
Abstract
Background and Objective
Tolebrutinib is a covalent inhibitor of Bruton’s tyrosine kinase, an enzyme expressed in B lymphocytes and myeloid cells including microglia, which are thought to be major drivers of inflammation in multiple sclerosis. This excretion balance and metabolism study evaluated the metabolite profile of tolebrutinib in healthy male volunteers.
Methods
Six healthy volunteers received a 60-mg oral dose of [14C]-tolebrutinib, and metabolite profiling of 14C-labeled metabolites was performed using a combination of liquid chromatography, mass spectrometry, and radioactivity assay methods.
Results
Tolebrutinib was rapidly and completely absorbed from the gastrointestinal tract, followed by rapid and extensive metabolism. Excretion via feces was the major elimination pathway of the administered radioactivity (78%). Tolebrutinib was highly metabolized, with 19 metabolites identified in human plasma. Phase 1 biotransformations were primarily responsible for the circulating metabolites in plasma. Seven metabolites that achieved exposure in plasma similar to or higher than the parent compound were characterized biochemically for inhibition of Bruton’s tyrosine kinase activity. Metabolite M8 exceeded the exposure threshold of 10% (18%) of the total radioactivity but had little if any pharmacological activity. Metabolite M2 (4% of circulating radioactivity) retained the ability to irreversibly and potently inhibit Bruton’s tyrosine kinase in vitro, similar to the parent compound. Tolebrutinib and metabolite M2 had short (3.5-h) half-lives but durable pharmacodynamic effects as expected for an irreversible antagonist.
Conclusions
Tolebrutinib was extensively metabolized to multiple metabolites. The hydroxylated metabolite M2 demonstrated similar inhibitory potency toward Bruton’s tyrosine kinase as the parent compound. Both tolebrutinib and metabolite M2 likely contributed to pharmacological activity in vivo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Tolebrutinib was rapidly and almost completely absorbed from the gastrointestinal tract. |
Results of the present study showed that tolebrutinib is extensively metabolized leading to the formation of one active metabolite in human, not observed at preclinical stage. |
It implied that pharmacokinetics of the active metabolite should be characterized in clinical studies to evaluate the impact of sources of pharmacokinetic variability and its correlation with safety and efficacy. |
1 Introduction
Bruton’s tyrosine kinase (BTK), a non-receptor tyrosine kinase, is an essential element in coupling certain cell surface receptors to downstream signaling to the nucleus of most hematopoietic cells [1, 2]. Bruton’s tyrosine kinase plays an essential role in activating B lymphocytes and myeloid cells, including central nervous system (CNS) microglia, in response to immunogenic stimuli [3, 4]. Each of these cell types contributes to the pathophysiology of multiple sclerosis [5, 6].
Tolebrutinib, a CNS-penetrant BTK inhibitor, has a dual mechanism of action, acting on both adaptive and innate immunity. Bruton’s tyrosine kinase mediates adaptive immunity by coupling the antigenic stimulation of the B-cell receptor to nuclear signaling that leads to acute inflammatory responses. Bruton’s tyrosine kinase also couples innate responses in CNS-resident microglia, mainly though Fc-like and Toll-like receptors, to invoke proinflammatory phenotypes that have been linked to chronic neuroinflammation in the brain and spinal cord. Because of this dual role for BTK, tolebrutinib is in development to treat multiple sclerosis [7]. A randomized, double-blinded, crossover, phase IIb trial of tolebrutinib 60 mg in patients with relapsing forms of multiple sclerosis showed a substantial reduction in contrast-enhancing T1 hyperintense lesions after 12 weeks of treatment [8].
A first-in-human phase I trial established several pharmacokinetic parameters of tolebrutinib [9]. Orally administered tolebrutinib was rapidly absorbed and cleared, with a half-life of 90–120 min. However, because tolebrutinib binds irreversibly, the enzyme occupancy measured 4 h after a single dose (60 and 120 mg) exceeded 96%. Upon repeated daily administration, trough (24 h post-administration) occupancy exceeded 75% for doses of 7.5–90 mg, with a linear recovery over the course of 5–7 days after the last dose administered. Exposure of tolebrutinib within the CNS was also confirmed, with mean exposure in cerebrospinal fluid (used as a surrogate for brain parenchyma) that exceeded the half-maximal inhibitory concentration 2 h after a single 120-mg dose. The purpose of the current study was to further investigate the pharmacokinetic properties of tolebrutinib and their relationship to the metabolites identified in healthy participants after a single oral dose.
2 Methods
2.1 Study Design and Participants
This single-center, open-label, single-dose, phase I trial (NCT04171310) was conducted in November and December 2019 at Quotient (Nottingham, UK). Healthy nonsmoking men aged 30–65 years with a body mass index of 18–32 kg/m2 and body weight of 50–100 kg were enrolled. Participants were excluded if they had clinically significant abnormalities in hematology, coagulation, platelet function, or biochemistry or a history or presence of clinically relevant disease. Poor metabolizers for cytochrome P450 2D6 and cytochrome P450 2C8 were excluded because of the involvement of these cytochrome isoforms in in vitro tolebrutinib metabolism (internal data). The independent London—Riverside Research Ethics Committee approved the protocol, and the study was conducted in accordance with consensus ethics principles, including the Declaration of Helsinki, the International Council for Harmonization Guideline for Good Clinical Practice, and all applicable laws, rules, and regulations. All participants provided written informed consent prior to the conduct of any study-related procedures.
2.2 Radiolabeled Drug and Formulation
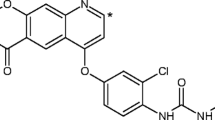
All participants received a single dose of [14C]-tolebrutinib 60 mg under fasting conditions on day 1. [14C]-tolebrutinib (Fig. 1) was synthesized at Pharmaron (Hoddesdon, UK) and administered as an oral solution comprising hydroxypropyl betadex, sucralose, and sterile water for injection, after preparation at the clinical site (not more than 3.7 MBq).

Tolebrutinib structure with 14C labeling. The asterisk denotes ring labeling
2.3 Metabolite Synthesis
Based on the preliminary data on human metabolism, the metabolites were structurally characterized and synthesized for a liquid chromatography/mass spectrometry (LC/MS) assay and to determine their pharmacological activities. The M2 and M18 metabolites were provided by Pharmaron (Beijing, China). The M5, M5a, M8, and M10 metabolites were provided by Sanofi (Vitry, France).
2.4 Sampling Schedule
Participants were monitored up to 15 days after receiving tolebrutinib, based on the extent of radioactivity recovered. The end-of-study visit was performed after the last collection of excreta, or on day 43. Blood samples (10–16 mL) were collected to determine total radioactivity (TR) in blood and plasma and to identify the concentrations of tolebrutinib and its metabolites in plasma at defined timepoints up to 168 h after isotope administration. Samples were stored at − 80 °C until analysis. Urine and feces were collected before administration and at 0–24 (0–12 and 12–24 for urine), 24–48, 48–72, 72–96, 96–120, 120–144, and 144–168 h after administration.
2.5 Sample Preparation and Assay Methods to Determine Radioactivity
Aliquots of blood, plasma, urine, and feces samples were mixed with scintillation fluid (Gold Star; Meridian Biotechnologies Ltd., Waterfield, UK). Radioactivity was determined using a PerkinElmer Tri-Carb® liquid scintillation counter with automatic external standard quench correction. The limits of quantification were 45.6 ng eq/mL (whole blood), 17.3 ng eq/mL (plasma), 1.51 ng eq/mL (urine), and 17.8 ng eq/g (feces).
For accelerator mass spectrometry (AMS) analysis, plasma samples were diluted with commercially obtained plasma, thawed, and then centrifuged prior to processing. Portions of each sample and carbon carrier (sodium benzoate) were placed in tubes containing pre-baked copper oxide powder. Oxalic acid II, synthetic graphite, and sodium benzoate were placed in separate tubes containing pre-baked copper oxide powder. Following 1 h combustion at 900 °C (oxidation) and 5 h graphitization at 550 °C (reduction) under vacuum, the graphite was left in the sealed graphitization tube before being packed into a cathode. Single-stage AMS was used to determine isotope ratios (12C:13C:14C) in the graphitized samples. The limit of quantification in plasma using AMS was 0.0312 ng eq/mL. Plasma concentrations of tolebrutinib and M5/M5a were measured by an existing validated LC–MS/MS method using deuterated internal standard (Sciex QTrap® 6500+), after liquid extraction from plasma, with a lower limit of quantification of 0.01 ng/mL for both analytes (not published). A fit-for-purpose LC–MS/MS method was used to assess plasma concentrations of the M2 and M8 metabolites, with lower limits of quantification of 0.02 and 0.1 ng/mL (not published), respectively.
2.6 Sample Preparation and Metabolite Profiling in Plasma, Urine, and Feces
Two types of pooled plasma samples were prepared. For each participant, individual plasma samples were pooled across 0–96 h post-dose using an area under the concentration–time curve (AUC) approach [10]. Timepoint plasma pools were also prepared for all participants at specific timepoints (pre-dose and 1, 8, 48, and 96 h post-administration) using equivalent portions of plasma from each participant. The 0-h to 96-h interval was chosen because it represented at least 70% of [14C]-tolebrutinib AUC in plasma, as measured by AMS.
To extract one volume of plasma samples, three volumes of acetonitrile were added to the plasma tube, followed by sonication, vortex mixing at room temperature, and centrifugation (3220×g at 4 °C for 10 min using a Beckman Coulter Allegra 6R centrifuge). The supernatant fraction was transferred to a microcentrifuge tube. Acetonitrile was added one more time to the pellet. Supernatants were combined, and samples were reduced until completely dry under a stream of nitrogen. The dried samples were reconstituted with ammonium acetate (0.75 g/L) and formic acid (1 mL/L) in water, then sonicated, vortex mixed, and centrifuged (3220×g at 4 °C for 10 min). The final samples were stored at − 80 °C.
Plasma metabolite profiles were generated by collecting ultra-performance liquid chromatography (UPLC) fractions followed by AMS analysis of each fraction or by pooling fractions across regions. The regions of interest in each radiochromatogram were identified as the parent compound or the metabolites based on the retention time of known metabolites (M18, M10, M8, M5/M5a, M2) using a 12C reference standard. Ultra-performance liquid chromatography coupled to high-resolution mass spectrometry (Waters QTof Xevo G2-XS) was used to elucidate the metabolite structure (including accurate mass determination and fragmentation experiments) in fractionated plasma samples that accounted for the ten most abundant regions of interest in the AUC pools. For this step, specific plasma pools were prepared to maximize the sensitivity of metabolite structure identification; the same LC conditions were used as in the pooled AUC from 0 to 96 h (AUC0–96h) profiling. Equal portions of the plasma samples were pooled per participant and timepoint (1, 2, 4, and 8 h), then across all participants.
Portions of the pooled feces homogenates were extracted for each participant with acetonitrile/formic acid (99.9 + 0.1, V/V). Aliquots of the urine pool samples were condensed under a stream of nitrogen at 37 °C, then diluted with ACN, methanol, and water (1 + 1 + 8, V/V/V). Metabolite profiling was performed using ultra-performance liquid chromatography coupled to high-resolution mass spectrometry coupled with radio detection via fraction collection and off-line radioactivity counting. Radiochromatograms were re-compiled from those data and used to identify the structure of metabolites that accounted for more than 5% of TR in urine and feces. For all matrices, the appropriate technology was used to evaluate recovery according to the radioactivity level.
2.7 Pharmacokinetic Evaluation
Pharmacokinetic parameters estimated from TR (plasma and blood) or unchanged drug and metabolite concentrations (plasma) included maximum concentration (Cmax); time to Cmax; AUC from 0 to 24 h; AUC from time 0 to time of the last quantifiable concentration; AUC from time 0 to infinite time, calculated as the sum of AUC from time 0 to time of the last quantifiable concentration and Clast/lambda z, where Clast is the last observed quantifiable concentration and lambda z is the slope of the terminal phase of elimination; and elimination half-life.
2.8 Safety Evaluation
Safety and tolerability were assessed via ongoing monitoring of adverse events and by a 12-lead electrocardiogram, physical examinations, vital signs, and clinical laboratory investigations assessed before and after drug administration and at the end of the study.
2.9 Investigation of the Pharmacological Activity of Metabolites
A biochemical BTK kinase assay was used to evaluate the activity of the parent and its metabolites. The assay combined BTK with a fluorescent peptide substrate probe that was phosphorylated in the presence of ATP, and the substrate and phosphosubstrate were separated chromatographically to determine relative enzyme activity. A GST-tagged full-length BTK enzyme was acquired from BPS Bioscience (San Diego, CA, USA). The peptide substrate for the BTK assay was FAMGEEPLYWSFPAKKK-NH2 (where FAM is carboxyfluorescein) synthesized by Nanosyn (Santa Clara, CA, USA).
The BTK enzyme was diluted in an assay buffer (100 mM HEPES, pH 7.5, 0.1% BSA, 1 mM DTT, 0.01% Triton X-100, and 5 mM MgCl2) and dispensed into 384-well plates. The test compounds were serially diluted in DMSO, added to the protein samples by acoustic dispensing (Labcyte Echo), and tested at 12 concentrations. The control samples of 0% inhibition (in the absence of inhibitor, DMSO only) and 100% inhibition (in the absence of enzyme) were assembled in replicates of four and used to calculate percent inhibition for the test compounds. The peptide substrate supplemented with ATP was added to each reaction. The final concentrations were 0.5 nM for BTK enzyme, 20 µM for ATP, and 1 µM for the peptide substrate. The kinase reactions lasted 3 h at room temperature and were quenched by the addition of termination/assay buffer, followed by microfluidic electrophoresis (Caliper LabChip® 3000).
The measured parameter consisted of a change in the relative fluorescence intensity of the peptide substrate and phosphorylated product peaks. Activity in each test sample was determined as the product to sum ratio (PSR), calculated as P/(S + P), where P is the peak height of the product and S is the peak height of the substrate. Percent inhibition (Pinh) was determined using the equation Pinh = (PSR0%inh − PSRcompound)/(PSR0%inh − PSR100%inh) × 100, where PSR0%inh is PSR in the absence of compound, PSRcompound is PSR in the presence of compound, and PSR100%inh is PSR in the absence of enzyme. To estimate a half-maximal inhibitory concentration value for each compound, Pinh versus compound concentrations were fitted via a four-parameter sigmoid dose-response model using IDBS XLfit software.
Time-dependent inhibition of BTK was evaluated using 11 concentrations of tolebrutinib and M2. Test compounds were serially diluted in the assay buffer and the 4-mM peptide substrate and dispensed into 384-well plates (35 mL per well). The kinase was diluted to 2× concentration (1.0 nM) in the assay buffer with 500 mM of ATP. Reactions were initiated by adding the enzyme solution to the wells and rapidly mixing with a compound/peptide substrate solution. Control samples (0% and 100% inhibition) were assembled in replicates of six and used to calculate the percent conversion in the presence of the test compounds. The final concentrations were 0.5 nM for the BTK enzyme and 500 mM for ATP. Immediately after mixing, progress curves [percent conversion over time (%-Conv)] were acquired for approximately 5 h using the Caliper Life Sciences LabChip 3000. The progress curves demonstrated time-dependent inhibition and were fitted using the equation %-Conv = Vs × t + (Vi − Vs)/Kobs app × (1 − exp (− Kobs app × t), where Vs is steady-state velocity, t is time, Vi is initial velocity, and Kobs is the observed rate constant. Curve fittings were performed using IDBS XLFit software.
Cellular activity was estimated using an immortalized B-lymphocyte cell line (Ramos cells; ATCC). Prior to the study, cells were plated (100,000 per well) in 1% serum containing buffer. The test articles were diluted serially at a ratio of 1:3 to achieve a defined concentration range from 6 μM to 33.9 pM, followed by incubating in an atmosphere of 5% CO2 at 37 °C for 30 min. Cells were stimulated by adding goat anti-human IgM F (Ab′)2 (Jackson Immunoresearch) at a concentration of 20 μg/mL and incubating for 18 h in 5% CO2 at 37 °C. Changes in CD69 expression (a marker for B-cell activation) was measured using an anti-human CD69 antibody (clone FN50; Biolegend) followed by flow cytometry (using a BD LSRII flow cytometer) and analysis (using FlowJo and Graphpad Prism software).
3 Results
3.1 Human Excretion Balance
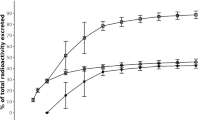
Six men were enrolled in the study (mean age of 50 years with a mean body weight of 78 kg and a body mass index < 30, with five Caucasian individuals and one Hispanic or Latino individual). Nearly complete recovery (> 90%) of the mean cumulative radioactivity occurred up to 216 h (day 9) post-administration (Fig. 2). Most of the radioactivity (85%) was excreted within 72 h. Excretion via feces was the major elimination pathway, as 78% of the dose was excreted in feces and 14% was excreted in the urine.

Mean ± standard deviation cumulative recovery of radioactivity in urine and feces at the indicated intervals following a single oral dose of [14C]-tolebrutinib (60 mg) in healthy male participants (n = 6) under fasting conditions
3.2 Metabolite Profiling in Plasma, Urine, and Feces
The extraction efficiencies for the individual AUC0–96h pooled plasma samples were 39.2%, 42.2%, 38.1%, 40.0%, 40.2%, and 41.6%. The extraction efficiencies for the 1-h, 8-h, 48-h, and 96-h timepoint pools, pooling all participants, were 94.3%, 61.5%, 5.2%, and 13.3%, respectively. Figure 3 shows a representative radiochromatogram from one individual plasma pool extract (AUC0–96h) overlaid against the ultraviolet response for a mixture containing 12C reference standard. Figure 4 shows the radiochromatogram generated from the plasma pool extract with annotations for the regions of interest. The metabolites were identified in ten fractions, with the most abundant radioactivity in plasma. Representative radiochromatograms from metabolite profiling in urine and feces pools are presented in Fig. 5A, B, respectively. Figure 6 shows the proposed metabolic scheme for tolebrutinib in human plasma, urine, and feces.

Representative radiochromatogram of an individual plasma pool extract (area under the concentration–time curve from 0 to 96 h) overlaid against the ultraviolet (UV) absorption of a mixture containing 12C reference standards for metabolites M18, M10, M8, M5/M5a, and M2. min minutes

Representative radiochromatogram from an individual plasma pool extract (area under the concentration–time curve from 0 to 96 h) processed using LAURA software, with time-normalized fraction data and regions of interest annotated. The ten fractions most abundant in radioactivity (P3, P5, P7, P9, P11, P12, P13, P14, P15, and P17) were used for metabolite identification. The relative distribution of co-eluting metabolites was assessed based on their relative mass spectrometry responses. DPM disintegrations per minute, min minutes

Pooled metabolite profiling in urine A over 0–48 h post-dose for an exemplar participant in feces B over 0–72 h post-dose for an exemplar participant. CPM count per minute, min minutes

Proposed combined metabolic pathways of tolebrutinib in plasma and excreta in healthy male participants. Boxes gather positional isomeric metabolites of the same functionalization and which cannot be individually affiliated to their downstream metabolites. F feces, P plasma, U urine
In total, the structures of 21 phase 1 and four phase 2 metabolites were fully or partially elucidated. The phase 1 metabolites were formed via single and multiple oxygenations and hydroxylations, epoxidation and hydrolysis of acrylamide, oxidative piperidine ring opening including further oxidation of an alcohol to the carboxylic acid, N-dealkylation, reduction of acrylamide, or multiple combinations thereof. The phase 2 metabolites were formed via glucuronidation of single-oxygenated phase 1 metabolites, direct cysteine conjugation of the acrylamide moiety, or sulfate conjugation of multiple-oxygenated tolebrutinib.
In plasma, the structures of 17 phase 1 and two phase 2 metabolites were partially or fully elucidated. Metabolites resulting from the phase 1 reactions were M2, M5/M5a, M8, M10, M13, M18, M19, M22, M26, M28, M29, M30, M31, M32, M33, M34, and M35. Metabolites resulting from conjugation were M25 and M27. Based on AMS results, the parent drug accounted for 1.33% of TR of the plasma AUC0–96h. The main circulating metabolites were M8 (9.14% of TR), M2 (4.38% of TR), M32 (3.45% of TR), M19 (2.99% of TR), M10 (1.92% of TR), M18 (1.30% of TR), M26 (1.17% of TR), and M27 (1.17% of TR) (Table 1). Other metabolites each accounted for less than 0.90% of TR.
In urine, 11.4–16.2% of the radioactive dose was excreted within the 0-h to 48-h pooling interval. The unchanged parent drug was not observed in urine. The ten metabolites detected in urine were elucidated, eight of which (M2, M5/M5a, M8, M10, M13, M18, M19, and M22) were formed by phase 1 reactions and two (M6 and M20) by phase 2 reactions. The most abundant metabolites excreted in urine were M8 (5.98% of the dose), M19 (1.37% of the dose), M2 (1.24% of the dose), and M5/M5a (0.672% of the dose) (Table 1). Other metabolites each accounted for up to 0.55% of the dose.
In feces, 66.4–78.3% of TR was excreted during the 0-h to 72-h pooling interval. Only minor amounts of unchanged parent drug (3.83% of the dose) were observed. In total, 14 metabolites were detected in feces and elucidated, 12 of which (M2, M4, M5/M5, M8, M10, M13, M18, M19, M21, M22, M23, and M24) were formed by phase 1 reactions and two (M6 and M20) formed by conjugation. M8 was the main fecal metabolite (11.9% of the dose), followed by M19 (4.29% of the dose), M4 (hydroxylation; 4.24% of the dose), M5/M5a (3.05% of the dose), M23 (2.86% of the dose), M10 (2.37% of the dose), M18 (2.30% of the dose), M24 (2.20% of the dose), and M13 (1.35% of the dose) (Table 1). Other metabolites, such as M20, were only observed in one participant or did not exceed 0.885% of the dose.
The main elimination pathways of tolebrutinib consisted of oxidative ring openings of the piperidine (~ 23% of the dose) followed by oxygenations/hydroxylations of the piperidine and/or “core” moiety (~ 13% of the dose). Modifications of the acrylamide warhead, such as cleavage, epoxidation, subsequent hydrolysis, and reductions, accounted for approximately 12% of the dose. The phase 2 metabolic pathways, comprising direct cysteine conjugation of the acrylamide and sulfation, were almost negligible (1.6% of the dose).
3.3 Pharmacokinetics
The time to Cmax values for tolebrutinib and key metabolites (M5/M5a, M2, and M8) were similar to TR at 1 h (Table 2; Fig. 7). Tolebrutinib and metabolites M5/M5a, M2, and M8, represented 0.75%, 1.30%, 3.80%, and 18.0% of TR in plasma, respectively, using the LC/MS assay (Table 1). The AUC from time 0 to infinite time ratios for M5/M5a, M2, and M8 versus tolebrutinib were 2.14, 5.65, and 25.1, respectively (Table 3). The ratio of blood to plasma exposure of TR ranged from 0.38 on AUC from time 0 to time of the last quantifiable concentration to 0.72 on Cmax, showing a limited distribution of radioactivity in blood cells.

Mean ± standard deviation concentrations of tolebrutinib and metabolites M2, M5/M5a, and M8 in plasma, and of radioactivity in plasma by accelerated mass spectrometry (AMS), following a single oral dose of [14C]-tolebrutinib (60 mg) in healthy male participants (n = 6) under fasting conditions A in linear scale to 24 h post-dose and B in semi-log scale to 168 h post-dose. conc. concentration, h hours
3.4 Safety
Overall, tolebrutinib was safe and well tolerated when administered as a 60-mg single-dose [14C]-radiolabeled oral solution in healthy men. Four participants reported at least one post-treatment adverse event. The common adverse events were mild diarrhea (n = 3), mild abdominal pain (n = 1), and moderate back pain (n = 1). No clinically significant changes were observed in vital signs, electrocardiogram, or laboratory parameters.
3.5 Pharmacological Characterization of the Metabolites
Metabolite M2 (which retains the acrylamide warhead) exhibited inhibitory activity similar to that of the parent compound in the biochemical kinase assay (Table 4). For covalent inhibitors, the Kinact/KI rate described the efficiency of the covalent bond formation that resulted from the first reversible binding event and the maximum potential rate (Kinact) of covalent inactivation [11]. The covalent binding potency of M2, represented by Kinact/KI, was about 62% that of the parent compound (Table 5). M2 was approximately two-fold less potent than the parent compound in Ramos cells (Table 6). M5/M5a, the diol resulting from oxidation of the acrylamide warhead, lacked the covalent interaction with the target but retained binding affinity with a half-maximal inhibitory concentration of 35 nM. Thus, the relative contributions of the parent and the active metabolites is a complex set of interactions with the relevant cell types expressing the BTK target.
4 Discussion
This open-label, phase I pharmacokinetic trial was designed to determine the absorption, metabolism, and routes of excretion for orally administered [14C]-tolebrutinib. After a single 60-mg dose, the radioactivity was mainly excreted in feces, with almost complete (> 90%) recovery over 8 days. Tolebrutinib was extensively metabolized (19 metabolites in plasma and 14 in excreta), and metabolites formed quickly based on a similar plasma time to Cmax (1 h) for tolebrutinib and its metabolites. The main elimination pathways from plasma were phase 1 metabolic biotransformations.
The pharmacokinetic elimination profile of TR in plasma was characterized by the first phase of a rapid elimination of the parent drug and metabolites (3.6–6.3 h) and the second slower phase of covalent and non-specific binding of moieties to plasma proteins. However, almost all radioactivity was eliminated from the body within 1 week, based on the near-complete recovery of the administered dose of radioactivity. A substantial portion of circulating radioactivity (60% on AUC0–96h pool) was not extracted and was likely covalently bound to plasma proteins [13]. Albumin was the main plasma target protein for tolebrutinib binding (> 85%).
The absence of unchanged tolebrutinib in urine and the limited excretion in feces (3.83%) strongly supports a near-complete absorption of tolebrutinib in the gastrointestinal tract. High levels of oxidized metabolites that formed quickly suggest substantial first-pass metabolism.
Metabolism profiling and specific LC/MS assays displayed consistent results, expressed as a percentage of TR, for tolebrutinib (1.3% and 0.77%, respectively) and M2 (4.3% and 3.8%) but not for M8 or M5/M5a. A degradation of M8 and M5/M5a during plasma storage at − 80 °C and/or during freeze/thaw cycles is a reasonable explanation for the lower percentage in the metabolism profiling (9.14% for M8 and trace for M5/M5a) compared with percentages from the LC/MS assays (18% and 1.3%, respectively). The time between sampling and processing (9 months for the metabolism profiling and 6 months for the LC/MS assay) and different sample handling conditions might account for these differences in degradation.
Tolebrutinib was extensively metabolized, representing less than 1% of TR in plasma, and metabolites were formed quickly. Seven of the 19 metabolites identified in plasma (M2, M5/M5a, M8, M10, M18, M19, and M32) had a percentage of TR greater or equal to that of the parent compound. Metabolite M8, which was above the 10% threshold of TR used for testing the safety of drug metabolites [12], was only active in the micromolar concentration range, indicating that an intact piperidine ring was necessary for nanomolar inhibitory potency. In contrast, M2 retained pharmacological activity with a nanomolar potency of covalent binding similar to tolebrutinib.
It is difficult to assess the relative contribution of tolebrutinib and the two relevant metabolites (M2, M5/M5a) to overall pharmacological activity because of the nature of their competition via covalent binding to the BTK target. However, based on the plasma exposure at early timepoints, M2 may represent a pharmacologically relevant contributor to the overall pharmacological activity of orally administered tolebrutinib [14]. Accordingly, the M2 metabolite will be assayed in all clinical studies of tolebrutinib.
5 Conclusions
In summary, in response to a single oral dose of 60 mg, tolebrutinib was rapidly and almost completely absorbed from the gastrointestinal tract, then underwent rapid and extensive oxidative metabolism. The majority of the administered radioactivity was excreted in feces (78%), with lesser amounts in the urine (14%). Tolebrutinib and the hydroxylated metabolites M2 and M5/M5a had similar short half-lives. Despite this short half-life, BTK occupancy was sustained for over 24 h, consistently with covalent binding. Because M2 retains an active warhead, it possessed a pharmacological profile (potency, selectivity) similar to the parent, while the non-covalent metabolite M5/M5a was a competitive inhibitor of BTK. Assessing the relative contributions of these active metabolites to the overall pharmacological properties is complex, and additional work is needed to establish a quantitative account.
References
Wang Q, Pechersky Y, Sagawa S, Pan AC, Shaw DE. Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc Natl Acad Sci USA. 2019;116(19):9390–9.
Garcia-Merino A. Bruton’s tyrosine kinase inhibitors: a new generation of promising agents for multiple sclerosis therapy. Cells. 2021;10:2560.
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:210–32.
Martin E, Aigrot M-S, Grenningloh R, Stankoff B, Lubetzki C, Boschert U, et al. Bruton’s tyrosine kinase inhibition promotes myelin repair. Brain Plast. 2020;5(2):123–33.
Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. B cells in multiple sclerosis: from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. 2021;17:399–414.
Lu HC, Kim S, Steelman AJ, Tracy K, Zhou B, Michaud D, et al. STAT3 signaling in myeloid cells promotes pathogenic myelin-specific T cell differentiation and autoimmune demyelination. Proc Natl Acad Sci USA. 2020;117(10):5431–41.
Contentti EC, Correale J. Bruton’s tyrosine kinase inhibitors: a promising emerging treatment option for multiple sclerosis. Expert Opin Emerg Drugs. 2020;25:377–81.
Reich DS, Arnold DL, Vermersch P, Bar-Or A, Fox RJ, Matta A, et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a randomized, double-blind, placebo-controlled trial. Lancet Neurol. 2021;20(9):729–38.
Owens TD, Smith PF, Redfern A, Xing Y, Shu J, Karr DE, et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin Transl Sci. 2022;15(2):442–50. https://doi.org/10.1111/cts.13162.
Hamilton RA, Garnett WR, Kline BJ. Determination of mean valproic acid serum level by assay of a single pooled samples. Clin Pharmacol Ther. 1981;29(3):408–13.
Strelow JM. A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discov. 2017;22(1):3–20.
US Food and Drug Administration. Safety testing of drug metabolites. Updated March 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/safety-testing-drug-metabolites. Accessed 22 Oct 2021.
Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, et al. Absorption, metabolism, and excretion of oral 14C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 2015;43(2):289–97.
US Food and Drug Administration. In vitro drug interaction studies: cytochrome P450 enzyme and transporter-mediated drug interactions; 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed 22 Oct 2021.
Acknowledgements
Critical review of the manuscript was provided by Eric Sultan, François Donat, John Newton, Timothy Owens, Ross Gruber, and Svend Geertsen of Sanofi. Editorial support (editing of the manuscript) was provided by Janna Afanasjeva of Elevate Scientific Solutions, and funded by Sanofi.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was funded by Sanofi.
Conflict of Interest
All authors are employees of, and may hold stock in, Sanofi.
Ethics Approval
This study was conducted in accordance with the consensus ethics principles, including the Declaration of Helsinki, the International Council for Harmonization Guidelines for Good Clinical Practice, and all applicable laws, rules, and regulations. The study protocol was approved by London—Riverside Research Ethics Committee (19/LO/1528) on 18 October, 2019.
Consent to Participate
All participants provided a written informed consent prior to the conduct of any study-related procedures.
Consent for Publication
Not applicable.
Data Availability
Datasets generated and/or analyzed during this study are available from the corresponding author upon reasonable request.
Code Availability
Not applicable.
Author Contributions
Participated in study concept or design: ON, MJC, PB. Performed data analysis or interpretation: ON, PB, PM, PS, OV, SM, CA, MJC, TT, SK. Drafted or reviewed the manuscript: ON, PB, PM, PS, OV, SR, MJC, TT. Approved the manuscript for submission: all authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Nicolas, O., Moliner, P., Soubayrol, P. et al. Absorption, Metabolism, and Excretion of [14C]-Tolebrutinib After Oral Administration in Humans, Contribution of the Metabolites to Pharmacological Activity. Clin Drug Investig 43, 653–665 (2023). https://doi.org/10.1007/s40261-023-01296-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-023-01296-1