
Abstract
Background and objective
Excessive daytime sleepiness (EDS) and fatigue are major complaints in patients with obstructive sleep apnoea (OSA) syndrome. Pitolisant is an orally active selective histamine H3 receptor (H3R) antagonist/inverse agonist, which enhances histaminergic transmissions in the brain and thereby elicits strong wake-promoting effects. This article assesses the efficacy and safety of pitolisant 20 mg in patients with OSA, based on existing randomised controlled studies.
Methods
An individual patient data (IPD) meta-analytical two-level (study-patient) hierarchical model was used assuming a random treatment effect. The Epworth Sleepiness Scale (ESS) and Oxford Sleep Resistance (OSleR) tests were co-primary endpoints.
Results
A total of 512 patients, including 384 treated with pitolisant and 128 with placebo, were included in the analysis. Compared with placebo, pitolisant reduced mean ESS by − 3.1 (95% CI [− 4.1; − 2.1]; p < 0.001) and improved OSleR by 1.18 (1.02; 1.35, p = 0.022); 30% more patients had reduced fatigue (risk ratio [RR] = 1.3, [1.11; 1.53]), p = 0.001) and 46% more patients had improved Clinical Global Impression (CGI) (RR = 1.46 [1.12; 1.89], p = 0.005). No significant differences in safety endpoints were found. These results proved homogeneous across studies and subgroups of the population.
Conclusion
The results provide evidence of a significant benefit of pitolisant in improving EDS and fatigue, irrespective of baseline conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Based on the available data, this analysis provides evidence of a clinically relevant benefit of pitolisant in improving EDS and fatigue in patients with OSA. |
This benefit was similar for patients using, or not using, continuous positive airway pressure, and was found homogeneous across any subgroups defined by baseline difference. |
1 Introduction
1.1 Description of the Condition
Excessive daytime sleepiness (EDS) and fatigue are major complaints in patients with obstructive sleep apnoea (OSA) syndrome, defined as sleep-related breathing disorder (SRBD) with full or partial occlusion of the upper airway during sleep.
The current prevalence estimates of moderate-to-severe sleep-disordered breathing (Apnoea Hypopnoea Index [AHI] ≥ 15) are 10% among men aged 30–49 years; 17% among men aged 50–70 years; 3% among women aged 30–49 years; and 9% among women aged 50–70 years [1].
Several mechanisms have been proposed for EDS in OSA, such as hypoxia, neuronal damage, brain structural changes [2], sleep fragmentation and micro-arousals produced by increased ventilatory effort [3, 4].
Patients may be personally unaware of their snoring and breathing pauses during sleep, but the consequences of daytime hyper-somnolence and fatigue include deficits in attention (road accidents, accidents at work). When used properly, nasal continuous positive airway pressure (nCPAP) reduces apnoea and hypopnoea rate, often normalises arterial blood oxygen saturation, decreases sleep fragmentation, improves sleep quality and often reduces EDS and fatigue [1], which are the major disabling symptoms of the disease. Nevertheless, a significant fraction of patients treated with nCPAP or alternatives, such as mandibular devices, still display EDS and fatigue, which are accompanied by loss of productivity at work, deterioration in quality of life, and increased risk of occupational and motor vehicle accidents [5]. In addition, CPAP effectiveness is limited due to suboptimal acceptance and adherence rates: 50% of individuals will discontinue CPAP treatment within 1 year and 25% terminate CPAP treatment within 3 years. Non-adherence to treatment, estimated at 30–40%, did not vary significantly over 20 years [6]. Hence, a significant proportion of OSA patients not benefiting from nCPAP therapy are exposed to risks related to car, professional or domestic accidents. The risk of car accidents associated with OSA in Europe was recently emphasised by Bonsignore et al, clearly highlighting the relevance of sleepiness on the ability to drive [7]. In the USA, an estimated 810,000 collisions per year and 1400 fatalities are attributable to OSA-related car accidents [8].
For all these OSA patients, pharmacotherapy of EDS and fatigue should be envisaged, and any treatment should be evaluated with and without nCPAP [9].
1.2 Description of the Intervention
Pitolisant is an orally active selective histamine H3 receptor (H3R) antagonist/inverse agonist, which enhances histaminergic transmissions in the brain and thereby elicits strong wake-promoting effects. It is the first compound of this class to be introduced in the clinic, initially to treat EDS and cataplexy in patients with narcolepsy [10,11,12,13].
Improvement of learning deficit and memory by H3R antagonists was also demonstrated experimentally in mice [14,15,16]. The effects of pitolisant 20 mg on sleep/wakefulness and electroencephalography (EEG) patterns were investigated in cats and mice. It was shown that pitolisant markedly increased the duration of waking in both species. Moreover, the EEG results suggest that pitolisant could improve the quality of arousal by reinforcing, for example, the level of attention in the absence of any noticeable psychomotor activation that is observed with psychostimulant drugs like amphetamines or modafinil acting via dopamine and noradrenaline release. Pitolisant does not increase dopamine release in the striatum including the nucleus accumbens, as observed for other wake-promoting agents. In healthy volunteers, pitolisant showed good clinical and biological tolerance following single oral doses of 1–240 mg [10,11,12,13].
1.3 Existing Studies, Review Justification and Objective
The efficacy and safety of pitolisant were assessed in a first prospective multicentre, double-blind (DB) randomised controlled trial (RCT) of pitolisant with a maximum dosage of 20 mg versus placebo (HAROSA-1, 17) in patients diagnosed with moderate or severe OSA using CPAP therapy. The duration of the trial was 3 months followed by an open-label follow-up of 9 months. This study was followed by a second RCT (HAROSA-2) in OSA patients who refused CPAP therapy [18]. Apart from the difference in patient selection, the two studies had the same design, duration and endpoints. Almost all the patients used a final dose of 20 mg.
There was no pharmacokinetic assessment in these studies, but a PET scan trial in healthy volunteers assessed the brain H3R occupancy of pitolisant [19].
This integrated analysis had been planned since the beginning of this research project with the objective of synthesising the results of the two studies in providing pooled estimates of the main and secondary endpoints based on a large sample, comparing the results between patients treated/not treated with CPAP, and assessing the homogeneity of the efficacy across possible subgroups defined by baseline characteristics. The two above-mentioned studies were the pivotal studies for submission of pitolisant approval to the European Medicine Agencies. As the studied drug just received the indication for OSA, these studies are the only two available, and constitute the exhaustive material for this meta-analysis concentrating on the effect of pitolisant in obstructive sleep apnoea.
2 Methods
2.1 Protocol and Guidelines
The study protocol and the statistical analysis plan defining this analysis were commonly agreed by a scientific review board and locked (15–12–2011) blind to treatment, at the early stage of the clinical research programme assessing the benefit of pitolisant in OSA. PRISMA guidelines were followed; information sources and risk of bias across studies do not apply in this analysis as all the studies are known.
2.2 Participants, Intervention
All randomised patients constitute the intention-to-treat population (full analysis set, FAS); no other selection was analysed.
Intervention was defined as treatment taken, fasting once daily, individual titration starting from 5 mg/day for 1 week, then 10 mg/day and 20 mg/day (studied in the present analysis) based on efficacy and tolerability. The best adapted and tolerated dose was administered for the 9-week stable dose period. Being an escalating dose, the lower dosages (5 mg and 10 mg) were not studied.
2.3 Outcomes
The Epworth Sleepiness Scale (ESS) and Oxford Sleep Resistance (OSleR) test, two commonly used measurement tools of EDS, constitute our main endpoints: ESS is a validated self-administered questionnaire for assessing EDS severity, and is a sensitive and specific assessment of sleepiness in OSA patients. Responses are summed to yield a score between 0 and 24, with higher scores representing greater sleepiness. A score ≥ 11 is considered to indicate abnormal sleepiness [20]. ESS was evaluated at each study visit and reviewed by the investigator. The OSleR test is an objective measurement of time wakefulness, consisting of 40 minutes of sleep-resistance challenges conducted in a darkened room isolated from external noise. The subject is asked to stay awake and must respond to a visual stimulus [21]. Improvement of EDS was evaluated according to increase in sleep latency and reduction in the number of errors.
Secondary endpoints were the Pichot Asthenia-Fatigue Subscale questionnaire (range 0–32) [22] and the Clinical Global Impression (CGI) scale, which assesses illness change. The study assessed safety by comparing the number of patients having experienced at least one treatment-emergent adverse event (TEAE) and at least one serious adverse event (SAE) or early trial interruption due to an AE.
2.4 Risk of Bias in Individual Studies
The risk of bias tool (RBT) [23] addresses six specific domains: sequence generation, allocation concealment, blinding, incomplete outcome data, selective outcome reporting, and other bias. Each item is measured on a 3-category risk of bias scale (low, high or unclear). The tool was first completed by the author, followed by the comparison with the European Medicines Agency review [24] and more recent appraisal [25].
2.5 Assessment of the Certainty of the Evidence
The certainty of the evidence was assessed using the GRADE approach [26]. The certainty rating of the studies has four levels: high, moderate, low, and very low. RCTs are initially categorised as high certainty but can be downgraded after assessment of five aspects: risk of bias, consistency, directness, imprecision, and publication bias.
2.6 Data Collection and Management
The research was conducted as an Individual Patient Data (IPD) meta-analysis. The raw data from each study were requested from the manufacturer as an SAS original database. The ESS endpoint was analysed by assuming a normal distribution (confirmed in earlier trials). OSleR values are not normally distributed, due to a truncation limiting the maximum value to 30 minutes and strong left-asymmetry; data were log-transformed, anti-logs of change reflecting the ratio of end-of-treatment/baseline values instead of their difference.
As ESS and OSleR constitute two alternative measurements of EDS, a unique summarised EDS score was defined as the mean of the ESS and OSleR Z-scoresFootnote 1 following the usual approach [27, 28] already proposed for similar purposes in sleep medicine [29].
CGI was collected on a 7-level ordinal scale coded 0–6, shown to be distributed according to a normal distribution. A post hoc analysis was added to compare the proportion of improving patients from the baseline.
A patient was considered as responding to therapy when the final ESS was ≤ 10 or the reduction in ESS was ≥ 3 and OSleR change was ≥ 0. The asthenia-fatigue Pichot subscale, comprises 8 questions (score 0–4), assimilated as a normal distribution, constituted our last endpoint.
For the assessment of safety, a comparison was made of the number of patients having experienced at least one TEAE with at least moderate intensity (thus ignoring TEAEs with mild intensity), as well as the number of patients having experienced at least one SAE or an early trial interruption due to an adverse event.
2.7 Synthesis of Results
The main analysis was the IPD two-level (study-patient) hierarchical model of Higgins et al. [30], assuming a random treatment effect, a random study effect and between-study variance heterogeneity (the error terms allowed to vary by study), but not by treatment group. For sensitivity purposes, results were compared following three alternative models: in fixing between-study variance, in considering the fixed study effect, and the fixed treatment effect. The study compared the relevance of these models using the Bayesian Information Criterion (BIC) [31]. All analyses were adjusted for their baseline values through mixed models featuring linear regression (for ESS, log-transformed OSleR and Z-scores) or logistic regression (for therapy response). For sensitivity purposes, the alternative meta-analytical calculation based on summary means from each study based on random model (summary means meta-analysis, SMM) [32] and multiple imputation averaged data were illustrated by Forest plots; between-study heterogeneity was tested and quantified by the tau2 and I2 statistics. Finally, as ESS values were reported at each visit, and the interaction effect of the treatment with time was calculated through a mixed model on repeated measures (MMRM).
Endpoints and multiplicity of tests: ESS and OSleR were the co-primary endpoints and tested at a two-sided 0.025 significance level. The strategy of testing the secondary endpoints was conducted in conformity with the fixed-sequence technique [33], to limit the overall family-wise type 1 error to a two-sided confidence level of 0.05. Secondary endpoints were only to be tested conditionally to the joint observed significance of the two co-primary endpoints, and the order of testing secondary endpoints was the ESS-OSleR Z-score, CGI, the three responder definitions and the Fatigue scale.
A secondary objective of this analysis was the identification of potential subgroups responding to the therapy defined by baseline conditions (age, gender, occupation, baseline severity of the illness). This objective was reached in adding the first-order interaction between the treatment and each baseline variable.
Multiple imputation was used in this analysis based on a longitudinal model of the full factorial time x treatment effect. Baseline observed carried forward missing allocation was used as a supportive technique for sensitivity purposes.
In what follows, estimates are always reported with their standard deviation (note ± SD) and 95% lower and upper limits of the confidence interval between brackets [LL, UL].
3 Results
3.1 Study Characteristics (Baseline, Treatment)
A total of 512 patients, including 384 treated with pitolisant and 128 with placebo, comprised the FAS. The sample sizes of the two studies were similar (HAROSA-1, n = 244; HAROSA-2, n = 268). Within each study, baseline profiles (gender, employment status, age, and baseline values for ESS, OSleR and Fatigue scale) were comparable (Table 1). Between-study differences in some baseline values were apparent: the HAROSA-1 study included patients treated with CPAP, and baseline values of ESS and OSleR test were better than the corresponding baseline values of the HAROSA-2 study, in which patients without CPAP were included. More women were enrolled in HAROSA-2 (Table 1). Overall, 486 patients completed the studies (5.1% dropout rate).
During the titration phase, 18% and 26.5% of patients in control and pitolisant 20 mg groups, respectively, were controlled before reaching 20 mg (thus, at 5 mg and 10 mg), (risk ratio RR = 1.78 [1.08, 2.94], p = 0.024) (Supp. 13.1 and 13.2).
3.2 Summary of Existing Studies
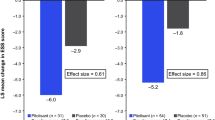
The HAROSA-1 (NCT01071876) [17] and HAROSA-2 (NCT01072968) [18] studies are briefly summarised below. HAROSA-1 was a prospective multicentre, randomised, DB study of pitolisant versus placebo, carried out in patients diagnosed with moderate or severe OSA, having received nCPAP therapy for ≥ 3 months and experiencing EDS. The study consisted of two parts, a 12-week DB phase starting with an escalating dose period followed by treatment with the selected dose. The primary endpoint was ESS; other endpoints were the OSleR test, the Pichot Fatigue scale, and CGI-Severity. A total of 244 patients were randomised to receive either pitolisant (n = 183) or placebo (n = 61) and were observed to match well on baseline characteristics. The change in ESS score between the start and end of DB treatment was − 5.52 in the pitolisant treatment group and − 2.75 in the placebo group, with a treatment effect of − 2.6 (− 3.9; − 1.4; p < 0.001).
The HAROSA-2 trial was a similar RCT to HAROSA-1, limited to patients not using CPAP (n = 268) The primary efficacy analysis showed a statistically significant treatment effect with mean changes in the ESS score of − 6.3 and − 3.6, respectively, with a difference of − 2.8 (− 4.0; − 1.5; p < 0.001). In both studies, results on secondary endpoints indicated beneficial effects of pitolisant compared with placebo. The statistical significance of these endpoints remains to be demonstrated.
3.3 Risk of Bias and Indirectness Within Studies
The RBT [23] for the two studies was first completed by the author, afterwards compared with the EMA review report [34], and finally with the RBT completed in a recent meta-analysis [25]. The three sources were almost concordant and did not suspect any methodological biases or indirectness within the study (patient selection, intervention, comparator, outcome) (Suppl. Section 12).
3.4 Assessment of the Certainty of Evidence
This assessment was completed for the two main endpoints. The four components of certainty of evidence according to GRADE [26] are summarised as: (a) Bias: only RCTs are included into this analysis and the RBT showed a lack of bias risk; (b) Indirectness: the external validity of the two trials was found to be acceptable; (c) Precision: for binary endpoints, according to Cochrane the MID is fixed to RR = 1.25 (thus relative risk increase [RRI] = relative risk reduction [RRR] = 25%). In this pathology, a responder rate of 0.50 in standard therapy comparator can be considered. Under these assumptions, a sample size of 2x246 or ≅492 patients should be needed. This meta-analysis included 508 patients; thus, the sample size was superior to the needed Important Information Size IOS; (d) Publication bias is de facto ruled out. These results provide evidence of high-certainty evidence based on these two studies.
3.5 Main Endpoints
3.5.1 ESS
Baseline ESS values between treatment groups were comparable in each study (Table 1). Overall, final ESS values for pitolisant and placebo were 9.4 ± 4.6 and 12.0 ± 5.7, respectively, with mean differences from baseline of − 5.9 ± 4.5 and − 3.2 ± 5.7 (Table 2). The pitolisant effect was found with a mean difference, pitolisant–placebo, of − 3.1 (− 4.1; − 2.1; p < 0.001) (Table 2; Suppl. 2.2.1). The alternative statistical models provided very similar results with less than 1% relative difference (Suppl. 2.2.2–2.2.4). The summary mean-based (SMB) alternative model provided an almost identical pooled mean ESS difference of − 3.1 (− 3.4; − 2.7; p < 0.001, Table 2), summarised by a Forest plot (Fig. 1). Alternative BCF multiple imputation provided a slightly reduced effect of − 2.7 (− 3.8; − 1.6, p < 0.001) (Suppl. 2.5). All these results were homogeneous among studies (p > 0.5, I2 ≅ 0).

Forest plots and estimates from the summary mean-based (SMB) meta-analyses assuming random effect (RE) model. 1 ESS: Epworth Sleepiness Scale, m(c), n(c) designate the mean and sample size for the control group in each study and m(p), n(p) the mean and sample size for the pitolisant group. 2 OSleR: Oxford Sleep Resistance test, gm(c),n(c) designate the geometric mean and sample size for the control group in each study and gm(p), n(p) the geometric mean and sample size for the pitolisant group. 3 Therapy responder proportion: responder defined as final value ESS ≤ 10 or a ESS decrease ≥ 3 and a positive OSleR change:% (c) and n(c) are the percent of responder and the sample size in the control group,% (p) and n (p) are the percent of responder and the sample size in the pitolisant group. 4 Patients improving on Fatigue scale defined as the proportion of patients for which the Pichot Fatigue decreased compared with their baseline, same notation as graphic 3. 5 Improving patients following the Clinical Global Impression reported at the end of the double-blind period (see graphic 3 for notations). 6 Proportion of patients with at least one treatment-emergent adverse event (TEAE), see notation graphic 3
Finally, through an MMRM using intermediate values of ESS at each visit (Suppl. Table 2.8), compared with placebo, a treatment effect of − 3.4 (− 4.3; − 2.6) (interaction time x treatment test, p < 0.001) was found corresponding to a treatment effect at the end of the 12 weeks. A supportive nonlinear longitudinal analysis (Suppl. Table 2.8.2,) provided some evidence of a faster decrease in the early phase of the treatment until Week 4, whereas change in time becomes more stable after this time.
3.5.2 OSleR Test
Geometric mean OSleR values at baseline were comparable (Table 1). Geometric mean final observed values for pitolisant and placebo were 18.9 (17.2; 20.7) and 17.6 (15.0; 20.8), respectively, with final/baseline ratios of 1.6 (1.4;1.7) and 1.3 (1.1;1.5), respectively (Table 2) and the resulting pitolisant effect on final/baseline ratio was 1.18 (1.02; 1.35; p = 0.022) (Table 2; Suppl. 3.2.1). Alternative IPD models provided very similar treatment effects with a relative difference not exceeding 1% (Table 3, Suppl. 3.2.1–3.2.4), as did the main model used with other missing data strategies (Suppl. 3.3.1–3.3.2). The SMB meta-analysis (Fig. 1, Suppl. 3.4) provided a very similar ratio of change of 1.19 (1.03; 1.37; p = 0.019, Fig. 1) and demonstrated homogeneity among studies.
3.6 Secondary Endpoints
3.6.1 Composite ESS and OSleR Z-Score
Baseline Z values were comparable for each study between treatment groups (Table 1). The mean observed benefit of pitolisant compared with placebo for the final/baseline difference was 0.71 (0.46 to 0.97; p < 0.001) (Table 2, Suppl. 4.2.1), confirmed using alternative BCF missing data imputation (Suppl. 4.3), and with very similar results by using the summary mean meta-analysis (Fig. 1).
3.6.2 Clinical Global Impression (CGI)
A highly significant treatment effect was found on the CGI scale, with a benefit of − 0.63 (− 0.84; − 0.47; p < 0.001, Suppl. 9.2.1) compared with the placebo, confirmed by the results from the SMM model − 0.67 (− 0.97; − 0.36, p < 0.001, Suppl. 9.3, Fig. 1), and found to be homogeneous among studies. The unadjusted proportion of patients having improvements was 54.9% and 78.1% in the control and pitolisant groups, respectively, with 46% more in the pitolisant group (RR = 1.46 [1.12;1.89]; p = 0.005, Suppl. 9.4.1), and confirmed by the analysis of summary means in the two studies (RR = 1.45 [1.23; 1.72]; p = 0.005), Fig. 1, Suppl. 9.4.2).
3.6.3 Fatigue Scale
A significant improvement was shown by a mean change from baseline of − 1.3 (− 2.3; − 0.2; p = 0.017) (Table 8; Suppl. 8.2.1) in the pitolisant group compared with the placebo group. Overall, 56.2% and 74.0% of patients improved on placebo and pitolisant, respectively, with 30% more patients improving in the pitolisant group (relative risk RR = 1.30 [1.11;1.53], p = 0.001, Suppl. 8.3).
3.6.4 Treatment Response
It was observed that 56.2% and 32% of patients in the pitolisant and placebo groups, respectively, responded to therapy (RR = 1.76, [1.39; 2.24], p < 0.001), defined as a final value of ESS ≤ 10 or an ESS decrease ≥ 3 and a positive OSleR change (Table 2, Suppl. 7.2). SBM results were similar (RR = 1.72 [1.36; 2.17], p < 0.001). Responder therapy definition based on separate conditions (ESS ≤ 10) or (ESS decrease ≥ 3) provided similar results (RR = 1.81 and 1.85, respectively; Suppl. 5.2 and 6.2).
3.6.5 Safety
Serious TEAEs were reported for two patients in HAROSA-2 (one prolonged QT interval on the ECG and one cardiopulmonary failure leading to death) during pitolisant treatment, considered unlikely to be related to treatment, and in no patient receiving placebo. No such SAEs were observed for HAROSA-1. In addition, there was no significant change in cardiovascular parameters (systolic or diastolic blood pressure and heart rate) in the two groups.
A non-significantly higher number of patients experienced at least one TEAE of at least moderate severity, RR = 1.27 (0.82; 1.97; p = 0.288), adjusting for age, gender, and baseline severity; (Suppl. Table 11.1). By comparing the proportion of patients in each study (Suppl. Table 11.2), a similar value of RR = 1.29, (0.87; 1.91, p = 0.198) was found.
Four types of TEAEs were found with an incidence of more than 2% in at least one of the two groups (Table 3): gastrointestinal (diarrhoea, nausea, 7.8% and 8.3% for the placebo and pitolisant groups, respectively, χ2, p = 0.971), influenza (6.2%, 7.5%, p = 0.761), insomnia (3.1%, 7.3%, p = 0.138), and headache (11.7%, 11.4%, p = 0.997).
3.6.6 Homogeneity Assessment Across Subgroups Defined by Baseline Characteristics
The significance of differences among subgroups was assessed by adding the interaction term between treatment and each available baseline characteristic: gender, job (yes/no), age (< > 50 years), BMI (< > 30 kg/m2), medical history (yes/no), Apnoea AHI index (< > 15), ESS baseline (< ≥ 15), and OSleR baseline (< ≥ 16 min). Results on ESS (Suppl. 10.2.1–10.2.33, summary Suppl. 10.2.34) and OSleR (Suppl. 10.3.1–10.3.33, summary Suppl. 10.3.34) based on the main model failed to find any significant interaction term.
4 Discussion
This integrated analysis of two RCTs compared pitolisant (pitolisant 20 mg) with placebo in the treatment of OSA syndrome with or without the use of CPAP.
During the preparation of this publication, a meta-analysis was published [25] based on literature findings based on trials in both narcolepsy and OSA on the ESS main endpoint. The objective of this study is much different: based on individual patient data from studies in OSA only, we assess pitolisant efficacy on a variety of main and secondary other endpoints, in accordance with a pre-determined statistical analysis plan, in adjusting for baseline conditions, in conducting various sensitivity analyses, in assessing the homogeneity of the effect across possible subgroups defined by baseline covariates, and testing the difference of efficacy between patients using or not using CPAP.
Compared with the results of each separate study, our analysis provides the following additional findings:
-
1.
Main endpoints The statistical analysis plan was based on ESS and OSleR tests considered as co-primary endpoints, tested at 0.025 two-sided significance. Our first co-primary endpoint was the ESS scale, for which the efficacy of pitolisant compared with placebo was a mean reduction of – 3.1 (− 4.1; − 2.1; p < 0.001). This result was completed by several sensitivity analyses showing an average mean reduction of ESS slightly above the value 3. Our second co-primary endpoint was the OSleR test considered as an objective laboratory assessment of EDS, for which a higher change from baseline was 18% compared with placebo (final/baseline ratio = 1.18, p = 0.022). As for ESS, sensitivity analyses provide an average mean value of 1.17 with a relative error of 2.5%. These two significant results allow us to conjecture a significant effect of pitolisant in reducing subjective and objective assessments of EDS in OSA patients.
-
2.
Secondary endpoints The overall significance based on the two co-primary endpoints enabled the assessment of secondary endpoints following a fixed sequence controlling the initially accepted type 1 risk.
Definition of therapy response was based on a final value < 10 or a decrease of > 3, under conditions of an increasing change in OSleR. We justify this definition as follows: a value of 10 was considered as normal value [20]; on the other hand, a difference of at least 3 was demonstrated to be the minimum clinically relevant difference for ESS [35]. Also, the minimum clinically significant difference in OSA was recently calculated to be between 2 and 3 [36, 37]. Finally, EDS response can only be considered when both alternative endpoints (ESS and OSLeR) are observed to improve. Based on this definition, the proportion of responders was 76% higher in the pitolisant arm compared with placebo RR = 1.76, (1.39; 2.24; p < 0.001).
For the EDS Z-score, encompassing both ESS and OSleR, a difference of 0.71 (p < 0.001). In addition to confirming the consistency of efficacy across the two main endpoints, the EDS-Z score provides an estimate of the clinical relevance of the efficacy of pitolisant. As this score is calculated as a standardised mean difference, the well-known Cohen rule [28] stipulates that those values above 0.2, 0.5 and 0.8 may be assimilated to clinically relevant small, medium and large effect sizes, respectively. From the lower limit (0.46), and with a mean value of 0.71, the effect of pitolisant on this index is thus assimilated with a medium effect size.
Results on Clinical Global Impression confirm the two co-primary endpoints, with 46% more patients improving in the pitolisant group, whereas our final secondary endpoint was the Fatigue scale with 30% more improving patients in the pitolisant group. Fatigue is, together with EDS, a major complaint of OSA patients. It is often confounded with EDS but, although the two symptoms are often displayed together by patients, they clearly differ. The Fatigue scale assesses the general level of excessive tiredness through scoring items like “I have no energy”, “Everything requires effort” and “I feel difficulty concentrating”, which clearly differ from those on the Epworth scale. Both parameters appear predictive of the increased risk of professional, home or car driving accidents among OSA patients [7, 8].
-
3.
The proportion of patients having experienced at least one TEAE of at least moderate severity was not significantly higher compared with the placebo arm.
-
4.
Possible differences in the pitolisant effect across various subgroups of the population were assessed: age, gender, and work conditions. For each of these variables, we failed to find a significant interaction with treatment, providing evidence that the efficacy of pitolisant remains constant, irrespective of the baseline conditions of severity of the patients.
-
5.
Combined effects of CPAP and pitolisant: HAROSA-1 study enrolled patients treated with CPAP, and baseline ESS and OSleR test values indicated lower baseline severity compared with those in the HAROSA-2 study, which included patients who did not receive CPAP. This result suggests a beneficial effect of CPAP. On the other hand, the highly significant pitolisant effect was found homogeneous across the two studies and allows one to conjecture an additional effect of pitolisant to CPAP in EDS.
These results have at least two important limitations.
The comparison of the studied drug with and without CPAP is based on two studies based on a selection of patients using CPAP (HAROSA-1) or not (HAROSA-2). These results provide evidence of the efficacy of pitolisant, irrespective of CPAP use. Although the overall effect of pitolisant irrespective of CPAP was demonstrated, the synergistic effect of pitolisant 20 mg with CPAP necessitates a randomised design on both CPAP and pitolisant. However, due to the observed homogeneity of the main endpoints across studies, the confounded effect of CPAP with the study treatment can be disregarded.
The duration of the double-blind placebo-controlled follow-up was 3 months. For the two studies, this part was followed by an open-label non-controlled period of 9 months, allowing us to study the long-term effect of the studied drug at one year. This part was voluntarily disregarded in this research and remains to be investigated.
A meta-analysis is also known for other limitations. In this study, however, the two used were known in advance to constitute the exhaustive material for assessing pitolisant in OSA, and the methodological quality and certainty of evidence were already assessed. This has substantially limited the arbitrary choice of study, and the risk of bias within and between studies.
5 Conclusions
This Individual Patient Data meta-analysis, based on two randomised controlled studies and 512 patients, constitutes the exhaustive material evidencing the efficacy and safety of pitolisant up to 20 mg in OSA. This study provides evidence that, compared with placebo, pitolisant has a clinically relevant effect in improving EDS and fatigue in OSA patients with or without CPAP. This effect was observed as consistent across endpoints, studies, and irrespective of baseline conditions.
Notes
Z-score of an endpoint X as the transformed value (X-m)/SD where m and SD are the mean and the standard deviation of X measured at baseline.
References
Peppard PE et al. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006–14. https://doi.org/10.1093/aje/kws342.
Macey PM, Kumar R, Woo MA, Valladares EM, Yan-Go FL, Harper RM. Brain structural changes in obstructive sleep apnea. Sleep. 2008;31(7):967–77.
Nieto F, Young T, Lind B, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. JAMA. 2000;283:1829–36. https://doi.org/10.1001/jama.283.14.1829.
Peppard P, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342:1378–84. https://doi.org/10.1056/NEJM200005113421901.
Rosenzweig I, Glasser M, Polsek D, et al. Sleep apnoea and the brain: a complex relationship. Lancet Respir Med. 2015;3(5):404–14. https://doi.org/10.1016/S2213-2600(15)00090-9.
Rotenberg BW, Murariu D, Pang KP. Trends in CPAP adherence over twenty years of data collection: a flattened curve. J Otolaryngol Head Neck Surg. 2016;45(1):43. https://doi.org/10.1186/s40463-016-0156-0.
Bonsignore MR, Randerath W, Schiza S, et al. European Respiratory Society statement on sleep apnoea, sleepiness and driving risk. Eur Respir J. 2021;57(2):2001272. https://doi.org/10.1183/13993003.01272-2020.
Rodenstein D. Sleep apnea: traffic and occupational accidents—individual risks, socioeconomic and legal implications. Respiration. 2009;78(3):241–8. https://doi.org/10.1159/000222811.
Schwartz JRL, Hirshkowitz M, Erman EK, et al. Modafinil as adjunct therapy for daytime sleepiness in obstructive sleep apnea: a 12-week, open-label study. Chest. 2003;124(6):2192–9. https://doi.org/10.1378/chest.124.6.2192.
Schwartz J-C. The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol. 2011;163(4):713–21. https://doi.org/10.1111/j.1476-5381.2011.01286.x.
European Medicines Agency: Committee for Medicinal Products for Human Use (CHMP). Assessment report, Wakix. International non-proprietary name: pitolisant. Procedure no. EMEA/H/C/002616/0000. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002616/WC500204749.pdf. Accessed 13 Apr 2021.
US Food and Drug Administration (FDA). WAKIX® (pitolisant) tablets, for oral use. Highlights of prescribing information. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211150s000lbl.pdf. Accessed 13 Apr 2021.
Lamb YN. Pitolisant in narcolepsy with or without cataplexy. CNS Drugs. 2020;34(2):207–18. https://doi.org/10.1007/s40263-020-00703-x.
Miyazaki S, Imaizumi M, Onodera K. Effects of thioperamide, a histamine H3-receptor antagonist, on a scopolamine-induced learning deficit using an elevated plus-maze test in mice. Life Sci. 1995;57(23):2137–44. https://doi.org/10.1016/0024-3205(95)02206-x.
Meguro K, Yanai K, Sakai N, et al. Effects of thioperamide, a histamine H3 antagonist, on the step-through passive avoidance response and histidine decarboxylase activity in senescence-accelerated mice. Pharmacol Biochem Behav. 1995;50(3):321–5. https://doi.org/10.1016/0091-3057(95)00248-u.
Onodera K, Miyazaki S, Imaizumi M, et al. Improvement by FUB 181, a novel histamine H3-receptor antagonist, of learning and memory in the elevated plus-maze test in mice. Naunyn Schmiedebergs Arch Pharmacol. 1998;357(5):508–13. https://doi.org/10.1007/pl00005200.
Pépin JL, Georgiev O, Tiholov R, et al. Pitolisant for residual excessive daytime sleepiness in OSA patients adhering to CPAP: a randomized trial. Chest. 2020;S0012–3692(20):35105–9. https://doi.org/10.1016/j.chest.2020.09.281.
Dauvilliers Y, Verbraecken J, Partinen M, et al. Pitolisant for daytime sleepiness in patients with obstructive sleep apnea who refuse continuous positive airway pressure treatment. A randomized trial. Am J Respir Crit Care Med. 2020;201(9):1135–45. https://doi.org/10.1164/rccm.201907-1284OC.
Rusjan P, Sabioni P, Di Ciano P, Mansouri E, Boileau I, Laveillé A, Capet M, Duvauchelle T, Schwartz JC, Robert P, Le Foll B. Exploring occupancy of the histamine H3 receptor by pitolisant in humans using PET. Br J Pharmacol. 2020;177(15):3464–72. https://doi.org/10.1111/bph.15067.
Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14(6):540–5. https://doi.org/10.1093/sleep/14.6.540.
Bennett LS, Stradling JR, Davies JO. A behavioral test to assess daytime sleepiness in obstructive sleep apnoea. J Sleep Res. 1997;6:142–5. https://doi.org/10.1046/j.1365-2869.1997.00039.x.
Pichot P, Brun JP. Brief self-evaluation questionnaire for depressive, asthenic and anxious dimensions. Ann Med Psychol (Paris). 1984;142(6):862–5.
Higgins JPT, Altman DG, Sterne JAC. Chapter 8: assessing risk of bias in included studies. In: Higgins JP, Green S, editors. Cochrane handbook for systematic reviews of interventions version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. http://handbook.cochrane.org.
European Medicines Agency: Committee for Medicinal Products for Human Use (CHMP). Assessment report, Wakix. International non-proprietary name: pitolisant. Procedure No. EMEA/H/C/002616/0000. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002616/WC500204749.pdf. Accessed 13 Apr 2019.
Wang J, Li X, Yang S, Wang T, Xu Z, Xu J, Gao H, Chen G. Pitolisant versus placebo for excessive daytime sleepiness in narcolepsy and obstructive sleep apnea: A meta-analysis from randomized controlled trials. Pharmacol Res. 2021;167:105522. https://doi.org/10.1016/j.phrs.2021.105522.
Guyatt G, Oxman AD, Akl EA, et al. GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. J Clin Epidemiol. 2011;64(4):383–94. https://doi.org/10.1016/j.jclinepi.2010.04.026.
O’Brien PC. Procedures for comparing samples with multiple endpoints. Biometrics. 1984;40:1079–87.
Cohen J. Statistical power analysis for the behavioral sciences. 2nd ed. New York: Academic Press; 1988.
Lehert P, Falissard B. Multiple treatment comparison in narcolepsy: a network meta-analysis. Sleep. 2018;41(12):zsy185. https://doi.org/10.1093/sleep/zsy185.
Higgins JP, Whitehead A, Turner RM, et al. Meta-analysis of continuous outcome data from individual patients. Stat Med. 2001;20(15):2219–41. https://doi.org/10.1002/sim.918.
Pinheiro JC, Bates DM. Linear mixed-effects models: basic concepts and examples. In: Mixed-effects models in S and S-PLUS. Statistics and computing. Springer, New York; 2000. pp. 3–56. https://doi.org/10.1007/0-387-22747-4_1.
Brockwell SE, Gordon IR. A comparison of statistical methods for meta-analysis. Stat Med. 2001;20(6):825–40. https://doi.org/10.1002/sim.650.
US Food and Drug Administration. Multiple endpoints in clinical trials guidance for industry. US Department of Health and Human Services, 2017. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/multiple-endpoints-clinical-trials-guidance-industry. Accessed 20 Apr 2021.
Ozawade. EMA. 2021. https://www.ema.europa.eu/en/documents/assessment-report/ozawade-epar-public-assessment-report_en.pdf. Accessed 30 Nov 2021.
Lehert P. Epworth sleepiness scale ESS: determination of a minimum clinically relevant difference. Sleep Med. 2017;40(Suppl 1):e186–7. https://doi.org/10.1016/j.sleep.2017.11.544.
Patel S, Kon SSC, Nolan CM, et al. The Epworth sleepiness scale: minimum clinically important difference in obstructive sleep apnea. Am J Respir Crit Care Med. 2018;197(7):961–3. https://doi.org/10.1164/rccm.201704-0672LE.
Crook S, Sievi NA, Bloch KE, et al. Minimum important difference of the Epworth Sleepiness Scale in obstructive sleep apnoea: estimation from three randomized controlled trials. Thorax. 2019;74(4):390–6. https://doi.org/10.1136/thoraxjnl-2018-211959.
Acknowledgements
I am grateful to Dr. Evelyne Deschamps de Paillette for her precious and relevant comments during this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data Availability Statement
The data and detailed results of the analyses that support the findings of this study are available in the supplementary material to this article. Any further detail on the used material, including the data, methods, statistical analysis plan, can be obtained by a simple request to the author.
Consent to participate
Not applicable.
Code availability
Not applicable.
Consent for publication
Not applicable.
Funding
This article was supported by Bioprojet. Bioprojet, No Number. Philippe LEHERT.
Author contribution statement
P. Lehert is the only author, contributed to the conception, design, acquisition of data, analysis and interpretation of data; participated in drafting, reviewing and revising the manuscript and approved its submission.
Conflict of interest
P. Lehert received an unconditional grant from Bioprojet for conducting this analysis.
Ethics approval
Ethics approval is not applicable to this article since it is a meta-analysis.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lehert, P. Efficacy of Pitolisant 20 mg in Reducing Excessive Daytime Sleepiness and Fatigue in Patients with Obstructive Sleep Apnoea Syndrome: An Individual Patient Data Meta-analysis. Clin Drug Investig 42, 65–74 (2022). https://doi.org/10.1007/s40261-021-01104-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-021-01104-8