the Japanese Society for Transplantation and Cellular Therapy and the Japanese Data Center for Hematopoietic Cell Transplantation were enlisted to support the transplant centers by partially drafting CRFs, using data elements in TRUMP, and the CRFs were then sent to the participating physicians to complete in order to reduce the burden placed on the physicians. Specifically, the data elements in TRUMP included the patient demographics, disease backgrounds, transplant sources, details of CMV treatment after transplantation, and outcomes.
Hematopoietic stem cell transplantation patients who received letermovir by an accredited physician at one of 136 centers across Japan between May 2018 and May 2022 were to be registered in the PMS. The database was locked on 5 October, 2022. In addition to CMV antibody-positive recipients, CMV antibody-negative recipients were also eligible for this PMS in accordance with the approved indication in Japan and the Japan Risk Management Plan (Japan-RMP) approved by the Pharmaceuticals and Medical Devices Agency, which requires evaluation of the effectiveness of letermovir in CMV antibody-negative recipients from antibody-positive donors [34]. Patients were followed up for 48 weeks after transplantation for safety and effectiveness outcomes. The Japanese prescribing information at the period of this PMS analyses [35, 36] recommends administering letermovir as an oral tablet or intravenously at a dose of 480 mg and reduced to 240 mg when given with cyclosporine, once daily, through 100 days post-transplantation.
2.2 Assessments
As previously described [30], the CRFs were designed to record information that included patient demographic characteristics, clinical history, transplantation information, and therapies administered prior to and post-transplantation. The CRFs primarily recorded adverse drug reactions (ADRs), defined as adverse events (AEs) for which a causal relationship with letermovir could not be ruled out by the attending physician, including the priority survey items (renal disorders with intravenous [IV] administration, cardiac disorders, and developmental and reproductive toxicity) described in the Japan-RMP [34]. The physicians also determined the seriousness of each AE/ADR. Adverse drug reactions were categorized according to the Medical Dictionary for Regulatory Activities version 25.0.
The clinical effectiveness of letermovir was also evaluated by the attending physician in terms of the following outcomes: CMV antigen status, the use of PET, and the development of CMV disease. To monitor CMV infection, polymerase chain reaction or CMV antigenemia were recommended at baseline, treatment initiation, and at 1, 8, 14, 24, and 48 weeks post-transplantation. The thresholds for PET and target therapy, as well as the treatment strategy in the event of CMV disease, were at the attending physician’s discretion.
2.3 Data Analyses
Two patient sets were evaluated in this PMS: the safety and effectiveness analysis set. The safety analysis set included all patients treated with letermovir within the surveillance period, excluding any duplicates and patients for whom the presence/absence of AEs was unknown to avoid overestimating the safety data. The effectiveness analysis set comprised all patients included in the safety analysis set, excluding those who received letermovir for a non-approved indication or at a non-approved dose. The baseline, safety, and effectiveness data were analyzed using relevant methods and are reported as the count/number of patients, percentages, mean, standard deviation, median, range, and interquartile range as appropriate. We also used the Kaplan–Meier method to plot CMV disease rates and survival rates over time. For this, patients were considered to have an event if they developed CMV disease at any time post-transplantation or their final outcome was survival. The effectiveness analyses were truncated at 48 weeks; patients whose last observation date was after 48 weeks were censored at this time. Univariate Cox regression analyses were performed to identify possible risk factors for CMV antigen positivity at weeks 14, 24, and 48, separately. Statistical Analysis System version 9.4 (SAS Institute Inc., Cary, NC, USA) was used for all data analyses.
3 Results
3.1 Patient Disposition
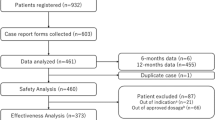
Overall, 932 patients were registered in this PMS (Fig. 1) across 136 centers. Because CRFs had not been collected for 94 patients at the time of the database lock, 838 patients were eligible for the analyses. Seventeen patients were excluded, resulting in 821 patients being eligible for the safety analysis set (Fig. 1). A further 151 patients were excluded from the effectiveness analysis set; 119 did not receive an approved dose of letermovir and 32 received letermovir for off-label indication. Thus, the effectiveness analysis set comprised 670 patients.

Patient flowchart. *Includes six patients who did not receive letermovir and one duplicate patient. †Includes five pediatric patients. AEs adverse events, CRF case report form
3.2 Patient Characteristics
The median age of the patients in the safety data set was 53 years, and 59.20% were male (Table 1). Most patients (81.61%) underwent one transplantation during the surveillance period. The median duration of hospital stay after the first transplantation was 87.0 days. The most common reason for HSCT was acute myeloid leukemia (37.88%). Over 60% of recipients were positive for CMV antibodies (R+/D+: 34.96%; R+/D−: 33.25%). The stem cells were derived from cord blood (36.42%), bone marrow (33.13%), and peripheral blood (30.45%). Regarding donor type, allogeneic transplantation was performed in nearly all of the patients (99.63%; 29.60% related and 70.04% unrelated), autologous transplantation was performed in one patient, and the donor type was unknown in two patients. Human leukocyte antigen status was matched in 32.40%, mismatched excluding haploidentical in 50.79%, and haploidentical in 13.40% of patients. The most common conditioning regimen was myeloablative (47.38%). Pre-transplant corticosteroids were administered to 47.50% of patients. Nearly all (96.83%) of the patients underwent graft versus host disease (GVHD) prophylaxis using tacrolimus (78.68%) and/or methotrexate (72.84%). Acute and chronic post-transplant GVHD occurred in 50.67% and 18.03% of patients, respectively, and engraftment was achieved in most patients (92.81%) [Table 1 of the Electronic Supplementary Material [ESM]). The characteristics of patients included in the effectiveness analysis set were similar to those included in the safety analysis set (Tables 1, 2), suggesting no evidence of bias in the terms of baseline factors in the effectiveness analysis set.
3.3 Letermovir Administration
Approximately three quarters of patients (76.00%) started letermovir at a dose of 480 mg without cyclosporine (Table 3), and 16.57% started at a dose of 240 mg combined with cyclosporine. Small proportions of patients were started on letermovir 240 mg without cyclosporine (6.46%) or letermovir 480 mg with cyclosporine (0.61%). While 23.87% of patient used a combination of an oral and IV formulation, the majority patients received oral tablets (72.35%) only and a few received IV (3.53%) only. The dosage form was switched from oral tablets to an IV infusion, or vice versa, in 23.87% of patients. The median time from transplantation to the first dose of letermovir was 2 days (range −10 to 100 days; interquartile range 1.0–8.0 days). Letermovir was administered for >70 days in 58.83%, and up to 100 days in 85.99% with a median of 83.0 days (range 1–521 days; interquartile range 43–98 days).
3.4 Safety
3.4.1 ADRs
Adverse drug reactions, as assessed by the attending physician, were reported in 93 patients (11.33%) in the safety analysis set (Table 4). Adverse drug reactions that occurred in ≥0.5% of patients were nausea (1.58%), renal impairment (1.46%), acute GVHD (0.61%), CMV test positive (0.61%), and hepatic function abnormal (0.61%). Serious ADRs, as assessed by the attending physician, occurred in 3.05% of patients; ascites, acute kidney injury, graft failure, hepatic function abnormal, and hepatic vein occlusion each occurred in 0.24% of patients (Table 4).
The frequency of ADRs did not differ among patients treated with letermovir 240 or 480 mg (9.52% and 11.92%, respectively), between the dosage forms (tablet: 11.11%; IV: 6.90%; switched between dosage form: 12.76%), or between patients treated with or without cyclosporine (11.35% and 11.32%, respectively) [Table 2 of the ESM].
3.4.2 Priority Survey Items Listed in the Japan-RMP
This PMS also assessed priority items based on the Japan-RMP, and the frequencies of these AEs are presented in Table 5. Adverse events related to a renal disorder during IV letermovir administration were reported in 15 patients (6.67%). These AEs comprised renal dysfunction (n = 8), blood creatinine increased (n = 4), acute kidney injury (n = 2), and renal failure (n = 1). Of these, one case each of renal dysfunction and acute kidney injury were considered serious ADRs.
Though not specified in the Japan-RMP, we have assessed the AEs related to a renal disorder observed in patients treated with oral letermovir, and found that they were reported in 34 patients (4.30%); renal impairment (n = 15), cystitis hemorrhagic (n = 6), acute kidney injury (n = 4), renal dysfunction (n = 3), blood creatinine increased (n = 2), chronic kidney disease (n = 2), renal failure (n = 2), and tubulointerstitial nephritis (n = 1). Only one case with acute kidney injury was recorded as a serious ADR.
Cardiac disorders were reported as AEs in 25 patients (3.05%) [IV or oral letermovir], none of which was recorded as serious ADRs. Non-serious ADRs related to cardiac disorders were cardiomyopathy (n = 2), atrial fibrillation (n = 1), and blood pressure increased (n = 1). Administration of letermovir in pregnant or lactating female patients and ADRs associated with reproductive or developmental toxicity in male patients were not reported.
3.4.3 Recovery of Blood Cell Counts After Transplantation
Most patients showed recovery of blood cells counts as noted after transplantation. Among 821 patients in the safety analysis set, the neutrophil count recovered to ≥ 500 cells/μL in 749 (91.23%), the reticulocyte count recovered to ≥ 10% in 683 (83.19%), the platelet count recovered to ≥ 20,000 cells/μL in 651 (79.29%) or to ≥ 50,000 cells/μL in 613 (74.67%), and the white blood cell count recovered to ≥ 1000 cells/μL in 745 (90.74%).
3.5 Effectiveness
The effectiveness of letermovir was evaluated in 670 patients for up to 48 weeks after HSCT by evaluating CMV antigen positivity, use of PET, CMV disease, and survival.
3.5.1 CMV Antigen Positivity
The CMV antigen positivity rate at weeks 14, 24, and 48 were 21.34%, 36.72%, and 38.36%, respectively (Fig. 2). We also performed univariate Cox regression analyses to identify potential risk factors for CMV antigen positivity at weeks 14, 24, and 48 (Tables 3, 4, 5 of the ESM). The risk factors at week 14 were CMV antibody status (recipient positive, donor negative), human leukocyte antigen status (mismatch, haploidentical), non-myeloablative transplantation, and post-transplant GVHD (acute and chronic). The same risk factors were also found at weeks 24 and 48, but corticosteroid use was identified as a risk factor only at week 48.

Cytomegalovirus (CMV) antigen positivity rate within 14, 24, and 48 weeks (N = 670). Values are n (%)
3.5.2 PET
Preemptive therapy was administered to 16.57%, 27.61%, and 28.66% of patients through weeks 14, 24, and 48, respectively (Fig. 3). Table 6 shows the proportions of patients without PET at weeks 14 and 24 for each subgroup. As might be anticipated, the proportions of patients without PET at 14 and 24 weeks were significantly greater if the recipient was CMV antibody negative. The proportion of patients without PET up to week 24 was similar between patients who received cord blood transplant (69.67%), bone marrow transplant (72.97%), or peripheral blood transplant (75.00%). The proportions of patients without PET differed significantly among some subgroups, including recipient CMV antibody status, human leukocyte antigen compatibility, acute or chronic GVHD after transplantation, engraftment, and comorbidities (Table 6), reflecting the risk factors for CMV antigen positivity described in the previous section.

Percentage of patients who received preemptive therapy up to weeks 14, 24, and 48 (N = 670). Values are n (%)
3.5.3 CMV Disease
According to the Kaplan–Meier analysis, the rates of CMV disease at weeks 14, 24, and 48 were 1.34%, 2.99%, and 3.28%, respectively. Of the 22 patients who developed CMV disease, there were 25 events through week 48 that included enterocolitis (n = 14), retinitis (n = 3), viremia (n = 3), hepatitis (n = 2), pneumonia (n = 2), and gastritis (n = 1).
3.5.4 Survival
Survival rates analyzed using the effectiveness analysis set were 89.28%, 81.79%, and 71.88% at weeks 14, 24, and 48, respectively (Fig. 1 of the ESM).
4 Discussion
Letermovir is a CMV DNA terminase inhibitor that disrupts viral replication and has been approved in Japan and other countries for the prevention of CMV infection and disease following allo-HSCT. Here, we report the final results of the PMS that serve as the first nationwide study with the largest number of patients of such studies. We provide the real-world data for the safety profile and effectiveness of letermovir in the Japanese clinical setting. The results obtained in this larger population of patients were essentially consistent with the interim analysis in terms of the safety profile and effectiveness data [30].
The present analysis accumulated safety data for almost double the number of patients, especially those treated with letermovir for >40 days (n = 611) compared with the interim analysis (n = 351), offering a greater opportunity to detect the priority survey items and infrequent ADRs although no notable infrequent ADRs were detected (Table 6 of the ESM). The frequency of ADRs in this PMS (11.33%) is consistent with that reported in the international phase III study (16.9%). Although direct comparisons are difficult to make, the most common AEs in the phase III trial were GVHD, diarrhea, nausea, fever, and rash [14], while common ADRs in this PMS were nausea (1.58%), renal impairment (1.46%), acute GVHD (0.61%), CMV test positive (0.61%), and hepatic function abnormal (0.61%). Regarding myelotoxicity, myelosuppression was reported as an AE in one patient (0.12%) and did not show evidence of myelotoxicity due to letermovir administration in this PMS as well as in the phase III trial. Renal disorders, as AEs, were specified as priority survey items because of the possibility of worsening of renal function from the accumulation of hydroxypropyl β-cyclodextrin in the IV formulation. The frequency of renal disorders in the IV formulation was 6.67%, while in the tablet formulation it was 4.30%. However, these values cannot be directly compared because of the different patient conditions and duration of administration, such as that patients who received the IV formulation were assumed to be in a worse condition than those who received oral tablets. Overall, the results of this PMS suggest that the safety profile of letermovir in real-world Japanese clinical practice is similar to that observed in the clinical trials. This PMS also assessed the effectiveness data with the results consistent with the phase III study in that the frequency of CMV antigen positivity and the proportion of patients initiating PET increased from week 14 (21.34% and 16.57%, respectively) to week 24 (36.72% and 27.61%, respectively).
In the study by Mizuno et al., the only independent risk factor for clinically significant CMV infection after allo-HSCT under letermovir prophylaxis was transplantation from a seronegative donor to a seropositive recipient (HR 2.76; 95% CI 1.14–6.68) [26]. However, the HR for CMV antigen positivity in R+/D− versus R+/D+ patients was 0.96 at week 14 (95% CI 0.66–1.40) in this PMS, reaching a comparable risk potentially because of successful letermovir prophylaxis. However, the risk of CMV antigen positivity tended to be higher in R+/D− patients than in R+/D+ patients, although it was not significantly different at week 24 (HR 1.30, 95% CI 0.97–1.75) or week 48 (HR 1.30, 95% CI 0.98–1.73) after letermovir prophylaxis was completed in most of the patients. This suggests that a longer duration of administration may be beneficial in patients who remain at high risk for CMV infection, even 100 days after HSCT, including R+/D− patients. It is important to consider that the Japanese prescribing information at the time of this PMS recommended letermovir prophylaxis for approximately 100 days [35, 36].
Consistent with the results of CMV antigen positivity, we found that the proportions of patients without PET through week 14 was comparable between the R+/D− (82.13%) and R+/D+ (82.43%) patients, and these proportions diverged slightly through 24 weeks (65.96% and 73.87%, respectively). In addition, we found that different HSCT donor sources did not affect the proportion of patients requiring PET, providing evidence that letermovir prophylaxis is effective to prevent CMV infection regardless of HSCT donor sources. Unlike the phase III study, R− patients were included in this PMS, and interestingly, 12.2% of R− patients showed positive CMV antigen conversion after week 14. These data suggest that CMV infection was prevented during letermovir administration, but CMV infection occurred after letermovir discontinuation, which indicates even R− patients were at risk of CMV infection after HSCT and letermovir prophylaxis may be beneficial in R− patients.
Generally, our results are consistent with those reported in the phase III trial, in which 17.5% of patients developed clinically significant CMV infection by week 24, and 16.0% of patients received PET, out of 325 patients in the letermovir group [14]. Our PMS reported effectiveness data through 48 weeks of prophylaxis, and the results suggest that the incidence of CMV infection remains low throughout this time period.
Last, dosing of letermovir for up to 200 days was approved in August 2023 in Japan based on the results of a recently completed phase III trial (NCT03930615) [37], in which letermovir was administered continuously for 200 days after allo-HSCT. In this PMS, 113 patients (within the safety analysis set) continued letermovir administration for >100 to ≤200 days. Favorably, administration of letermovir for this extended period was not associated with an increased incidence of ADRs, which indicates that the safety profile of letermovir administration for over 100 days is consistent with the phase III trial of extended letermovir administration. Overall, the present PMS have provided valuable evidence showing the safety profile and effectiveness of letermovir for preventing CMV infection after allo-HSCT.
4.1 Limitations
This PMS has some limitations that should be considered when interpreting the results. The PMS utilized retrospective data and the follow-up period is limited in which reporting individual healthcare providers made decisions on AEs and causality. This PMS, which included the completions of CRFs and submission of reports to TRUMP, did not occur for all patients despite being encouraged in all centers that prescribed letermovir during the surveillance period. Furthermore, because of the absence of a control group and no formal comparison conducted, the interpretation of effectiveness is limited.
5 Conclusions
This final analysis of the PMS in Japan of letermovir provides valuable evidence to support the safety profile and effectiveness of letermovir for preventing CMV infection and disease in patients undergoing allo-HSCT throughout the 48-week follow-up period. The findings of this PMS are expected to be useful for guiding clinical practice, considering the prior phase III study comprised relatively few Asian patients and this PMS contains the largest amount of data on real-word letermovir treatment in this setting.
References
Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol Oncol Clin North Am. 2011;25(1):151–69. https://doi.org/10.1016/j.hoc.2010.11.011.
Green ML, Leisenring W, Xie H, Mast TC, Cui Y, Sandmaier BM, et al. Cytomegalovirus viral load and mortality after haemopoietic stem cell transplantation in the era of pre-emptive therapy: a retrospective cohort study. Lancet Haematol. 2016;3(3):e119–27. https://doi.org/10.1016/s2352-3026(15)00289-6.
Giménez E, Torres I, Albert E, Piñana JL, Hernández-Boluda JC, Solano C, et al. Cytomegalovirus (CMV) infection and risk of mortality in allogeneic hematopoietic stem cell transplantation (Allo-HSCT): a systematic review, meta-analysis, and meta-regression analysis. Am J Transplant. 2019;19(9):2479–94. https://doi.org/10.1111/ajt.15515.
Cho SY, Lee DG, Kim HJ. Cytomegalovirus infections after hematopoietic stem cell transplantation: current status and future immunotherapy. Int J Mol Sci. 2019;20(11):2666. https://doi.org/10.3390/ijms20112666.
Takenaka K, Nishida T, Asano-Mori Y, Oshima K, Ohashi K, Mori T, et al. Cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation is associated with a reduced risk of relapse in patients with acute myeloid leukemia who durvived to day 100 after transplantation: the Japan Society for Hematopoietic Cell Transplantation Transplantation-related Complication Working Group. Biol Blood Marrow Transplant. 2015;21(11):2008–16. https://doi.org/10.1016/j.bbmt.2015.07.019.
Schmidt-Hieber M, Labopin M, Beelen D, Volin L, Ehninger G, Finke J, et al. CMV serostatus still has an important prognostic impact in de novo acute leukemia patients after allogeneic stem cell transplantation: a report from the Acute Leukemia Working Party of EBMT. Blood. 2013;122(19):3359–64. https://doi.org/10.1182/blood-2013-05-499830.
Teira P, Battiwalla M, Ramanathan M, Barrett AJ, Ahn KW, Chen M, et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood. 2016;127(20):2427–38. https://doi.org/10.1182/blood-2015-11-679639.
Goodrich JM, Bowden RA, Fisher L, Keller C, Schoch G, Meyers JD. Ganciclovir prophylaxis to prevent cytomegalovirus disease after allogeneic marrow transplant. Ann Intern Med. 1993;118(3):173–8. https://doi.org/10.7326/0003-4819-118-3-199302010-00003.
Winston DJ, Ho WG, Bartoni K, Du Mond C, Ebeling DF, Buhles WC, et al. Ganciclovir prophylaxis of cytomegalovirus infection and disease in allogeneic bone marrow transplant recipients. Results of a placebo-controlled, double-blind trial. Ann Intern Med. 1993;118(3):179–84. https://doi.org/10.7326/0003-4819-118-3-199302010-00004.
Einsele H, Ljungman P, Boeckh M. How I treat CMV reactivation after allogeneic hematopoietic stem cell transplantation. Blood. 2020;135(19):1619–29. https://doi.org/10.1182/blood.2019000956.
Green ML, Leisenring W, Stachel D, Pergam SA, Sandmaier BM, Wald A, et al. Efficacy of a viral load-based, risk-adapted, preemptive treatment strategy for prevention of cytomegalovirus disease after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012;18(11):1687–99. https://doi.org/10.1016/j.bbmt.2012.05.015.
Ljungman P, de la Camara R, Robin C, Crocchiolo R, Einsele H, Hill JA, et al. Guidelines for the management of cytomegalovirus infection in patients with haematological malignancies and after stem cell transplantation from the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis. 2019;19(8):e260–72. https://doi.org/10.1016/s1473-3099(19)30107-0la.
Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. The novel anticytomegalovirus compound AIC246 (letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol. 2011;85(20):10884–93. https://doi.org/10.1128/jvi.05265-11.
Marty FM, Ljungman P, Chemaly RF, Maertens J, Dadwal SS, Duarte RF, et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017;377(25):2433–44. https://doi.org/10.1056/NEJMoa1706640.
Marty FM, Ljungman PT, Chemaly RF, Wan H, Teal VL, Butterton JR, et al. Outcomes of patients with detectable CMV DNA at randomization in the phase III trial of letermovir for the prevention of CMV infection in allogeneic hematopoietic cell transplantation. Am J Transplant. 2020;20(6):1703–11. https://doi.org/10.1111/ajt.15764.
Anderson A, Raja M, Vazquez N, Morris M, Komanduri K, Camargo J. Clinical, “real-world” experience with letermovir for prevention of cytomegalovirus infection in allogeneic hematopoietic cell transplant recipients. Clin Transplant. 2020;34(7): e13866. https://doi.org/10.1111/ctr.13866.
Derigs P, Radujkovic A, Schubert ML, Schnitzler P, Schöning T, Müller-Tidow C, et al. Letermovir prophylaxis is effective in preventing cytomegalovirus reactivation after allogeneic hematopoietic cell transplantation: single-center real-world data. Ann Hematol. 2021;100(8):2087–93. https://doi.org/10.1007/s00277-020-04362-2.
Johnsrud JJ, Nguyen IT, Domingo W, Narasimhan B, Efron B, Brown JW. Letermovir prophylaxis decreases burden of cytomegalovirus (CMV) in patients at high risk for CMV disease following hematopoietic cell transplant. Biol Blood Marrow Transplant. 2020;26(10):1963–70. https://doi.org/10.1016/j.bbmt.2020.07.002.
Sassine J, Khawaja F, Shigle TL, Handy V, Foolad F, Aitken SL, et al. Refractory and resistant cytomegalovirus after hematopoietic cell transplant in the letermovir primary prophylaxis era. Clin Infect Dis. 2021;73(8):1346–54. https://doi.org/10.1093/cid/ciab298.
Sharma P, Gakhar N, MacDonald J, Abidi MZ, Benamu E, Bajrovic V, et al. Letermovir prophylaxis through day 100 post transplant is safe and effective compared with alternative CMV prophylaxis strategies following adult cord blood and haploidentical cord blood transplantation. Bone Marrow Transplant. 2020;55(4):780–6. https://doi.org/10.1038/s41409-019-0730-y.
Vyas A, Raval AD, Kamat S, LaPlante K, Tang Y, Chemaly RF. Real-world outcomes associated with letermovir use for cytomegalovirus primary prophylaxis in allogeneic hematopoietic cell transplant recipients: a systematic review and meta-analysis of observational studies. Open Forum Infect Dis. 2023;10(1): ofac687. https://doi.org/10.1093/ofid/ofac687.
Kim ES. Letermovir: first global approval. Drugs. 2018;78(1):147–52. https://doi.org/10.1007/s40265-017-0860-8.
MSD K.K., Tokyo, Japan. Prevymis® (letermovir) tablet 240mg, anti-cytomegalovirus chemotherapeutic agent. Pharmaceutical interview form. Revised May 2024, 10th edition [in Japanese]. Available from: https://www.msdconnect.jp/wp-content/uploads/sites/5/2024/05/if_prevymis_tab.pdf . [Accessed 6 Jun 2024].
Japanese Society for Transplantation and Cellular Therapy. Prevention and treatment of viral infections: cytomegalovirus infection, 5th edition (June 2022) [in Japanese]. Available from: https://www.jstct.or.jp/uploads/files/guideline/01_03_01_cmv05.pdf. [Accessed 3 Nov 2023].
Olson AL, Politikos I, Brunstein C, Milano F, Barker J, Hill JA. Guidelines for infection prophylaxis, monitoring and therapy in cord blood transplantation. Transplant Cell Ther. 2021;27(5):359–62. https://doi.org/10.1016/j.jtct.2021.01.024.
Mizuno K, Sakurai M, Kato J, Yamaguchi K, Abe R, Koda Y, et al. Risk factor analysis for cytomegalovirus reactivation under prophylaxis with letermovir after allogeneic hematopoietic stem cell transplantation. Transpl Infect Dis. 2022;24(6): e13904. https://doi.org/10.1111/tid.13904.
Mori Y, Jinnouchi F, Takenaka K, Aoki T, Kuriyama T, Kadowaki M, et al. Efficacy of prophylactic letermovir for cytomegalovirus reactivation in hematopoietic cell transplantation: a multicenter real-world data. Bone Marrow Transplant. 2021;56(4):853–62. https://doi.org/10.1038/s41409-020-01082-z.
Terao T, Matsuoka KI, Narita K, Tsushima T, Yuyama S, Kuzume A, et al. Letermovir administration to prevent cytomegalovirus reactivation is the potential risk of chronic graft-versus-host disease in patients who received haploidentical stem-cell transplantation with post-transplant cyclophosphamide. Front Oncol. 2021;11: 666774. https://doi.org/10.3389/fonc.2021.666774.
Yoshimura H, Satake A, Ishii Y, Ichikawa J, Saito R, Konishi A, et al. Real-world efficacy of letermovir prophylaxis for cytomegalovirus infection after allogeneic hematopoietic stem cell transplantation: a single-center retrospective analysis. J Infect Chemother. 2022;28(9):1317–23. https://doi.org/10.1016/j.jiac.2022.05.019.
Hiraishi I, Ueno R, Watanabe A, Maekawa S. Safety and effectiveness of letermovir in allogenic hematopoietic stem cell transplantation recipients: interim report of post-marketing surveillance in Japan. Clin Drug Investig. 2021;41(12):1075–86. https://doi.org/10.1007/s40261-021-01096-5.
Atsuta Y. Introduction of Transplant Registry Unified Management Program 2 (TRUMP2): scripts for TRUMP data analyses, part I (variables other than HLA-related data). Int J Hematol. 2016;103(1):3–10. https://doi.org/10.1007/s12185-015-1894-x.
Atsuta Y, Suzuki R, Yoshimi A, Gondo H, Tanaka J, Hiraoka A, et al. The unification of hematopoietic stem cell transplantation registry in Japan and establishment of the TRUMP system [In Japanese]. Rinsho Ketsueki. 2007;48(11):1462–9.
Transplant Registry Unified Management Program (TRUMP). Available from: http://www.jdchct.or.jp/trump/. [Accessed 6 Jun 2024].
MSD K.K., Tokyo, Japan. Prevymis® (letermovir) tablets 240mg, intravenous infusion 240mg. Pharmaceutical risk management plan [in Japanese]. Available from: https://www.msdconnect.jp/wp-content/uploads/sites/5/2021/02/rmp_prevymis.pdf. [Accessed 6 Jun 2024].
MSD K.K., Tokyo, Japan. Prevymis® (letermovir) tablets 240mg, anti-cytomegalovirus chemotherapeutic agent. Prescribing information. Revised January 2022, 2nd edition [in Japanese. Available from: https://www.msdconnect.jp/wp-content/uploads/sites/5/2022/01/pi_prevymis_tab.pdf. [Accessed 6 Jun 2024].
MSD K.K., Tokyo, Japan. Prevymis® (letermovir) intravenous infusion 240mg, anti-cytomegalovirus chemotherapeutic agent. Prescribing information. Revised May 2024, 4th edition [in Japanese]. Available from: https://www.msdconnect.jp/wp-content/uploads/sites/5/2024/05/pi_prevymis_tab.pdf. [Accessed 6 Jun 2024].
Extension of letermovir (LET) from day 100 to day 200 post-transplant for the prevention of cytomegalovirus (CMV) infection in hematopoietic stem cell transplant (HSCT) participants (MK-8228-040), study results. Available from: https://clinicaltrials.gov/ct2/show/results/NCT03930615. [Accessed 6 Jun 2024].
Acknowledgments
The authors thank the Japanese Society for Transplantation and Cellular Therapy and the Japanese Data Center for Hematopoietic Cell Transplantation for their invaluable support with providing the CRFs, as well as all physicians and associated staff who cooperated in this PMS. The authors also thank CMIC Holdings Co., Ltd. for data management and statistical analyses, and Nicholas D. Smith (EMC K.K.) for medical writing support, which were funded by MSD K.K., Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The PMS and publication of this article were funded by MSD K.K., Tokyo, Japan.
Conflicts of Interest
Masaki Fukuda, Junko Hattori, Rika Ohkubo, Asuka Watanabe, and Shinichiroh Maekawa are employees of MSD K.K., Tokyo, Japan.
Ethics Approval
This survey was carried out in accordance with Good Post-Marketing Study Practice for Drugs as specified by the Ministry of Health, Labour and Welfare in Japan. The research protocol was approved by the Japan Conference of Clinical Research Institution Review Board (approval date 21 January, 2021).
Consent to Participate
Informed consent was not required for this PMS in accordance with Good Post-Marketing Study Practice in Japan. Informed consent was obtained voluntarily at some facilities. All facilities agreed with the use of the survey data under a contract/agreement with MSD K.K., Tokyo, Japan.
Consent for Publication
Not applicable.
Availability of Data and Material
The data sharing policy, including restrictions, of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA is available at https://engagezone.msd.com/ds_documentation.php. Requests for access to the study data can be submitted through the Engage Zone site or via e-mail to the Data Access mailbox (dataaccess@merck.com).
Code Availability
Not applicable.
Authors’ Contributions
All authors met ICMJE authorship criteria; their specific contributions are as follows: conceptualization: all authors; data curation: AW, SM; formal analysis: AW, SM; investigation: all authors; methodology: AW, SM; project administration: MF; supervision: MF; validation: AW, SM; visualization: MF, JH, RO; writing: original draft: MF, JH, RO; writing: review and editing: all authors. All authors approved the final version of this manuscript and agree to be accountable for all aspects of the work.
Additional information
Prior publication: Interim results were published as: Hiraishi I, Ueno R, Watanabe A, Maekawa S. Safety and effectiveness of letermovir in allogenic hematopoietic stem cell transplantation recipients: interim report of post-marketing surveillance in Japan. Clin Drug Investig. 2021;41(12):1075–86. https://doi.org/10.1007/s40261-021-01096-5.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fukuda, M., Hattori, J., Ohkubo, R. et al. Real-World Safety and Effectiveness of Letermovir in Patients Undergoing Allogenic Hematopoietic Stem Cell Transplantation: Final Results of Post-Marketing Surveillance in Japan. Clin Drug Investig (2024). https://doi.org/10.1007/s40261-024-01376-w
Accepted:
Published:
DOI: https://doi.org/10.1007/s40261-024-01376-w