
Abstract
Background
Naldemedine is a peripherally acting μ-opioid receptor antagonist that is indicated to treat opioid-induced constipation.
Objectives
To assess the potential for drug-drug interactions between a single oral dose of naldemedine and the oral P-glycoprotein inhibitor cyclosporine, cytochrome P450 (CYP) 3A inhibitors itraconazole and fluconazole, and CYP3A inducer rifampin.
Methods
Three Phase 1, open-label studies were conducted in healthy subjects. In the P-glycoprotein inhibitor study, subjects received naldemedine 0.4 mg alone or coadministered with cyclosporine 600 mg. In the CYP3A inhibitors study, subjects in separate cohorts received naldemedine 0.2 mg alone or with itraconazole or fluconazole. In the CYP3A inducer study, subjects received naldemedine 0.2 mg alone or with rifampin 600 mg. Geometric mean ratios and 90 % confidence intervals were used to evaluate the effects of coadministered drugs on naldemedine maximum plasma concentration (Cmax) and the area under the concentration-time curve (AUC). Safety assessments included occurrence of adverse events (AEs), laboratory parameters, vital signs, and electrocardiography results.
Results
A total of 56 subjects were enrolled (n = 14 in each cohort). Cyclosporine increased naldemedine AUC0–inf 1.78-fold and Cmax 1.45-fold. Itraconazole and fluconazole increased naldemedine AUC0–inf 2.91-fold and 1.90-fold, and Cmax 1.12-fold and 1.38-fold, respectively. Rifampin decreased naldemedine AUC0–inf by 83% and Cmax by 38%. Across studies, AEs were generally mild. Laboratory, vital sign, or electrocardiogram assessments produced no clinically significant findings.
Conclusions
Coadministration of naldemedine with a P-glycoprotein inhibitor or a strong/moderate CYP3A inhibitor increases naldemedine exposure; coadministration with a strong CYP3A inducer decreases its exposure. Coadministration of naldemedine with cyclosporine, itraconazole, fluconazole, or rifampin was generally safe and well tolerated.
Plain Language Summary
Naldemedine is a targeted medication approved in the USA, Europe, and Japan for the treatment of opioid-induced constipation. Symptoms of constipation may include passing fewer stools than usual, having lumpy or hard stools, and/or straining to have bowel movements. In some cases, these symptoms are side effects of regular opioid use, which is often medically necessary for the management of moderate-to-severe pain. For naldemedine to be prescribed safely, doctors must know what other medications a patient is taking and how these medications may affect one another. This is commonly known as drug-drug interactions. Some drug-drug interactions may decrease how well a medication works, while other drug-drug interactions may increase the side effects experienced by a patient. In this paper, researchers report the results of three Phase 1 studies in healthy subjects examining how naldemedine interacts with other drugs. The drugs chosen for investigation are commonly evaluated in DDI studies and may affect the transport or metabolic pathway of naldemedine, including the P-glycoprotein inhibitor cyclosporine, the CYP3A inhibitors itraconazole and fluconazole, and the CYP3A inducer rifampin. These studies demonstrate that co-administration of naldemedine with each of these drugs impacted the pharmacokinetics of naldemedine. Cyclosporine, itraconazole, or fluconazole all increased naldemedine exposure, while rifampin decreased naldemedine exposure. For all drug combinations, observed side effects were generally mild and well tolerated. Additional testing, including vital signs and heart monitoring, did not reveal any other safety concerns. In conclusion, these findings support the cautious use of naldemedine in combination with cyclosporine, itraconazole or fluconazole. Concomitant use with rifampin should be avoided.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Coadministration of naldemedine with a P-gp inhibitor (cyclosporine) or CYP3A inhibitors (itraconazole or fluconazole) increased naldemedine exposure |
Coadministration of naldemedine with a CYP3A inducer (rifampin) decreased naldemedine exposure |
Single doses of naldemedine were generally safe and well tolerated in healthy subjects when naldemedine was administered alone or in combination with cyclosporine, itraconazole, fluconazole, or rifampin |
1 Introduction
Opioid-induced constipation (OIC) is a side effect that occurs in 40% to > 90% of patients taking opioid analgesics for the management of moderate-to-severe chronic pain [1,2,3]. The features of OIC include reduced bowel movement frequency, increased straining to pass bowel movements, sensations of incomplete rectal evacuation, or harder stool consistency [4]. The symptoms of OIC can be debilitating, often having profoundly negative effects on patients’ quality of life and causing many patients to discontinue opioid analgesics or reduce their dose of opioid medication [5, 6].
Unlike other side effects, such as nausea, vomiting, and somnolence, that diminish over time, OIC is persistent, and can increase in severity and prevalence with duration of treatment [1, 5, 7, 8]. Although many patients attempt to manage their OIC using lifestyle modifications and over-the-counter laxatives and stool softeners, they may find that these therapies have limited efficacy. Conventional treatments do not target the underlying pathophysiology of enteric µ-opioid receptor activation, which can increase fluid absorption, delay gastric emptying, and prolong intestinal transit time [7,8,9,10].
Naldemedine (Symproic®: Shionogi & Co., Ltd., Osaka, Japan, and BioDelivery Sciences International, Inc. Raleigh, NC; Rizmoic®: Shionogi & Co., Ltd. Osaka, Japan) is a peripherally acting μ-opioid receptor antagonist (PAMORA) approved for the treatment of OIC in adults with chronic noncancer pain (USA and Japan), in adults with cancer pain (Japan), and in adults who have previously been treated with a laxative (EU) [11,12,13,14]. Naldemedine is an amide derivative of the opioid receptor antagonist naltrexone that has increased molecular weight and polar surface area and acts as a substrate of the P-glycoprotein (P-gp) efflux transporter [15, 16]. These molecular features of naldemedine limit its potential to cross the blood–brain barrier and penetrate the central nervous system to disrupt opioid analgesia [17, 18].
Naldemedine is primarily metabolized by cytochrome P450 3A4 (CYP3A4) to form nor-naldemedine [15, 17]. In a Phase 1 study in 12 healthy male subjects who received single oral 2-mg doses of [oxadiazole-14C]-naldemedine or [carbonyl-14C]-naldemedine, nor-naldemedine accounted for 9–13% of the systemic exposure of unchanged naldemedine [16].
Given the clinical importance of the P-gp transporter and CYP3A in drug-drug interactions [19] and the knowledge that naldemedine is a substrate of P-gp and is metabolized by CYP3A [15, 16], clinicians should understand the potential for drug-drug interactions between naldemedine and P-gp inhibitors, CYP3A inhibitors, and CYP3A inducers. The current study evaluated the effects of coadministration of the P-gp inhibitor cyclosporine, the CYP3A inhibitors itraconazole and fluconazole, and the CYP3A inducer rifampin on the pharmacokinetics of a single oral dose of naldemedine as a substrate in healthy subjects.
2 Methods
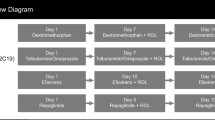
Three Phase 1, open-label, drug-drug interaction studies were conducted between February 2012 and June 2015 to determine the effects of a P-gp inhibitor, CYP3A inhibitors, and a CYP3A inducer on the pharmacokinetics of naldemedine in healthy adult subjects. The protocols and amendments for all three studies were reviewed and approved by appropriate institutional review boards in accordance with US Food and Drug Administration guidelines, International Conference on Harmonization (ICH) Good Clinical Practice (GCP) guidelines, and local requirements, as applicable. All three studies were conducted in accordance with the Declaration of Helsinki and GCP according to ICH guidelines. Prior to enrollment, all subjects were informed of and provided written consent.
2.1 Participants
The cyclosporine P-gp inhibitor study included healthy male subjects aged 18–55 years with a body mass index (BMI) of ≥ 22.0 and < 30 kg/m2 and bodyweight ≥ 50 kg. The itraconazole and fluconazole CYP3A inhibitors study included Japanese healthy male and female subjects aged 20–55 years with a BMI of ≥ 18.0 and < 25 kg/m2. The rifampin CYP3A inducer study included healthy male and female subjects aged 18–55 years with a BMI of ≥ 18.0 and ≤ 30 kg/m2.
All three studies included normotensive subjects with no medical histories of significant metabolic, hepatic, renal, hematological, pulmonary, cardiovascular, gastrointestinal, urological, neurological, or psychiatric disorders; gastrointestinal surgeries; or clinically significant abnormal laboratory test results or electrocardiogram (ECG) measurements. Subjects were excluded for medical histories of anaphylaxis or significant food or drug allergies; alcoholism or substance abuse within 6 months (for CYP3A inhibitor or inducer studies) or 1 year (P-gp inhibitor study) prior to screening; testing positive for human immunodeficiency virus, hepatitis B surface antigen, or hepatitis C virus; or positive screening results for drugs of abuse or cotinine use. Subjects were excluded if they used tobacco or nicotine-containing products within 6 months prior to admission; medications that were potential P-gp and/or CYP3A inducers within 28 days prior to admission or potential P-gp and/or CYP3A inhibitors within 28 days (P-gp inhibitor study) or 14 days (CYP3A inhibitors and inducer studies); any prescription or non-prescription drugs or dietary supplements within 14 days prior to admission; use of alcohol- or caffeine-containing products or acetaminophen within 72 h prior to admission; or grapefruit-containing products within 72 h (P-gp inhibitor study) or 7 days (CYP3A inhibitors and inducer studies) prior to admission.
2.2 Study Designs
2.2.1 P-gp Inhibitor Study
This was a single-center, open-label, 2-way crossover study in the USA to evaluate the effects of cyclosporine on the pharmacokinetics of naldemedine in fasting, healthy, male subjects. The study consisted of a 28-day screening, after which subjects were assigned to 1 of 2 treatment sequences. In Sequence 1, subjects received 0.4 mg of oral naldemedine alone on Day 1, and 0.4 mg of oral naldemedine coadministered with 600 mg of cyclosporine (Cyclosporine Oral Solution US Pharmacopeia Modified, TEVA Pharmaceuticals USA, Inc., North Wales, PA) on Day 15. In Sequence 2, subjects received 0.4 mg of naldemedine coadministered with 600 mg of cyclosporine on Day 1, and 0.4 mg of naldemedine alone on Day 15. Follow-up safety assessments were performed on Day 16 to Day 18, and an end-of-study visit (Day 28 ± 2 days) occurred 10 ± 2 days after the date of discharge. Pharmacokinetic blood sampling for naldemedine was performed pre-dose and at 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, 8.0, 10.0, 12.0, 24.0, 36.0, 48.0, 60.0, and 72.0 hours post-dose on Day 1 and Day 15. Plasma samples were stored at ≤ − 70 °C.
2.2.2 CYP3A Inhibitors Study
This was a single-center, open-label, 1-sequence, 2-period crossover study in Japan to evaluate the effects of itraconazole (strong CYP3A inhibitor; Cohort 1) and fluconazole (moderate CYP3A inhibitor; Cohort 2) on the pharmacokinetics of naldemedine in fasting, healthy subjects. The study consisted of a 28-day screening followed by 2 treatment periods (Day 1 to Day 4 and Day 5 to Day 12, respectively).
In Cohort 1, subjects received a single oral dose of naldemedine 0.2 mg on Day 1. On Day 5, subjects received oral itraconazole (Itrizole®, Janssen Pharmaceutical K.K., Tokyo, Japan) 200 mg twice daily dose (BID). On Day 6 through Day 8, subjects received itraconazole 200 mg once daily dose (QD). On Day 9, subjects received naldemedine 0.2 mg coadministered with itraconazole 200 mg. On Day 10 and Day 11, subjects received itraconazole 200 mg QD.
In Cohort 2, subjects received a single oral dose of naldemedine 0.2 mg on Day 1. On Day 5, subjects received oral fluconazole (Diflucan®, Pfizer Japan Inc., Tokyo, Japan) 400 mg QD. On Day 6 through Day 8, subjects received fluconazole 200 mg QD. On Day 9, subjects received naldemedine 0.2 mg coadministered with fluconazole 200 mg. On Day 10 and Day 11, subjects received fluconazole 200 mg QD.
In both cohorts, safety assessments were performed throughout the study until an end-of-study visit on Day 26 ±2. Pharmacokinetic whole-blood sampling (3 mL in sodium heparin tubes) for naldemedine was performed pre-dose and at 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, 8.0, 10.0, 12.0, 24.0, 36.0, 48.0, 60.0, and 72.0 h post-dose on Day 1 and Day 9. Plasma samples were stored at ≤ − 70 °C.
2.2.3 CYP3A Inducer Study
This was a single-center, open-label, 1-sequence, crossover study in the USA to evaluate the effects of repeated oral rifampin administration on the pharmacokinetic of naldemedine in healthy subjects. Following a 28-day screening, all subjects received a single oral dose of naldemedine 0.2 mg on Day 1 and Day 18 and once-daily doses of rifampin 600 mg (RIFADIN®, Lannett Company Inc., Philadelphia, PA) consecutively on Day 4 through Day 20. On Day 18, rifampin was co-administered with naldemedine. All doses were administered to subjects in the morning in the fasted state. On Day 1 and Day 18, subjects remained fasted through 4 hours post-dose. Safety assessments were performed throughout the study until the end-of-study visit on Day 35 ± 2. Whole-blood samples (4 mL in sodium heparin tubes) for the determination of naldemedine plasma concentrations were obtained pre-dose and at 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, 8.0, 10.0, 12.0 (Days 1 and 18), 24.0, 36.0 (Days 2 and 19), 48.0, 60.0 (Days 3 and 20), and 72.0 hours (Days 4 and 21) post-Day 1 and Day 18 doses. Plasma samples were stored at ≤ − 70 °C.
2.3 Bioanalytical Assessments
Plasma concentration levels of naldemedine were determined by liquid chromatography coupled with tandem mass spectrometry methods using a SCIEX API 5000 mass spectrometer (AB SCIEX, Framingham, Massachusetts) systems by Syneos Health, Inc. (Morrisville, NC, USA) for the drug-drug interactions studies with cyclosporine and rifampin. Stable isotope‐labeled internal standard solution and 500 mmol/L ammonium formate (pH level: 8.5) were added to plasma samples (200 μL) and applied to a solid‐phase extraction cartridge (Bond Elute Plexa, 30 mg, Varian, St-Laurent, Canada). Naldemedine was extracted using methanol, evaporated under nitrogen stream, and dissolved with 2 mmol/L ammonium formate/methanol/formic acid (85:15:2). Aliquots of plasma (30 μL) extracts were injected into the high‐performance liquid chromatography system and separated by a reversed-phase column (Atlantis d C18 column, 4.6 mm × 50 mm; 3 μm) and reversed phase using a gradient elution of mobile phase A (3.3 mmol/L ammonium formate/methanol/2-propanol/formic acid [60:35:5:2]) and mobile phase B (40 mmol/L ammonium formate/methanol/2-propanol/formic acid [5:85:10:2]) at a flow rate of 1.0 mL/min. Mass spectrometry was performed in the positive electrospray ionization mode, using multiple reaction monitoring with m/z transitions of 571 → 368 for naldemedine and 577 → 368 for naldemedine‐15Nd5. The analytical method was validated at the concentration range from 0.01 to 10 ng/mL and dilution reproducibility was confirmed up to 20-fold. Precision and accuracy of the method were 2.4–7.1 % and 98.6–105.5%, respectively. Plasma naldemedine concentration was determined using a previously published method by Shin Nippon Biomedical Laboratories, Ltd. (Tokyo, Japan) for the drug-drug interaction studies with itraconazole and fluconazole [15]. The analytical method was validated at the concentration range from 0.01 to 10 ng/mL and dilution reproducibility was confirmed up to 1000-fold. Precision and accuracy of the method were 4.2–9.7% and 94.7–104.8%, respectively. Plasma samples, with concentrations expected to be higher than the upper limit of quantification, were diluted with blank plasma within the confirmed dilution reproducibility.
2.4 Safety Assessments
Safety assessments performed in all three studies included a medical review of adverse events (AEs), treatment-emergent AEs (TEAEs), serious AEs (SAEs), AEs leading to drug withdrawal, clinical laboratory test results, vital sign measurements, 12-lead ECG measurements, and physical examination findings.
2.5 Pharmacokinetic Assessments and Statistical Analyses
In all three studies, the pharmacokinetic parameters of naldemedine were based on measured naldemedine concentrations and calculated using Phoenix® WinNonlin® (Certara, St Louis, MO, USA) software. Calculated parameters included maximum observed plasma concentration (Cmax), time to Cmax (Tmax), area under the concentration-time curve from 0 to the last measurable concentration (AUC0–last) and from 0 to infinity (AUC0–inf), apparent terminal elimination half-life (t1/2,z), apparent elimination rate constant (λz), and apparent total clearance (CL/F). AUC was calculated by log-linear trapezoidal approach. Statistical analyses were conducted using SAS® (SAS institute, Cary, NC, USA) software.
An analysis of variance model was used to compare the pharmacokinetic properties of naldemedine when administered alone versus when administered in combination with cyclosporine, itraconazole or fluconazole, or rifampin in each of the three studies, respectively. This model considered treatment as a fixed effect and subject as a random effect for the parameters of the logarithm of Cmax, AUC, t1/2,z, and CL/F. The ratio of geometric least squares (LS) means and the corresponding 90% confidence interval (CI) were estimated by exponentiating the mean differences in the logarithm. In all studies, 90% CIs for the ratio of geometric LS means were used to evaluate the effects of coadministered drugs on naldemedine Cmax and AUC.
Missing data were not replaced; all analyses were based on observed cases. Naldemedine plasma concentrations that were below the lower limit of quantification were treated as zero in the mean concentration-time profiles and as missing for the calculation of the geometric mean value and geometric mean coefficient of variation.
3 Results
3.1 Subjects
In the P-gp inhibitor study, 14 subjects were enrolled, and 12 subjects completed the study. One subject withdrew from the study for personal reasons prior to period 2 (naldemedine only), and 1 subject was excluded for no longer meeting inclusion criteria (i.e. had diastolic blood pressure > 90 mmHg) prior to period 2 (naldemedine coadministered with cyclosporine). In the CYP3A inhibitors study, 28 subjects (14 in each cohort) were enrolled, and all 28 subjects completed the study. In the CYP3A inducer study, 14 subjects were enrolled, and all 14 subjects completed the study. Demographic and clinical characteristics of the subjects included in each study are shown in Table 1.
3.2 Pharmacokinetics and Safety
Linear and semi-logarithmic plots show mean plasma concentrations of naldemedine over time when administered as a single oral dose with and without coadministration of cyclosporine (Fig. 1), itraconazole (Fig. 2), fluconazole (Fig. 3), and rifampin (Fig. 4). Table 2 shows the pharmacokinetic parameters of naldemedine alone and when coadministered with each other drug. Statistical comparisons of pharmacokinetic parameters are provided in Table 3. A forest plot illustrating the Cmax and AUC ratios of naldemedine after administration of a single oral dose with and without coadministration of cyclosporine, itraconazole, fluconazole, and rifampin is shown in Fig. 5.

Linear and semi-logarithmic plots of mean (± SD) plasma concentration-time profiles of naldemedine after administration of a single oral dose with and without coadministration of cyclosporine

Linear and semi-logarithmic plots of mean (± SD) plasma concentration-time profiles of naldemedine after administration of a single oral dose with and without coadministration of itraconazole

Linear and semi-logarithmic plots of mean (± SD) plasma concentration-time profiles of naldemedine after administration of a single oral dose with and without coadministration of fluconazole

Linear and semi-logarithmic plots of mean (± SD) plasma concentration-time profiles of naldemedine after administration of a single oral dose with and without coadministration of rifampin

Forest plot of Cmax and AUC ratios of naldemedine after administration of a single oral dose with and without coadministration of cyclosporine, itraconazole, fluconazole, and rifampin. AUC0–inf area under the concentration-time curve from 0 to infinity, CI confidence interval, Cmax maximum observed plasma concentration, LS least squares
3.2.1 Effect of P-gp Inhibitor Coadministration on Naldemedine Pharmacokinetics and Safety
The coadministration of cyclosporine increased naldemedine Cmax by 1.45-fold, AUC0–last by 1.79-fold, and AUC0–inf by 1.78-fold, compared with administration of naldemedine alone (Table 3). Median naldemedine Tmax was not notably affected by coadministration of cyclosporine (Table 2). The semi-logarithmic naldemedine plasma concentration-time profiles after Cmax was reached have similar slopes for both treatments (Fig. 1), suggesting that the observed changes in Cmax and AUC are mainly due to increased oral bioavailability of naldemedine when coadministered with cyclosporine.
In this study, 7 (50.0 %) of 14 subjects experienced 20 TEAEs; 19 of these TEAEs were considered drug related. Most observed TEAEs occurred in subjects who received naldemedine 0.4 mg plus cyclosporine 600 mg (7 [53.8%] in 13) compared with naldemedine alone 1 (7.7%) of 13. The only TEAE reported in subjects who received naldemedine alone was headache (1 [7.7%] of 13). TEAEs reported in subjects who received naldemedine plus cyclosporine included diarrhea (6 [46.2%] of 13), abdominal pain (3 [23.1%] of 13), nausea (3 [23.1%] of 13), flushing (3 [23.1%] of 13), frequent bowel movements (1 [7.7%] of 13), chills (1 [7.7%] of 13), hunger (1 [7.7%] of 13), and dizziness (1 [7.7%] of 13). All TEAEs except hunger were considered drug related.
No SAEs or AEs led to withdrawal. In addition, there were no clinically significant findings from clinical laboratory, vital sign, ECG, or physical examination measurements.
3.2.2 Effect of CYP3A Inhibitor Coadministration on Naldemedine Pharmacokinetics and Safety
The coadministration of a single oral dose of 0.2 mg naldemedine on Day 9 after administration of 200-mg doses of itraconazole BID on Day 5 and QD on Day 6 to Day 11 resulted in higher-plasma naldemedine concentrations and a slower rate of elimination compared with a single oral dose of 0.2 mg naldemedine administered alone on Day 1 (Fig. 2). The coadministration of itraconazole increased naldemedine Cmax by 1.12-fold, AUC0–last by 2.65-fold, and AUC0–inf by 2.91-fold compared with values after administration of naldemedine alone (Table 3). Naldemedine median Tmax was not notably affected by coadministration of itraconazole (Table 2).
The coadministration of a single oral dose of 0.2 mg naldemedine on Day 9 after administration of fluconazole 400 mg QD on Day 5 and 200 mg QD on Day 6 to Day 11 resulted in higher plasma naldemedine concentrations and a slightly slower rate of elimination compared with a single oral dose of 0.2 mg naldemedine administered alone on Day 1 (Fig. 3). The coadministration of fluconazole increased naldemedine Cmax by1.38-fold, AUC0–last by 1.88-fold, and AUC0–inf by 1.90-fold compared with values after administration of naldemedine alone (Table 3). Naldemedine median Tmax was not notably affected by coadministration of fluconazole (Table 2).
In Cohort 1 (coadministration of naldemedine with itraconazole), 5 (35.7%) of 14 subjects experienced at least 1 TEAE. TEAEs were reported in 1 (7.1%) subject after administration of naldemedine alone, 2 (14.3%) subjects after administration of itraconazole alone, 1 (7.1%) subject after coadministration of naldemedine and itraconazole, and 5 (35.7%) subjects during the follow-up period (after Day 12). The most frequent TEAEs were blood urine present in 4 (28.6%) subjects and upper respiratory tract inflammation in 2 (14.3%) subjects. Each of the other TEAEs (diarrhea, blood creatine phosphokinase increased, and urine ketone body present) was reported in 1 (7.1%) subject. Only 1 TEAE (diarrhea) was considered related to naldemedine.
In Cohort 2 (coadministration of naldemedine with fluconazole), 2 (14.3%) of 14 subjects experienced at least 1 TEAE. TEAEs were reported in 1 (7.1%) subject after administration of naldemedine alone (abdominal pain lower, diarrhea, and blood urine present) and in 1 (7.1 %) subject during the follow-up period after Day 12 (C-reactive protein increased). No TEAE was considered drug related.
In both cohorts, no SAEs or AEs led to withdrawal. There were no abnormal findings from clinical laboratory tests, and mean values from vital sign measurements and ECGs remained within normal limits at all time points, with minimal variations from baseline.
3.2.3 Effect of CYP3A Inducer Coadministration on Naldemedine Pharmacokinetics and Safety
The coadministration of a single oral 0.2-mg dose of naldemedine on Day 18 after repeated administration of 600-mg doses of rifampin QD on Day 4–Day 20 resulted in lower plasma naldemedine concentrations and a more rapid rate of elimination compared with a single oral dose of 0.2 mg naldemedine administered alone on Day 1 (Fig. 4). Coadministration of rifampin with naldemedine decreased naldemedine Cmax by 38 %, AUC0–last by 83 %, and AUC0–inf by 83% compared with values after administration of naldemedine alone (Table 3). Naldemedine median Tmax was not notably affected by coadministration of rifampin (0.51 h compared with 1.00 h; Table 2).
In this study, there were no TEAEs reported by any of the 14 subjects following administration of naldemedine alone. TEAEs were reported by all (100%) 14 subjects following administration of rifampin alone, and in 1 (7.1%) of 14 subjects when naldemedine was coadministered with rifampin. The most frequently reported TEAE was mild chromaturia, considered to be related to rifampin (and expected), which was reported by all (100%) 14 subjects following administration of rifampin alone. No TEAEs were considered related to naldemedine, either when naldemedine was administered alone or coadministered with rifampin. No SAEs, AEs led to withdrawal, or abnormal findings on clinical laboratory tests, vital sign measurements, or ECG measurements.
4 Discussion
Overall, results from the three Phase 1 studies reported herein identified pharmacokinetic drug-drug interactions between naldemedine and the P-gp inhibitor cyclosporine, the strong CYP3A inhibitor and P-gp inhibitor itraconazole, the moderate CYP3A inhibitor fluconazole, and the strong CYP3A inducer rifampin. Specifically, coadministration of cyclosporine increased naldemedine Cmax by 1.45-fold and AUC0-inf by 1.78-fold. Coadministration of itraconazole increased naldemedine Cmax by 1.12-fold and AUC0–inf by 2.91-fold. Coadministration of fluconazole increased naldemedine Cmax by 1.38-fold and AUC by 1.90-fold. Coadministration of rifampin decreased naldemedine Cmax by 38% and AUC by 83%.
Although the studied P-gp inhibitor, CYP3A inhibitors, and CYP3A inducer all affected the pharmacokinetic properties of naldemedine, coadministration of naldemedine with cyclosporine, itraconazole, fluconazole, or rifampin was generally well tolerated. The safety profile of naldemedine observed across all three drug-drug interaction studies reported here is consistent with that observed in a Phase 1 single- and multiple-ascending dose study [15] and in a Phase 1 absorption, distribution, metabolism, and excretion study [16] of a single dose of naldemedine in healthy subjects. In these studies, naldemedine doses of 0.1 to 100 mg were safe and well tolerated [15], and all TEAEs were mild and resolved without intervention [16].
In the cyclosporine drug-drug interaction study, coadministration of naldemedine 0.4 mg with cyclosporine 600 mg resulted in more observed TEAEs than with administration of naldemedine alone. Most TEAEs that occurred when naldemedine was coadministered with cyclosporine were mild; all of these TEAEs resolved without intervention and are consistent with the known safety profile of cyclosporine [20, 21]. Although the gastrointestinal TEAEs could have been caused by the pharmacologic actions of naldemedine, these types of TEAEs (diarrhea and abdominal pain) were not observed when naldemedine 0.4 mg was administered alone in this study. It is notable that naldemedine is a P-gp substrate; however, preclinical studies demonstrate that brain distribution of naldemedine is low [18].
In the CYP3A inhibitors study, the incidence of TEAEs was low and similar in all treatment groups, none of the observed TEAEs were considered treatment related except one TEAE of diarrhea in the naldemedine plus itraconazole group and all TEAEs were mild in severity and resolved without intervention. In the CYP3A inducer study, no TEAEs were observed when naldemedine was administered alone. One TEAE occurred when naldemedine was coadministered with rifampin (fatigue), which was not considered to be related to coadministration of naldemedine and rifampin.
Based on standard DDI study methodology, naldemedine was administered as a single dose because it was being evaluated as a substrate in these studies (either of CYP3A or the P-gp efflux transporter). The naldemedine 0.4 mg dose was selected in the P-gp inhibitor study because 0.4 mg was one of the candidate doses for the naldemedine therapeutic dose. The naldemedine 0.2 mg dose was selected in both the CYP3A inhibitors study and the CYP3A inducer study because 0.2 mg is the recommended therapeutic dose for naldemedine [11, 12]. Naldemedine exhibits linear pharmacokinetics in the dose range of 0.2–0.4 mg [15]. As naldemedine was being evaluated as a substrate, we reasoned that DDI would be adequately examined with the selected doses (0.2 mg and 0.4 mg of naldemedine).
The naldemedine 0.2- and 0.4-mg doses in these drug-drug interaction studies are relevant from the perspective of clinical efficacy. Results from a Phase 2b randomized controlled study showed that naldemedine 0.2- and 0.4-mg doses were associated with significant clinical benefits, including increased spontaneous bowel movements compared with placebo, whereas no significant differences were observed between naldemedine 0.1 mg and placebo [22]. The once-daily 0.2-mg dose is recommended for the treatment of OIC in adults with chronic pain [11, 12].
Exposure-response analyses of naldemedine in Phase 2b (naldemedine 0.1, 0.2, and 0.4 mg) and Phase 3 studies (naldemedine 0.2 mg) suggest the probability of TEAE would increase as AUC increases [22, 23]. The results of these drug-drug interaction studies and exposure-response analyses are consistent with guidance provided in the naldemedine prescribing information [11, 12]. Specifically, clinicians should monitor patients treated with or using P-gp inhibitors (e.g. cyclosporine), strong CYP3A inhibitors (e.g. itraconazole, grapefruit juice, ketoconazole, ritonavir, indinavir, saquinavir, telithromycin, and clarithromycin), or moderate CYP3A inhibitors (e.g. fluconazole) for AEs because these drugs increase naldemedine concentrations. Concomitant use of naldemedine with strong CYP3A inducers (e.g. rifampin, St. John’s wort, carbamazepine, phenobarbital, and phenytoin) should be avoided because these drugs decrease naldemedine concentrations [11, 12].
A limitation of these Phase 1 studies was the small sample size (14 subjects in each drug-drug interaction evaluation). In addition, in the cyclosporine drug-drug interaction study, the dose of naldemedine studied (0.4 mg) was twice the recommended dose of naldemedine for the treatment of OIC [11]. Using a higher naldemedine dose than that prescribed in clinical practice may have affected the magnitude of the observed pharmacokinetic effects of coadministration with cyclosporine.
4.1 Conclusions
Results of these studies showed that coadministration of a P-gp inhibitor, CYP3A inhibitors, and a CYP3A inducer had notable effects on the pharmacokinetics of naldemedine. The P-gp inhibitor cyclosporine increased naldemedine AUC0–inf by 1.78-fold. The strong CYP3A inhibitor (and P-gp inhibitor) itraconazole increased naldemedine AUC0–inf by 2.91-fold, and the moderate CYP3A inhibitor fluconazole increased naldemedine AUC0–inf by 1.90-fold. The strong CYP3A inducer rifampin decreased naldemedine AUC by 83%. However, coadministration of these drugs with naldemedine was not associated with any new safety signals. Single doses of naldemedine were generally safe and well tolerated in healthy subjects when naldemedine was administered alone or in combination with cyclosporine, itraconazole, fluconazole, or rifampin.
References
Tuteja AK, Biskupiak J, Stoddard GJ, Lipman AG. Opioid-induced bowel disorders and narcotic bowel syndrome in patients with chronic non-cancer pain. Neurogastroenterol Motil. 2010;22(424–30):e96.
Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg. 2001;182:11S–8S.
Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med. 2014;370:2387–96.
Camilleri M, Drossman DA, Becker G, Webster LR, Davies AN, Mawe GM. Emerging treatments in neurogastroenterology: a multidisciplinary working group consensus statement on opioid-induced constipation. Neurogastroenterol Motil. 2014;26:1386–95.
Holzer P. Methylnaltrexone for the management of unwanted peripheral opioid effects. Therapy. 2008;5:531–43.
Bell T, Annunziata K, Leslie JB. Opioid-induced constipation negatively impacts pain management, productivity, and health-related quality of life: findings from the National Health and Wellness Survey. J Opioid Manag. 2009;5:137–44.
Coyne KS, Margolis MK, Yeomans K, King FR, Chavoshi S, Payne KA, et al. Opioid-induced constipation among patients with chronic noncancer pain in the United States, Canada, Germany, and the United Kingdom: laxative use, response, and symptom burden over time. Pain Med. 2015;16:1551–65.
Bell TJ, Panchal SJ, Miaskowski C, Bolge SC, Milanova T, Williamson R. The prevalence, severity, and impact of opioid-induced bowel dysfunction: results of a US and European Patient Survey (PROBE 1). Pain Med. 2009;10:35–42.
Brenner DM, Chey WD. An evidence-based review of novel and emerging therapies for constipation in patients taking opioid analgesics. Am J Gastroenterol Suppl. 2014;2:38–46.
Raffa RB, Taylor R Jr, Pergolizzi JV Jr. Treating opioid-induced constipation in patients taking other medications: avoiding CYP450 drug interactions. J Clin Pharm Ther. 2019;44:361–71.
Symproic (naldemedine) [package insert]. Raleigh, NC: BioDelivery Sciences International, Inc.; 2019.
Rizmoic 200 micrograms film-coated tablets [summary of product characteristics]. 2019. Amsterdam, The Netherlands: Shionogi B.V.
Report on the Deliberation Results: Symproic tablets 0.2 mg. 2017. https://www.pmda.go.jp/files/000222647.pdf. Accessed 18 Dec 2019.
Food and Drug Administration. Symproic (naldemedine) 0.2 mg oral tablets, NDA approval 208854. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/208854Orig1s000ltr.pdf. Accessed 18 Dec 2019.
Fukumura K, Yokota T, Baba Y, Arjona Ferreira JC. Phase 1, randomized, double-blind, placebo-controlled studies on the safety, tolerability, and pharmacokinetics of naldemedine in healthy volunteers. Clin Pharmacol Drug Dev. 2018;7:474–83.
Ohnishi S, Fukumura K, Kubota R, Wajima T. Absorption, distribution, metabolism, and excretion of radiolabeled naldemedine in healthy subjects. Xenobiotica. 2019;49:1044–53.
Stern EK, Brenner DM. Spotlight on naldemedine in the treatment of opioid-induced constipation in adult patients with chronic noncancer pain: design, development, and place in therapy. J Pain Res. 2018;11:195–9.
Watari R, Matsuda A, Ohnishi S, Hasegawa H. Minimal contribution of P-gp on the low brain distribution of naldemedine, a peripherally acting μ-opioid receptor antagonist. Drug Metab Pharmacokinet. 2019;34:126–33.
Sandson N. Drug-drug interactions: the silent epidemic. Psychiatr Serv. 2005;56:22–4.
Sandimmune (cyclosporine) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. 2015.
Neoral (cyclosporine) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. 2015.
Webster LR, Yamada T, Arjona Ferreira JC. A Phase 2b, randomized, double-blind placebo-controlled study to evaluate the efficacy and safety of naldemedine for the treatment of opioid-induced constipation in patients with chronic noncancer pain. Pain Med. 2017;18:2350–60.
Kubota R, Fukumura K, Wajima T. Population pharmacokinetics and exposure-response relationships of naldemedine. Pharm Res. 2018;35:225.
Acknowledgements
We would like to thank all participants of the study. Editorial assistance was provided by Eli Berdougo of Oxford PharmaGenesis Inc., Newtown, PA, and was funded by Shionogi & Co., Ltd.
Author information
Authors and Affiliations
Contributions
KF, NK, TI, RK, YT, and EO contributed to the study conception and design. Material preparation, data collection, and analysis were performed by all authors. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Funding
This work was supported by Shionogi & Co., Ltd., Osaka, Japan.
Conflicts of Interest
Kazuya Fukumura, Nao Kawaguchi, Toru Ishibashi, Ryuji Kubota, Yukio Tada, and Eriko Ogura are employees of Shionogi & Co, Ltd.
Data Availability
The data used in this manuscript are confidential and not accessible to the public.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (IntegReview [P-gp inhibitor study], IRB#00003657; Chesapeake IRB [CYP3A inducer study], IRB#00000790; Medical Corporation Heishinkai OPHAC [CYP3A inhibitor study], Hospital IRB) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual study participants included in the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fukumura, K., Kawaguchi, N., Ishibashi, T. et al. Clinical Drug-Drug Interaction Studies to Evaluate the Effects of a P-Glycoprotein Inhibitor, CYP3A Inhibitors, and a CYP3A Inducer on the Pharmacokinetics of Naldemedine in Healthy Subjects. Clin Drug Investig 40, 529–540 (2020). https://doi.org/10.1007/s40261-020-00902-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-020-00902-w