
Abstract
Background and Objectives
Co-suspension Delivery™ Technology has been developed for the administration of albuterol sulfate pressurised inhalation suspension via metered-dose inhaler (AS MDI, PT007). We assessed the efficacy and safety of AS MDI versus Proventil® in order to determine the optimal dose of AS MDI to take to Phase III clinical trials.
Methods
ASPEN (NCT03371459) and ANTORA (NCT03364608) were Phase II, randomised, crossover, multicentre studies of AS MDI versus Proventil® in patients with persistent asthma. In ASPEN, 46 patients received cumulative-dose treatments (90 μg/inhalation using 1 + 1 + 2 + 4 + 8 inhalations at 30-minute intervals) in 1 of 2 possible sequences: AS MDI/Proventil or Proventil/AS MDI. In ANTORA, 86 patients were randomised to one of 10 treatment sequences of AS MDI (90 μg or 180 μg), placebo MDI, or Proventil (90 μg or 180 μg). The primary endpoints were baseline-adjusted forced expiratory volume in 1 second (FEV1) 30 minutes after each cumulative dose (ASPEN) and change from baseline in FEV1 area under the curve from 0 to 6 h (ANTORA). Safety was assessed in both studies.
Results
In ASPEN, AS MDI was equivalent to Proventil (within pre-specified bounds of ± 200 mL) following cumulative doses of albuterol up to 1440 μg for the primary endpoint. In ANTORA, 90 μg and 180 μg doses of AS MDI and Proventil were significantly superior to placebo MDI (p < 0.0001), and AS MDI was non-inferior to Proventil at both doses, based on a margin of 100 mL. No new safety concerns were identified.
Conclusion
The effects of albuterol delivered via AS MDI and Proventil on bronchodilation were equivalent, supporting the selection of AS MDI 180 µg to be taken into Phase III clinical trials, either alone or in combination with an inhaled corticosteroid.
Trial Registration number
ASPEN (NCT03371459); Date of registration: 29/12/2017. ANTORA (NCT03364608); Date of registration: 15/12/2017.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The effects on lung function of albuterol sulfate delivered either via a novel co-suspension delivery technology pressurised metered-dose inhaler (AS MDI) or via the Proventil inhaler were equivalent at doses of albuterol up to 1440 μg. |
At doses of 90 μg and 180 μg, AS MDI was more effective than placebo MDI and non-inferior to Proventil in improving lung function. |
The data support the selection of AS MDI 180 µg to be taken into Phase III clinical trials. |
1 Introduction
Asthma is a heterogeneous inflammatory airway disease [1, 2] defined by a history of variable respiratory symptoms including wheeze and cough and with variable expiratory airflow obstruction [1, 2] across all asthma severities [1]. All patients are at risk of preventable and potentially serious exacerbations, irrespective of asthma severity.
Short-acting β2-agonists (SABAs), such as albuterol (salbutamol), have been prescribed worldwide for over 50 years, to provide quick relief of asthma symtoms as ‘rescue’ medication [1]. Albuterol can be combined with inhaled corticosteroids (ICS) or other anti-inflammatory medications to help patients receive an anti-inflammatory benefit each time they utilise a rescue inhaler. Combining rescue and anti-inflammatory therapies is now recommended for all asthma patients in the most recent update of the Global Initiative for Asthma (GINA) guidelines [1].
An innovative Co-suspension Delivery™ Technology has been developed and utilised in several mono- and combination inhalation therapy products (e.g. Bevespi Aerosphere™, Breztri Aerosphere™), including albuterol sulfate (hereafter referred to as albuterol) pressurised inhalation suspension delivered via metered-dose inhaler (AS MDI, PT007). Micronised albuterol sulfate, a racemic salt of albuterol, is co-suspended with spray-dried porous particles (i.e., phospholipid, 1,2-distearoyl-sn-glycero-3-phosphocholine and calcium chloride) in a hydrofluoroalkane (HFA) propellant. These porous particles form strong, nonspecific associations with albuterol to enable consistent delivery of single or combination products with respirable particle sizes after each inhalation to potentially treat the entire lung.
Phase II studies were required to select the optimal dose of albuterol in AS MDI, which then determined the albuterol dose to use in subsequent SABA/ICS combination (PT027) Phase III studies (NCT03769090, NCT03847896). This paper details the results of two such Phase II trials in patients with persistent asthma: ASPEN (NCT03371459) and ANTORA (NCT03364608).
ASPEN was a cumulative-dose study investigating the efficacy and safety of cumulative doses up to 1440 μg of AS MDI versus Proventil® HFA, in addition to characterising its pharmacokinetic/relative systemic bioavailability and extrapulmonary pharmacodynamic properties in adults with persistent asthma.
ANTORA was a dose-ranging study comparing the bronchodilatory efficacy and safety of 90 μg and 180 μg doses of AS MDI versus a matching placebo MDI and open-label Proventil in adults and adolescents with persistent asthma. The objectives of ANTORA were (1) to confirm that the dose of albuterol from AS MDI delivered comparable efficacy relative to Proventil; and (2) to assess the safety and tolerability of AS MDI relative to Proventil and placebo.
2 Methods
2.1 Study Designs and Patients
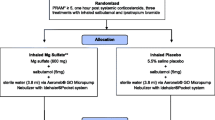
Both ASPEN (PT007002; NCT03371459) and ANTORA (PT007001; NCT03364608) were Phase II, randomised, crossover, multicentre studies. ASPEN was an open-label, two-period, cumulative dose trial, whereas ANTORA was a single-dose, five-period, dose-ranging study. Both studies consisted of screening/run-in periods, followed by a sequence of treatment periods separated by 3- to 7-day washouts (Supplementary Figure S1). AS MDI and placebo were administered as blinded treatments and Proventil was administered as an open-label treatment.
Patients eligible for randomisation were aged between 18 and 45 years (ASPEN) or 12 and 65 years (ANTORA); had stable, physician-diagnosed persistent asthma for at least 6 months prior to screening; had previously been treated with as-needed SABA-alone, or with stable, low-to-medium dose ICS alone or in combination with a long-acting β2-agonist (LABA) for ≥ 30 days prior to screening; had pre-bronchodilator forced expiratory volume in 1 s (FEV1) of ≥ 40% and < 90% (ASPEN) and ≥ 50% and < 80% (ANTORA) of predicted normal value; had reversibility of ≥ 15% following albuterol (Ventolin®) administration; and had to be willing to come in for morning treatment visits, and to withhold caffeine for 6 h, rescue albuterol for at least 6 h (ANTORA), and rescue ipratropium for at least 8 h (ASPEN) prior to visits.
Exclusion criteria included a diagnosis of chronic obstructive pulmonary disease (COPD) or other significant lung disease; oral corticosteroid for any use ≤ 6 months prior to screening; current or former smoker with a history of > 10 pack-years, former smokers who had stopped smoking < 6 months before screening; hospitalisation due to asthma ≤ 6 months prior to screening; and historical or current evidence of a clinically significant disease including cardiovascular (e.g., congestive heart failure), hepatic, renal, haematological, neuropsychological, endocrine (e.g., uncontrolled diabetes mellitus), or gastrointestinal disease. Significant disease was defined as any that would have put the subject at risk during the study or one that could have affected the efficacy or safety endpoints, should the condition have worsened during the study.
After signing informed consent, patients receiving ICS (with or without LABA) prior to study entry were switched to budesonide (Pulmicort Flexhaler®) 180 μg one or two inhalations twice daily. At each visit, pulmonary function tests (PFTs) were used to assess bronchodilation activity. To reduce bias, dosing and spirometry assessments were performed separately so that medical staff conducting the spirometry assessments were blinded to the identity of the study treatment administered.
2.1.1 ASPEN
All patients were allowed to take sponsor-provided rescue medication; albuterol (Ventolin) 90 μg/inhalation as needed (only up to 48 h before Visit 2) and ipratropium bromide 17 μg/inhalation as needed (from 48 h before Visit 2 through the remainder of the study). Ipratropium bromide was used as rescue to ensure that it would not confound the measurement of albuterol pharmacokinetics.
Eligible patients were randomised to receive one of two treatment sequences, A/B or B/A, where A is AS MDI 90 µg and B is Proventil 90 µg. Subjects received five doses of AS MDI and Proventil during Visits 2 and 3 for a total cumulative dose of 1440 µg each; cumulative doses were given as 1 + 1 + 2 + 4 + 8 inhalations (doses given 30 minutes apart), equalling doses of 90, 90, 180, 360, and 720 µg, respectively.
Treatment periods were separated by a 3- to 7-day washout period, with a follow-up telephone assessment 3–7 days after the final treatment period.
2.1.2 ANTORA
The screening/run-in period in the ANTORA study was identical to that in the ASPEN study, except ipratropium was not administered prior to Visit 2. Throughout the study, all patients were allowed to take rescue medication in the form of sponsor-provided albuterol (Ventolin®) 90 μg/inhalation as needed (up to 6 h prior to each treatment visit).
Eligible patients were randomised to one of 10 possible treatment sequences, based on a standard Williams design [3], and received a single dose of treatment at each visit. Each treatment sequence comprised all five of the following treatments: AS MDI 90 μg (2 inhalations of 45 μg/inhalation); AS MDI 180 μg (2 inhalations of 90 μg/inhalation); placebo MDI (2 inhalations); Proventil 90 μg (1 inhalation of 90 μg/inhalation); Proventil 180 μg (2 inhalations of 90 μg/inhalation).
2.2 Study Endpoints
2.2.1 ASPEN
The primary efficacy endpoint of ASPEN was change from baseline in FEV1 30 minutes after each cumulative dose up to 1440 µg. Key secondary efficacy endpoints included change from baseline in FEV1 area under the curve from 0 to 6 h (AUC0–6) after the last cumulative dose; extrapulmonary pharmacodynamic parameters (change, maximum change, and time-weighted average change from baseline); pharmacokinetic properties (albuterol systemic bioavailability); and adverse events (AEs) and serious adverse events (SAEs). AEs were determined by the primary investigator at each site. An AE was defined as any unfavourable and unintended sign (e.g., an abnormal laboratory finding), symptom, or disease temporally associated with the use of a drug. An SAE was defined as an AE resulting in death, inpatient hospitalisation or prolongation of existing hospitalisation, a persistent or significant incapacity to conduct normal life functions, or a congenital anomaly/birth defect.
Approximately 35 mL of blood per subject was collected during the study for clinical laboratory tests. Extrapulmonary pharmacodynamic parameters included Fridericia-corrected QT interval (QTcF), heart rate, serum glucose, serum potassium, diastolic blood pressure (DBP) and systolic blood pressure (SBP). They were all measured pre-dose, and at 12 times post-dose, at 15 minutes after each cumulative dose, and then at 30, 45, 60, 120, 180, 240 and 360 minutes after the final dose. For the pharmacokinetic analysis, blood samples to determine the plasma concentrations of albuterol were drawn during the treatment period at Visits 2 and 3 at the following time points: 30 minutes pre-dose, at 15 minutes after each cumulative dose, and then at 15, 30, 60, 120, 180, 240, 300, 360, 480, 600 and 720 minutes after the final dose. Approximately 220 mL of blood was collected at Visits 2 and 3 for subjects participating in the pharmacokinetic analysis (~ 255 mL of blood was collected in these subjects over the course of the study). Samples were collected, handled, labelled, stored, and shipped as detailed in the laboratory manual, and plasma samples were analysed for albuterol by a central laboratory according to standardised, validated assays. Albuterol systemic bioavailability parameters included plasma concentration 15 minutes after each cumulative dose (C15min); area under the concentration-time curve from time 0 (pre-dose) to the time of the last measurable plasma concentration (AUC0–t); and maximum plasma concentration (Cmax) and time to maximum plasma concentration (Tmax). Further details on the pharmacokinetic parameters are provided in the Supplementary Material.
2.2.2 ANTORA
The primary efficacy endpoint of ANTORA was change from baseline in FEV1 AUC0–6. Secondary endpoints were change from baseline in FEV1 AUC0–4 and peak change from baseline in FEV1. Other additional endpoints investigated included percentage of subjects achieving 12% improvement in FEV1 from baseline within 30 minutes of dose (FEV1% predicted > 12% is a well-recognised cut-off for determining a positive bronchodilator response [4, 5]), time to onset of this response, and safety, including AEs (defined the same as in ASPEN), laboratory parameters, vital signs, and electrocardiogram recordings. Approximately 10 mL of blood was collected per subject during the study for clinical laboratory tests.
2.3 Statistical Analyses
Data processing, descriptive reporting, and analysis of the efficacy, extrapulmonary pharmacodynamic, pharmacokinetic, and safety data were performed using SAS version 9.4. All efficacy and safety parameters were summarised by treatment. Categorical variables were summarised by treatment with frequency counts and percentages. Efficacy analyses in both studies were performed for the Modified Intent-to-Treat (mITT) analysis set (all patients who received study treatment and had post-treatment efficacy data at all treatment periods for ASPEN and at least two treatment periods for ANTORA). Pharmacokinetic and extrapulmonary pharmacodynamic analyses in ASPEN were performed for the Pharmacokinetic and Per Protocol Analysis Sets (all randomised patients who received ≥ 1 dose of the study drug for whom ≥ 1 primary pharmacokinetic parameter could be calculated, and all randomised patients who received ≥ 1 dose of the study drug and had post-treatment extrapulmonary pharmacodynamic data, respectively).
No formal analysis for safety was planned, and safety results were listed by treatment.
2.3.1 ASPEN
Change from baseline in FEV1 after each cumulative dose was analysed using a linear mixed effects model with a random subject effect for the correlation across periods and an unstructured covariance matrix for the repeated measures within subject-periods. Fixed effects were as follows: treatment, cumulative dose level, treatment-by-cumulative dose interaction, and period as categorical covariates and baseline FEV1 as a continuous covariate.
FEV1 AUC0-6 was defined as the AUC for the change from baseline in FEV1 calculated using the trapezoidal rule for all subjects with at least 1 non-missing data point during the first 2 h post-dose. For AUC calculations, the value of the spirometry parameter at time 0 was the change from baseline in pre-bronchodilator FEV1 at 30 minutes after the 4th cumulative dose (720 µg) at the visit. All AUC values were normalised for length of follow-up (typically 6 h). FEV1 AUC0-6 was also analysed using a linear mixed model with a random subject effect, with fixed effects of treatment and period as categorical covariates and baseline FEV1 as a continuous covariate.
Equivalence was analysed by comparing the 90% confidence interval (CI) for the between-treatment differences after each cumulative dose with predefined bounds of ± 200 mL. This margin was selected based on precedence from a similarly designed cumulative-dose study comparing ProAir Respiclick versus ProAir HFA [6], from which a mean change from baseline in FEV1 on the order of 700 mL was observed, thus the ± 200 mL cut-off accounts for greater variability. FEV1 AUC0–6 after the last cumulative dose was analysed using a linear mixed-effects model with a random subject effect; fixed effects were treatment and period as categorical covariates and baseline FEV1 as a continuous covariate.
Pharmacokinetic parameters were estimated by non-compartmental analysis using Phoenix® WinNonlin® Version 6.4. AUC0–t and Cmax values for albuterol were natural log-transformed and were each analysed with a linear mixed effects model containing fixed effects of treatment and period and a random effect of subject. Estimated geometric mean ratios with 90% CIs were provided.
Extrapulmonary pharmacodynamic parameters were analysed using a model similar to that described for the primary efficacy endpoint.
2.3.2 ANTORA
As in ASPEN, change from baseline in FEV1 AUC0-6 was analysed using a linear mixed model with a random subject effect, with fixed effects of treatment, treatment sequence, and period as categorical covariates and baseline FEV1 as a continuous covariate.
FEV1 area under the curve from 0 to 4 h (AUC0–4) was calculated in a similar manner to FEV1 AUC0–6, with the trapezoidal rule implemented through the 4-h nominal time point and AUC values normalised accordingly. The largest FEV1 value measured during the 6 h after dosing was used to calculate the peak change from baseline in FEV1. An approach similar to that used for the primary endpoint was used to analyse the secondary endpoints (FEV1 AUC0–6 and peak change from baseline in FEV1); linear mixed models with a random subject effect, with fixed effects of treatment, treatment sequence, baseline FEV1, and period.
Superiority comparisons for the active formulations versus placebo were conducted using a dose-ordered approach in the following order (with subsequent comparisons performed only after detection of statistical significance for earlier comparisons): Proventil 180 μg versus placebo; AS MDI 180 μg versus placebo; Proventil 90 μg versus placebo; AS MDI 90 μg versus placebo.
If both AS MDI 180 µg and Proventil 180 µg were statistically superior to placebo MDI, the non-inferiority of AS MDI 180 µg versus Proventil 180 µg for FEV1 AUC0–6 was assessed as an exploratory endpoint using a non-inferiority margin of 100 mL. This margin was selected as the known minimal perceivable improvement for FEV1 has been previously reported to be 230 mL [7, 8]. A 100 mL non-inferiority margin is appropriate for this magnitude of change and represents slightly less than half the minimal perceptible FEV1 value. Secondary efficacy analyses also used linear mixed models with a random subject effect; fixed effects included treatment, treatment sequence, baseline FEV1, and period. Peak change from baseline in FEV1 was calculated using the highest FEV1 value measured during the 6 h after dosing.
3 Results
3.1 Patient Demographics
Of the 69 patients screened in ASPEN, 46 were randomised and received ≥ 1 dose of study treatment, with 45 completing both treatment periods (mITT analysis set). One patient was lost to follow-up (Table 1). Patients’ mean age was 34.2 years (range 20–45), and just under half were female (48.9%). Patients had a mean pre-bronchodilator FEV1 63.6% predicted. Before screening, 42.2% and 51.1% of patients were receiving as-needed SABA alone and ICS/LABA, respectively, with the remainder treated with ICS (Table 2).
In ANTORA, 116 patients were screened, 86 were randomised and received ≥ 1 dose of study treatment, and 80 completed at least two treatment periods (mITT analysis set). A total of 78 patients completed all five treatment periods (Table 1). The mean age of patients was 42.6 years (range 13–64), and just over half were female (53.8%). Patients had a mean pre-bronchodilator FEV1 68.0% predicted. Prior to screening, patients were receiving as-needed SABA alone (55.0%), ICS/LABA (36.3%), or ICS (8.8%) (Table 2).
Per protocol, all patients demonstrated at least 15% reversibility to albuterol (Ventolin) during screening. The mean reversibility post-albuterol in ASPEN and ANTORA was 29.3% (standard deviation [SD] 13.1%) and 25.8% (SD 10.6%), and the median reversibility was 23.7% and 22.0%, respectively. Full patient baseline demographics for both trials are presented in Table 2.
3.2 ASPEN
3.2.1 Efficacy Results
Cumulative doses of both AS MDI and Proventil were associated with increases in the least squares (LS) mean change from baseline in FEV1 (30 minutes after each dose), peaking at approximately 2.5 h post-administration of the first cumulative dose (Fig. 1). AS MDI was equivalent to Proventil for the LS mean change from baseline in FEV1 after each cumulative dose, as the 90% CIs for the treatment differences were contained within the pre-specified bounds of ± 200 mL (Table 3).

Adjusted mean change from baseline in FEV1 (± SE) (L) 30 minutes after each cumulative dose and 6 h after the final cumulative dose (mITT analysis population) in the ASPEN study. Arrows indicate when each of the 5 doses was given. AS albuterol sulfate, FEV1 forced expiratory volume in 1 second, MDI metered-dose inhaler, mITT modified intent-to-treat, SE standard error
AS MDI and Proventil were equivalent for the change from baseline in FEV1 AUC0–6 after the last cumulative dose, with LS mean improvements of 561 mL (95% CI 448, 674) and 602 mL (95% CI 489, 715) for AS MDI and Proventil, and an LS mean difference of − 40 mL (90% CI − 77, − 4), which was contained within the predefined equivalence bounds of ± 200 mL (Table 3).
3.2.2 Extrapulmonary Pharmacodynamic Results
AS MDI was associated with numerically lower maximum LS mean changes from baseline and time-weighted average changes from baseline after the last cumulative dose than Proventil for the following extrapulmonary pharmacodynamic parameters: QTcF (maximum LS mean difference [LSMD]: − 5.71 ms; 90% CI − 11.07, − 0.35; time-weighted LSMD: − 1.13 ms; 90% CI − 3.55, 1.28), heart rate (maximum LSMD: − 3.81 beats per minute [bpm]; 90% CI − 6.26, − 1.37; time-weighted LSMD: − 2.27 bpm; 90% CI − 3.73, − 0.81), and serum glucose (maximum LSMD: − 1.56 mg/dL; 90% CI − 7.93, 4.81; time-weighted LSMD: − 1.73 mg/dL; 90% CI − 5.43, 1.97) (Table 4). Maximum LS mean changes from baseline and time-weighted average changes from baseline were slightly higher with AS MDI versus Proventil for both SBP (maximum LSMD: 0.28 mm Hg; 90% CI − 1.86, 2.42; time-weighted LSMD: 0.92 mm Hg; 90% CI − 0.69, 2.53) and DBP (maximum LSMD: 0.47 mm Hg; 90% CI − 1.28, 2.22; time-weighted LSMD: 1.01 mm Hg; 90% CI − 0.15, 2.16) (Table 4). Maximum LS mean changes from baseline and time-weighted average changes from baseline after the last cumulative dose for serum potassium were similar between AS MDI and Proventil (maximum: LSMD: 0.07 mmol/L; 90% CI 0.00, 0.13; time-weighted: LSMD: − 0.01 mmol/L; 90% CI − 0.08, 0.07) (Table 4).
3.2.3 Pharmacokinetic Results
Mean C15min of plasma albuterol for both AS MDI and Proventil increased with increasing cumulative doses until the final dose (~2 h after the first cumulative dose) (Fig. 2). However, following the final cumulative dose, a sharp increase in plasma albuterol concentration was observed with Proventil, which was followed by a steep decline, while in contrast, mean plasma albuterol concentration remained stable during the hour after the final cumulative dose of AS MDI, indicating a longer absorption time. At all time points, mean albuterol concentrations were lower with AS MDI versus Proventil (Fig. 2).

Mean (± SE) plasma albuterol concentration-time profile (pharmacokinetic analysis set) in the ASPEN study. Arrows indicate when each of the 5 doses was given. AS albuterol sulfate, MDI metered-dose inhaler, mITT modified intent-to-treat, SE standard error
The systemic exposure of albuterol was numerically lower for AS MDI versus Proventil following inhalation of cumulative doses (Table 5). The mean Cmax and AUC0-t of albuterol (measured to 14 h after the first cumulative dose) were numerically lower for AS MDI than Proventil (Table 5). The median Tmax was similar between treatments, at 2.6 h after the first cumulative dose for AS MDI and 2.4 h after the first cumulative dose for Proventil (Table 5).
The bioavailability of albuterol was ~21% to 27% lower with AS MDI than with Proventil. The geometric mean ratio of AS MDI to Proventil for Cmax was 72.61% (90% CI 66.48%, 79.30%) and for AUC0-t was 78.83% (90% CI 73.39%, 84.67%).
3.3 ANTORA
3.3.1 Efficacy Results
Both doses of AS MDI and Proventil were statistically superior to placebo for the primary efficacy endpoint (change from baseline in FEV1 AUC0–6) and secondary efficacy endpoints (change from baseline in FEV1 AUC0–4 and peak change from baseline in FEV1) (all p < 0.0001; Table 6). The LS mean differences of AS MDI 90 μg and 180 μg versus placebo were 134 mL (p < 0.0001) and 196 mL (p < 0.0001) for change from baseline in FEV1 AUC0–6 and 183 mL (p < 0.0001) and 250 mL (p < 0.0001) for change from baseline in FEV1 AUC0–4, respectively (Table 6). For Proventil 90 μg and 180 μg, LS mean differences versus placebo were 171 mL (p < 0.0001) and 212 mL (p < 0.0001) (FEV1 AUC0–6) and 217 mL (p < 0.0001) and 269 mL (p < 0.0001) (FEV1 AUC0–4), respectively (Table 6). For both endpoints, AS MDI was non-inferior to Proventil at both the 90 μg and 180 μg dose levels, as demonstrated by the 95% CIs for the difference (AS MDI vs Proventil) in FEV1 AUC0–6 and AUC0–4 being above the predefined non-inferiority margin of 100 mL (Table 6). For peak change from baseline in FEV1, AS MDI 90 μg and 180 μg doses were both statistically superior to placebo, with LS mean differences of 199 mL and 275 mL (both p < 0.0001) for the respective doses (Table 7). Similarly, Proventil 90 μg and 180 μg doses were also superior to placebo for this endpoint, with LS mean differences of 239 mL and 282 mL (both p < 0.0001), respectively (Table 7). Changes from baseline in FEV1 over the 6 h post-administration of treatment were greater with 180 μg versus 90 μg for both AS MDI and Proventil (Fig. 3).

Mean change from baseline in FEV1 (L) over time (mITT analysis set) in the ANTORA study. Error bars represent SE. AS albuterol sulfate, FEV1 forced expiratory volume in 1 second, MDI metered-dose inhaler, mITT modified intent-to-treat, SE standard error
Median times to onset of response (12% improvement in FEV1) were comparable across the dose levels and formulations, at approximately 7 minutes for all active treatments. The percentages of subjects achieving a 12% improvement in FEV1 were greater with all active treatment arms versus placebo. This percentage was slightly higher with the 180 μg doses compared with the 90 μg doses for both AS MDI and Proventil (Table 7). Of the patients treated with AS MDI and Proventil, 61%–75% achieved ≥ 12% improvement, versus only 12.8% of patients receiving placebo.
3.4 Safety and Tolerability
3.4.1 ASPEN
No deaths, serious treatment-emergent adverse events (TEAEs) or serious treatment-related AEs were reported during the study. Overall, the incidence of TEAEs was low and similar following cumulative doses of AS MDI (8/45; 17.8%) and Proventil (10/46; 21.7%) (Table 8). The majority of TEAEs were mild in severity. The most frequently reported treatment-related AEs included ‘feeling jittery’ and tremor and were reported during both AS MDI and Proventil treatment periods, with a greater proportion of patients reporting feeling jittery with Proventil (13.0% vs 6.7%) (Table 8). The overall incidence of TEAEs deemed to be treatment-related was 13.3% for AS MDI and 19.6% for Proventil (Table 8).
3.4.2 ANTORA
AS MDI and Proventil were well tolerated; safety findings were consistent with the known safety profile for albuterol [6, 9, 10]. Overall, the incidence of TEAEs was low across treatment groups, with no dose-related trends (Table 8). Only four TEAEs were reported in three patients all taking Proventil (Table 8). All TEAEs were mild in severity and none were considered treatment-related. No deaths, serious TEAEs or serious treatment-related AEs were reported.
4 Discussion
The results from ASPEN and ANTORA demonstrate that AS MDI is comparable to Proventil in improving lung function, and ANTORA also showed that both treatments are superior to placebo.
ASPEN demonstrated AS MDI was equivalent to Proventil for lung function improvement following cumulative doses of albuterol up to 1440 μg in patients with asthma, as measured by the primary and secondary efficacy endpoints. The bioavailability of albuterol was ~21%–27% lower with AS MDI versus Proventil. Extrapulmonary pharmacodynamic effects of albuterol were smaller in magnitude with AS MDI versus Proventil for changes from baseline in QTcF, heart rate and serum glucose, but were slightly higher with AS MDI for changes from baseline in SBP and DBP.
ANTORA demonstrated that the 90 μg and 180 μg doses of AS MDI and Proventil were significantly superior to placebo MDI for all primary and secondary efficacy endpoints. AS MDI was non-inferior to Proventil at both the 90 μg and 180 μg dose levels for primary and all secondary efficacy endpoints, with the 95% CIs for the observed differences between treatments all within the pre-specified non-inferiority limit of 100 mL. With both treatments, the magnitude of change from baseline for FEV1 AUC0-4 was greater than for the primary endpoint of FEV1 AUC0-6; however, this was expected, as albuterol is more pharmacodynamically active during the first 4 h after inhalation. Similar findings to the primary and secondary efficacy endpoints were observed with the other efficacy endpoints included in this study. In both ASPEN and ANTORA, all study treatments were well tolerated, and safety findings were consistent with the known safety profile of albuterol [6, 9, 10].
In ASPEN, treatment with Proventil demonstrated greater effects on systemic exposure, extrapulmonary pharmacodynamic and safety parameters versus AS MDI; a possible explanation could be that Proventil contains oleic acid and ethanol. Oleic acid has been shown to increase pulmonary absorption of protein, while the inclusion of ethanol may dissolve some of the albuterol in suspension, thus resulting in more rapid absorption [11, 12]. These additives may explain the increased systemic exposure, thus increasing extrapulmonary effects of Proventil versus AS MDI. This could also explain the sharp peak in albuterol plasma concentrations following the final cumulative dose from Proventil, while AS MDI had a more prolonged absorption period. Proventil is currently the only formulation of albuterol to contain these additional compounds/absorption enhancers [13].
The ASPEN results were compared to a study with a similar trial design investigating two albuterol products, ProAir RespiClick® and ProAir® HFA [7]. Evaluation of lung function, measured after each cumulative dose, found that AS MDI was comparable to both ProAir products. At a cumulative dose of 90 µg, LS mean (standard error) FEV1 was 421 (60) mL for AS MDI, 420 (76) mL for ProAir RespiClick® and 480 (76) mL for ProAir® HFA, and at a cumulative dose of 1440 µg, FEV1 was 721 (59) mL, 690 (76) mL and 720 (76) mL, respectively. AS MDI was similarly comparable to both ProAir products following evaluation of the pharmacokinetic results; the geometric LS mean AUC0-t was 21.5 ng·h/mL with AS MDI, 23.2 ng·h/mL for ProAir RespiClick® and 20.9 ng·h/mL for ProAir® HFA. The geometric LS mean Cmax was 3.71 ng/mL, 4.42 ng/mL and 3.30 ng/mL for AS MDI, ProAir RespiClick® and ProAir® HFA, respectively.
AS MDI 180 µg was well tolerated and produced similar bronchodilator effects to Proventil 180 µg in adult and adolescent patients with asthma. The data presented on the efficacy and safety of AS MDI support the selection of AS MDI 180 µg for a Phase III drug trial.
5 Conclusions
The bronchodilator effects of albuterol delivered via AS MDI using novel Co-Suspension Delivery™ Technology and Proventil were equivalent, both following dose-ranging and cumulative doses of albuterol up to 1440 μg. AS MDI was well tolerated, with numerically smaller extrapulmonary effects and lower systemic exposure versus Proventil. The findings from these Phase II efficacy and safety studies support selection of AS MDI 180 µg to be taken into Phase III clinical trials, either alone or in combination with ICS (SABA/ICS; PT027) and may facilitate development of novel inhalers combining rescue and controller therapies to benefit patients with asthma.
References
Global Initiative for Asthma (GINA). Global Strategy for Asthma Management and Prevention. 2021. www.ginasthma.org/reports. Accessed 5 May 2021.
Busse WW, Lemanske RFJ. Asthma. N Engl J Med. 2001;344(5):350–62.
Williams EJ. Experimental designs balanced for the estimation of residual effects of treatments Aust J Sci Res. 1949;2(3):149–68.
Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948–68.
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–38.
Kemp JP, Furukawa CT, Bronsky EA, Grossman J, Lemanske RF, Mansfield LE, et al. Albuterol treatment for children with asthma: a comparison of inhaled powder and aerosol. J Allergy Clin Immunol. 1989;83(3):697–702.
Kerwin EM, Taveras H, Iverson H, Wayne D, Shah T, Lepore MS, et al. Pharmacokinetics, pharmacodynamics, efficacy, and safety of albuterol (salbutamol) multi-dose dry-powder inhaler and ProAir® hydrofluoroalkane for the treatment of persistent asthma: Results of two randomized double-blind studies. Clin Drug Investig. 2016;36(1):55–65.
Santanello NC, Zhang J, Seidenberg B, Reiss TF, Barber BL. What are minimal important changes for asthma measures in a clinical trial? Eur Respir J. 1999;14(1):23–7.
Bronsky E, Bucholtz GA, Busse WW, Chervinsky P, Condemi J, Ghafouri MA, et al. Comparison of inhaled albuterol powder and aerosol in asthma. J Allergy Clin Immunol. 1987;79(5):741–7.
Proair HFA [package insert]. PA: Teva Respiratory, LLC; 2012.
Hussain A, Arnold JJ, Khan MA, Ahsan F. Absorption enhancers in pulmonary protein delivery. J Control Release. 2004;94(1):15–24.
Green PG, Guy RH, Hadgraft J. In vitro and in vivo enhancement of skin permeation with oleic and lauric acids. Int J Pharm. 1988;48:103–11.
Sheth P, Sandell D, Conti DS, Holt JT, Hickey AJ, Saluja B. Influence of formulation factors on the aerosol performance of suspension and solution metered dose inhalers: a systematic approach. AAPS J. 2017;19(5):1396–410.
Acknowledgements
This manuscript was supported by AstraZeneca. The authors would like to thank the health care providers, research staff, patients, and caregivers who participated in these trials, and Samantha Blakemore of inScience Communications, Springer Healthcare Ltd, UK, for providing medical writing support, which was funded by AstraZeneca in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
These two studies were solely funded by AstraZeneca.
Role of the Funding Source
AstraZeneca funded the study and had a role in study design, data collection, data analysis, data interpretation, and writing of the report. The corresponding author had full access to all the data and had final responsibility to submit for publication.
Conflict of Interest
AM, CC, CR, KR and MG are employees of, and own stock in, AstraZeneca. CLF has received payments from 4D Pharma PLC, Amphastar, Boehringer Ingelheim, Bond Avillion 2 Development LP, Bosch Healthcare Solutions GmbH, Cipla Ltd, GlaxoSmithKline, Knopp Biosciences LLC, Novartis Research and Development, Romark, Sanofi US Services Inc, Teva Pharmaceuticals Ltd and Vorso Corp. EK has performed consulting, served on advisory boards, and received travel reimbursement from Amphastar, AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Mylan, Novartis, Sunovion and Theravance. He has conducted multicentre clinical research trials for some 40 pharmaceutical companies.
Data Availability Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Ethics Approval
This study was performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with International Conference for Harmonisation (ICH), Good Clinical Practice (GCP), and applicable regulatory requirements.
Consent to Participate
The Investigator was responsible for obtaining written informed consent (and assent, where applicable) from potential subjects before any study-specific screening and entry into the study. After the subject signed the ICF (and/or assent form, where applicable), the original was retained by the Investigator; the subject retained a copy.
Consent for Publication
Not applicable.
Author Contributions
CC: Conceptualisation; data curation; investigation; methodology; supervision; validation; visualisation; roles/writing—original draft; writing—review and editing. AM: Conceptualisation; investigation; methodology; supervision; validation; visualisation; roles/writing—original draft; writing—review and editing. KR: Conceptualisation; investigation; methodology; project administration; resources; supervision; validation; roles/writing—original draft; writing—review and editing. MG: Conceptualisation; investigation; methodology; supervision; validation; visualisation; writing—review and editing. CLF: Data curation; methodology; writing—review and editing. EK: Data curation; investigation; methodology; writing—review and editing. CR: Conceptualisation; funding acquisition; methodology; supervision; validation; writing—review and editing.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cappelletti, C., Maes, A., Rossman, K. et al. Dose-Ranging and Cumulative Dose Studies of Albuterol Sulfate MDI in Co-Suspension Delivery™ Technology (AS MDI; PT007) in Patients with Asthma: the ASPEN and ANTORA Trials. Clin Drug Investig 41, 579–590 (2021). https://doi.org/10.1007/s40261-021-01040-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-021-01040-7