
Abstract
Purpose
Rolapitant is a neurokinin-1 receptor antagonist indicated in combination with other antiemetic agents in adults for the prevention of delayed chemotherapy-induced nausea and vomiting. We evaluated the effects of rolapitant oral on the pharmacokinetics of probe substrates for cytochrome P450 (CYP) 2D6 (dextromethorphan), 2C9 (tolbutamide), 2C19 (omeprazole), 2B6 (efavirenz), and 2C8 (repaglinide) in healthy subjects.
Methods
This open-label, multipart, randomized, phase 1 study assessed cohorts of 20–26 healthy subjects administered dextromethorphan, tolbutamide plus omeprazole, efavirenz, or repaglinide with and without single, oral doses of rolapitant. Maximum plasma analyte concentrations (Cmax) and area under the plasma analyte concentration–time curves (AUC) were estimated using noncompartmental analysis, and geometric mean ratios (GMRs) and 90% confidence intervals for the ratios of test (rolapitant plus probe substrate) to reference (probe substrate alone) treatment were calculated.
Results
Rolapitant significantly increased the systemic exposure of dextromethorphan in terms of Cmax and AUC0–inf by 2.2- to 3.3-fold as observed in GMRs on days 7 and 14. Rolapitant did not affect systemic exposure of tolbutamide, and minor excursions outside of the 80–125% no effect limits were detected for omeprazole, efavirenz, and repaglinide.
Conclusions
Inhibition of dextromethorphan by a single oral dose of rolapitant 180 mg is clinically significant and can last at least 7 days. No clinically significant interaction was observed between rolapitant and substrates of CYP2C9, CYP2C19, CYP2B6, or CYP2C8. CYP2D6 substrate drugs with a narrow therapeutic index may require monitoring for adverse reactions if given concomitantly with rolapitant.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chemotherapy-induced nausea and vomiting (CINV) are the major adverse effects of cancer chemotherapy associated with substantial reductions in health-related quality of life (QoL) [1,2,3]. In the absence of antiemetic prophylaxis, > 90% of patients receiving highly emetogenic chemotherapy and 30–90% of patients receiving moderately emetogenic chemotherapy experience emesis. Some combination therapies using receptor antagonists (RAs), such as 5-hydroxytryptamine-3 receptor antagonists (5-HT3-RAs), have been found effective against CINV in patients, but the effect was greatest in the acute phase (≤ 24 h post chemotherapy) [4]. Delayed-phase (> 24–120 h post chemotherapy) CINV has been found to have a greater impact on QoL than CINV experienced only in the acute phase [1, 3]. Rolapitant (VARUBI®) is a potent (Ki 0.66 nmol/L), selective, and competitive substance P/neurokinin-1 (NK-1) receptor antagonist approved for use in the USA as an adjunct to other antiemetic agents to prevent delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy in adults [5, 6]. Approval of the oral formulation by the US Food and Drug Administration (FDA) in 2015 was based on data from three global, randomized, double-blind phase 3 trials, in which rolapitant was well tolerated and provided, in combination with a 5-HT3-RA and dexamethasone, superior protection over a 5-HT3-RA and dexamethasone alone against CINV during the delayed phase [7, 8].
Oral rolapitant is formulated as a tablet and is administered approximately 1–2 h prior to the start of chemotherapy [6]. Following administration, rolapitant is highly bioavailable, attains rapid and extensive brain penetration, and is slowly cleared (plasma half-life [t1/2], 169–183 h) [5, 8, 9]. Findings from a positron emission tomography study demonstrated that a single oral 180-mg rolapitant dose provided at least 90% NK-1 receptor occupancy in the cortex for up to 5 days [10]. These attributes allow for administration of a single dose of rolapitant during 5 days at-risk period of each cycle of chemotherapy, which is highly suitable for an antiemetic drug used along with complex anticancer treatment regimens. Rolapitant undergoes extensive oxidative metabolism to yield a single major active metabolite SCH720881 (M19; C4-pyrrolidine-hydroxylated rolapitant), the formation and elimination of which are slow and predictable [6].
Unlike other NK-1 receptor antagonists (aprepitant and netupitant), rolapitant does not induce or inhibit cytochrome P450 (CYP) 3A4; hence, no dosage adjustment for dexamethasone (a CYP3A4 substrate) is needed when co-administered with rolapitant in an antiemetic treatment regimen [9, 11, 12]. Findings from in vitro liver microsomal studies suggested that rolapitant inhibited the activity of human CYP2D6 enzyme at high concentrations (IC50 > 7 μM) and had minimal inhibitory effects on CYP3A4, CYP2B6, CYP2C8, CYP2C9, and CYP2C19. The metabolite M19 is not an inhibitor of these CYP enzymes. Patients receiving emetogenic cancer chemotherapy may receive multiple concomitant medications, and several may have narrow therapeutic indices. Given the role of the CYP pathway in the metabolism of numerous drug classes [13,14,15], it is important to identify and quantify any drug–drug interactions in vivo to reduce the risk of adverse events (AEs) associated with specific drug combinations that include rolapitant.
Here we demonstrate a multipart, clinical, drug–drug interaction study to evaluate the potential effects of rolapitant and M19 on the pharmacokinetics (PK) of dextromethorphan (CYP2D6), tolbutamide (CYP2C9), omeprazole (CYP2C19), efavirenz (CYP2B6), and repaglinide (CYP2C8). These drugs are the commonly used probe substrates for the specific CYP enzymes [16]. Safety and tolerability of rolapitant in combination with the CYP probes were also evaluated.
Materials and methods
Subjects
Studies were approved by the appropriate institutional or national research ethics committee (Aspire Institutional Review Board, Santee, CA, USA; protocol number, PR-10-5001-C). The study was conducted at the Parexel Early Phase Clinical Unit (Baltimore, MD, USA) between November 2011 and November 2012 and performed in compliance with the ethical standards as laid down in the 1964 Declaration of Helsinki (including amendments in effect up to the time the study was conducted), Guidelines of the International Conference on Harmonization on Good Clinical Practice, requirements of the Health Insurance Portability and Accountability Act of 1996, privacy regulations, and all other applicable regulatory requirements. Written informed consent was obtained from all subjects before enrollment in the study.
A total of 86 healthy subjects (dextromethorphan, n = 26; tolbutamide and omeprazole, n = 20; efavirenz, n = 20; repaglinide, n = 20) were enrolled in these studies. Inclusion criteria were healthy, nonsmoking male and female subjects aged 18 to 45 years with a body mass index (BMI) between 18.5 and 29.9 kg/m2 and weight ≥ 50 kg at screening. All subjects were determined to be in general good health, based on medical history, a physical examination, and vital signs. Female subjects had to have a negative serum pregnancy test prior to visit 1 and agree to use a medically accepted method of birth control throughout the study and for at least 30 days after dosing. All subjects were required to refrain from caffeine consumption and discontinue intake of alcohol, other beverages, or foods known to interfere with CYP metabolic enzymes. None of the subjects received continuous medication or used nicotine products.
For inclusion in the dextromethorphan part of the study, subjects were required to be identified as dextromethorphan normal or intermediate metabolizers. For inclusion in all of the studies, subjects had to have normal laboratory test results for serum aminotransferase concentrations and negative results for human immunodeficiency virus, hepatitis B virus, hepatitis C virus, urine drug screen, and serum alcohol screen at screening. At the screening visit, blood samples were collected and sent to LabCorp (Research Triangle Park, North Carolina) for CYP2D6 genotyping (dextromethorphan study) and CYP2C9/2C19 genotyping (tolbutamide and omeprazole study). Subjects known to have a relevant CYP genotype allele combination predicting the phenotype as a poor metabolizer were excluded in the dextromethorphan and tolbutamide plus omeprazole studies to obtain relatively homogenous and drug-sensitive populations for study and to minimize variability for PK analysis.
Study design
These were phase 1, open-label, drug–drug interaction studies. Subjects were admitted to the study center prior to the study and remained in-house until completion of assessments. Investigational drugs were administered to the subjects in the morning on day 1 with approximately 240 mL (8 fluid ounces) of room temperature water after the subjects had fasted overnight for a minimum of 10 h. Subjects remained in a semi-recumbent position for at least 4 h after dosing. Each study part consisted of three dosing and sample collection periods.
In the dextromethorphan study, subjects (n = 26) received a single oral dose of 30 mg dextromethorphan on days 1 and 14 and a single dose of 180 mg rolapitant (four 45 mg rolapitant capsules) plus 30 mg dextromethorphan on day 7 (Fig. 1). In the tolbutamide and omeprazole study, subjects (n = 20) received a single oral cocktail of 500 mg tolbutamide and 40 mg omeprazole on days 1 and 14 and a single dose of 180 mg rolapitant (4 × 45 mg rolapitant capsules) plus the drug cocktail on day 7. Blood samples for PK analysis of rolapitant were collected at pre-dose and at multiple time points up to 72 h post dose on days 1, 7, and 14.

Drug–drug interaction study in cohorts of 20 to 26 healthy subjects of orally administered CYP probe substrates (part A 30 mg dextromethorphan; part B 500 mg tolbutamide plus 40 mg omeprazole; part C 600 mg efavirenz; part D 0.25 mg repaglinide) in the absence and presence of single oral dose 180 mg rolapitant free base. ROL rolapitant
In the efavirenz study, subjects (n = 20) received a single oral dose of 600 mg efavirenz on days 1 and 17 and a single dose of 180 mg rolapitant (four 45 mg rolapitant capsules) and 600 mg efavirenz on day 10. Blood samples for PK analysis of rolapitant were collected at pre-dose and at multiple time points up to 120 h post dose on days 1, 10, and 17.
In the repaglinide study, subjects (n = 20) received a single oral dose of 0.25 mg repaglinide on days 1 and 10 and a single dose of 180 mg rolapitant (four 45 mg rolapitant capsules) and 0.25 mg repaglinide on day 3. Blood samples for PK analysis of rolapitant were collected at pre-dose and at multiple time points up to 12 h post dose on days 1, 3, and 10.
Determination of plasma drug concentrations
Following collection into K2-EDTA tubes, blood samples were immediately placed on ice. Plasma was separated by centrifugation at 4 °C, transferred to appropriately labeled polypropylene specimen containers, and frozen at − 20 °C until analysis (XenoBiotic Laboratories, Plainsboro, NJ, USA). Plasma samples were analyzed using fully validated liquid chromatography coupled with tandem mass spectrometry detection (LC/MS/MS) method for determination of the concentrations of dextromethorphan, tolbutamide, omeprazole, efavirenz, repaglinide, and rolapitant in ESI positive ion mode (API-4000, Applied Biosystem, MA) except for tolbutamide, to which negative ion mode was applied. The LC columns were Luna 5 μ HILIC 200A (100 × 2.0 mm) for dextromethorphan; Luna PFP (2) 50 × 2.0 mm for tolbutamide, omeprazole, and efavirenz; Luna C8 (2) 5 μ 50 × 2.0 mm for repaglinide; and an ACE (Milford, MA) C18-AR column (2.1 × 50 mm, 3μm) for rolapitant. The multiple reaction monitoring (MRM) transitions were 272.2/147.0 for dextromethorphan, 268.9/169.9 for tolbutamide, 346/198 for omeprazole, 314/244 for efavirenz, 453.4/230.5 for repaglinide, and 501.4/198.2 for rolapitant (SCH619734). The MRM transitions for respective internal standards were dextromethorphan-d3 275.2/215.2, tolbutamide-d9 at 277.9/169.6, omeprazole-d3 349/197.9, efavirenz-d4 317.9/247.9, repaglinide-ethyl-d5 458.2/230.2, and 13C3-SCH619734 504/201. The validated ranges were 0.1 to 50 ng/mL for dextromethorphan, 100 to 50,000 ng/mL for tolbutamide, 2.0 to 1000 ng/mL for omeprazole, 10.0 to 5000 ng/mL for efavirenz, 0.02 to 10 ng/mL for repaglinide, and 2.0 to 2000 ng/mL for rolapitant.
Pharmacokinetic evaluations
The following pharmacokinetic parameters were calculated: maximum observed concentration (Cmax), time of observed maximum concentration (Tmax), area under the plasma concentration–time curve from time 0 to the time of the last quantifiable concentration (AUC0–last), area under the plasma concentration–time curve from time 0 extrapolated to infinity (AUC0–inf), and terminal elimination half-life (t1/2).
Safety assessments
Safety and tolerability were assessed by physical examinations, vital signs, laboratory safety tests, and reported AEs throughout the studies. The following safety parameters were recorded: clinical laboratory tests (clinical chemistry, hematology, and urinalysis), glucose monitoring for subjects receiving tolbutamide, 12-lead electrocardiograms (ECGs), vital signs (supine and orthostatic blood pressure, heart rate, and body temperature), physical examination, and concomitant medication monitoring.
Pharmacokinetic and statistical analysis
Individual plasma analyte concentration–time data were used to derive drug PK parameters using noncompartmental analyses performed with Phoenix® WinNonlin® 6.2 (Pharsight/Certara, Sunnyvale, CA, USA). SAS® (SAS Institute, Cary, NC) software was used for the statistical analysis. To determine the effect of rolapitant on the PK of the investigational drugs, the log-transformed primary endpoints (AUC and Cmax) were analyzed using a repeated-measures ANOVA. Ninety percent confidence intervals (CIs) were constructed for the ratio of geometric least squares means.
Results
Patients
A total of 86 subjects were enrolled. Most of the subjects (97.7%) completed the studies. Two subjects in the tolbutamide plus omeprazole part withdrew early for reasons unrelated to study drugs (one subject had an anxiety attack and the other left due to a family emergency). The majority of subjects were male (73.6%) and not Hispanic or Latino (78.8%; Table 1). Mean age ranged from 29.4 to 32.1 years and mean BMI from 24.1 to 26.1 kg/m2.
Interaction of dextromethorphan with rolapitant
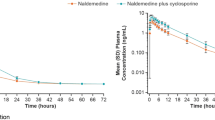
Median Tmax for dextromethorphan alone was 2.0 h and for dextromethorphan and rolapitant was 3.0 h. The mean t1/2 for dextromethorphan alone was 7.5 h and for dextromethorphan and rolapitant was 9.6 h (Table 2). Single-dose administration of rolapitant with dextromethorphan resulted in an increase in dextromethorphan exposure (increase by 2.3-fold for Cmax and 2.6-fold for AUC0–inf) on day 7 and in the presence of rolapitant (increase by 2.8-fold for Cmax and 3.3-fold for AUC0–inf) on day 14 (Table 3). Similar increase in AUC0–last was observed. Plasma concentration–time profiles for dextromethorphan demonstrated that dextromethorphan PK increased on day 7 when co-administered with rolapitant and increased on day 14 around peak metabolite concentration (Fig. 2a).

Mean concentration–time profiles for (a) dextromethorphan, (b) tolbutamide, (c) omeprazole, (d) efavirenz, and (e) repaglinide with and without a single oral dose of rolapitant 180 mg administration
Statistical analysis demonstrated that rolapitant inhibited dextromethorphan metabolism resulting in higher exposure (Cmax and AUC) of dextromethorphan following administration of rolapitant on day 7 and in the presence of M19 on day 14 (Table 3). Geometric least square mean ratios for Cmax and AUC0–inf ranged between 2.2 and 3.3 for the comparisons of day 7/day 1 and day 14/day 1 parameter values, and the 90% CIs for the ratio of geometric least squares means were significantly greater than the 80 to 125% limits for Cmax and AUC0–inf (Table 3).
Interaction of tolbutamide and omeprazole with rolapitant
Exposure (Cmax and AUC) of tolbutamide and omeprazole following concomitant administration with rolapitant on day 7 and prolonged exposure to rolapitant on day 14 were similar to those on day 1, suggesting rolapitant does not impact the PK of tolbutamide and omeprazole. Plasma concentration–time profiles demonstrate there was no significant effect of rolapitant on tolbutamide and omeprazole PK (Fig. 2b, c). Statistical analysis confirmed that exposure to tolbutamide was not impacted by co-administration with 180 mg rolapitant. The 90% CI of geometric least square mean ratios for Cmax, AUC0–last, and AUC0–inf of tolbutamide were contained within the 80 to 125% no effect limit (Table 3). For omeprazole, statistical analysis demonstrated a minimal effect of rolapitant on the PK of omeprazole. Geometric least square mean ratios were 1.44 and 1.37 for Cmax and 1.23 and 1.15 for AUC0–inf for the comparisons of day 7/day 1 and day 14/day 1, respectively (Table 3). However, these are unlikely to be clinically relevant.
Interaction of efavirenz with rolapitant
The PK parameters for efavirenz following administration with or without rolapitant on days 10 and 17, respectively, were comparable to those obtained on day 1, indicating that rolapitant is not an inhibitor of efavirenz exposure. The mean plasma efavirenz concentration–time profile demonstrates there was no significant effect of rolapitant on efavirenz PK (Fig. 2d). The 90% CI of geometric least square mean ratios for AUC0–last of efavirenz was contained within the 80 to 125% no effect limit (Table 3). Minor excursions outside the no effect limit were observed for Cmax and AUC0–inf but are not expected to be clinically significant.
Interaction of repaglinide with rolapitant
PK parameters of repaglinide on day 3 were similar to those on day 1. Co-administration of a single 180 mg dose of rolapitant with repaglinide resulted in a minimal increase in exposure of repaglinide on day 14 (Table 3). Median Tmax for repaglinide was unchanged for day 3 and day 10. The mean t1/2 for repaglinide alone on day 3 was 3.6 h, and repaglinide and rolapitant at day 10 was 3.8 h. Plasma concentration–time profile shows co-administration of rolapitant with repaglinide did not impact the PK of repaglinide (Fig. 2e). The 90% CIs for the ratio of geometric least squares means were contained within the 80 to 125% no effect limits for both Cmax and AUC0–inf for the day 3 and day 1 comparison but were slightly higher for day 10 and day 1 comparison. This is not expected to be clinically significant.
Safety
In general, rolapitant was safe and well tolerated when co-administered with 30 mg dextromethorphan, 500 mg tolbutamide plus 40 mg omeprazole, 600 mg efavirenz, or 0.25 mg repaglinide. There were no serious treatment-emergent AEs (TEAEs) or deaths. The majority of TEAEs were mild and resolved spontaneously. In the dextromethorphan study, 4 out of 26 subjects experienced at least one TEAE that was judged to be related or probably related to study treatment. All TEAEs were of mild intensity and resolved by the end of study. During the tolbutamide and omeprazole study, 9 out of 26 subjects experienced at least one TEAE with most of the events being mild and resolved by the end of the study. One subject reported anxiety on day 14 leading to withdrawal from the study. During the efavirenz study, 15 out of 20 subjects experienced at least one TEAE, including dizziness, somnolence, and headaches. All TEAEs were treated and resolved during the study. In the repaglinide study, 9 out of 20 subjects experienced at least one TEAE, including migraines, upper respiratory infection, and headache. Additionally, no clinically significant changes in blood chemistry, vital signs, or ECGs occurred in the studies.
Discussion
CINV has a major deleterious effect on the functional capacity and quality of life of patients with cancer undergoing chemotherapy and has the potential to compromise adherence to life-prolonging treatment regimens [17, 18]. Administration of an NK-1 receptor antagonist, such as rolapitant, along with a 5-HT3-RAand dexamethasone has been incorporated into international antiemesis guidelines for control of CINV in patients receiving highly emetogenic chemotherapy and select patients receiving moderately emetogenic therapy [4, 19, 20].
This is the first study to simultaneously assess the effects of oral rolapitant on the activity of multiple CYP enzymes in healthy subjects. Our findings indicate that IV rolapitant does not alter the activity of CYP2C9, CYP2C19, CYP2B6, or CYP2C8 to a clinically relevant extent, as evidenced by no effect or a weak effect of oral rolapitant on the Cmax and AUC0–inf of the substrates of these enzymes. Hence, oral rolapitant is not expected to alter appreciably the PK of concomitantly administered medications that are substrates of CYP2C9, CYP2C19, CYP2B6, or CYP2C8. However, oral rolapitant did alter the activity of CYP2D6 over a period of at least 7 days post oral rolapitant dose, as evidenced by a 3.3-fold increase in AUC0–inf for dextromethorphan 7 days post rolapitant dose. Therefore, oral rolapitant is expected to increase systemic exposure of CYP2D6 substrates. For this reason, AE monitoring is required if oral rolapitant is given with CYP2D6 substrates that have a narrow therapeutic index if their concomitant use cannot be avoided. These findings are consistent with in vitro metabolism studies showing that rolapitant did not inhibit CYP3A4, CYP2C9, and CYP2C19 but did act as an inhibitor of CYP2D6 in vitro.
A single oral dose of 180 mg rolapitant was well tolerated when co-administered with all CYP probe substrates. Most TEAEs were of mild intensity, and there were no serious adverse events reported.
In conclusion, rolapitant was well tolerated when co-administered with CYP2D6, CYP2C9, CYP2C19, CYP2B6, or CYP2C8 probe substrates in this study. Inhibition of dextromethorphan by a single oral dose of 180 mg rolapitant is clinically significant and can last at least 7 days. No clinically significant interaction was observed between rolapitant and substrates of CYP2C9, CYP2C19, CYP2B6, or CYP2C8. CYP2D6 substrate drugs with a narrow therapeutic index may require monitoring for adverse reactions if given concomitantly with rolapitant.
References
Ballatori E, Roila F, Ruggeri B, Betti M, Sarti S, Soru G, Cruciani G, Di Maio M, Andrea B, Deuson RR (2007) The impact of chemotherapy-induced nausea and vomiting on health-related quality of life. Support Care Cancer 15(2):179–185. https://doi.org/10.1007/s00520-006-0109-7
Bloechl-Daum B, Deuson RR, Mavros P, Hansen M, Herrstedt J (2006) Delayed nausea and vomiting continue to reduce patients’ quality of life after highly and moderately emetogenic chemotherapy despite antiemetic treatment. J Clin Oncol 24(27):4472–4478. https://doi.org/10.1200/JCO.2006.05.6382
Cohen L, de Moor CA, Eisenberg P, Ming EE, Hu H (2007) Chemotherapy-induced nausea and vomiting: incidence and impact on patient quality of life at community oncology settings. Support Care Cancer 15(5):497–503. https://doi.org/10.1007/s00520-006-0173-z
NCCN Clinical practice guidelines in oncology version 1 antiemesis. National Comprehensive Cancer Network (NCCN) [online]. www.nccn.org/professionals/physician_gls/PDF/antiemesis.pdf. Accessed 24 May 2018
Duffy RA, Morgan C, Naylor R, Higgins GA, Varty GB, Lachowicz JE, Parker EM (2012) Rolapitant (SCH 619734): a potent, selective and orally active neurokinin NK1 receptor antagonist with centrally-mediated antiemetic effects in ferrets. Pharmacol Biochem Behav 102(1):95–100. https://doi.org/10.1016/j.pbb.2012.03.021
VARUBI™ (rolapitant) tablets for oral use Highlights of prescribing information. Tesaro, Inc., Waltham, MA; 2015. http://www.tesarobio.com/varubi. Accessed 24 May 2018
Rapoport B, Chua D, Poma A, Arora S, Wang Y, Fein LE (2015) Study of rolapitant, a novel, long-acting, NK-1 receptor antagonist, for the prevention of chemotherapy-induced nausea and vomiting (CINV) due to highly emetogenic chemotherapy (HEC). Support Care Cancer 23(11):3281–3288. https://doi.org/10.1007/s00520-015-2738-1
Schwartzberg LS, Modiano MR, Rapoport BL, Chasen MR, Gridelli C, Urban L, Poma A, Arora S, Navari RM, Schnadig ID (2015) Safety and efficacy of rolapitant for prevention of chemotherapy-induced nausea and vomiting after administration of moderately emetogenic chemotherapy or anthracycline and cyclophosphamide regimens in patients with cancer: a randomised, active-controlled, double-blind, phase 3 trial. Lancet Oncol 16(9):1071–1078. https://doi.org/10.1016/S1470-2045(15)00034-0
Poma A, Christensen J, Pertikis H (2013) Rolapitant and its major metabolite do not affect the pharmacokinetics of midazolam, a sensitive cytochrome P450 3A4 substrate [Abstract 441]. Support Care Cancer 21:S154
Poma A, Christensen J, Davis J, Kansra V, Martell RE, Hedley ML (2014) Phase 1 positron emission tomography (PET) study of the receptor occupancy of rolapitant, a novel NK-1 receptor antagonist [abstract e20690]. J Clin Oncol 32 (suppl)
Aapro MS, Walko CM (2010) Aprepitant: drug-drug interactions in perspective. Ann Oncol 21(12):2316–2323. https://doi.org/10.1093/annonc/mdq149
Calcagnile S, Lanzarotti C, Rossi G, Henriksson A, Kammerer KP, Timmer W (2013) Effect of netupitant, a highly selective NK(1) receptor antagonist, on the pharmacokinetics of palonosetron and impact of the fixed dose combination of netupitant and palonosetron when coadministered with ketoconazole, rifampicin, and oral contraceptives. Support Care Cancer 21(10):2879–2887. https://doi.org/10.1007/s00520-013-1857-9
Donato MT, Castell JV (2003) Strategies and molecular probes to investigate the role of cytochrome P450 in drug metabolism: focus on in vitro studies. Clin Pharmacokinet 42(2):153–178. https://doi.org/10.2165/00003088-200342020-00004
Hisaka A, Kusama M, Ohno Y, Sugiyama Y, Suzuki H (2009) A proposal for a pharmacokinetic interaction significance classification system (PISCS) based on predicted drug exposure changes and its potential application to alert classifications in product labelling. Clin Pharmacokinet 48(10):653–666. https://doi.org/10.2165/11317220-000000000-00000
Lin JH, Lu AY (1998) Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet 35(5):361–390. https://doi.org/10.2165/00003088-199835050-00003
Huang SM, Temple R, Throckmorton DC, Lesko LJ (2007) Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin Pharmacol Ther 81(2):298–304. https://doi.org/10.1038/sj.clpt.6100054
Aapro M, Molassiotis A, Dicato M, Pelaez I, Rodriguez-Lescure A, Pastorelli D, Ma L, Burke T, Gu A, Gascon P, Roila F, investigators P (2012) The effect of guideline-consistent antiemetic therapy on chemotherapy-induced nausea and vomiting (CINV): the Pan European Emesis Registry (PEER). Ann Oncol 23(8):1986–1992. https://doi.org/10.1093/annonc/mds021
Jordan K, Gralla R, Jahn F, Molassiotis A (2014) International antiemetic guidelines on chemotherapy induced nausea and vomiting (CINV): content and implementation in daily routine practice. Eur J Pharmacol 722:197–202. https://doi.org/10.1016/j.ejphar.2013.09.073
Basch E, Prestrud AA, Hesketh PJ, Kris MG, Feyer PC, Somerfield MR, Chesney M, Clark-Snow RA, Flaherty AM, Freundlich B, Morrow G, Rao KV, Schwartz RN, Lyman GH, American Society of Clinical O (2011) Antiemetics: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol 29(31):4189–4198. https://doi.org/10.1200/JCO.2010.34.4614
MASCC/ESMO antiemetic guidelines http://www.mascc.org/antiemetic-guidelines. Accessed 24 May 2018
Funding
Medical writing and editorial assistance was funded by TESARO Inc.
Author information
Authors and Affiliations
Contributions
Study design (V Kansra)
Data collection (X Wang, Zhi-Yi Zhang, V Kansra)
Data analysis (X Wang, Zhi-Yi Zhang, V Kansra)
Manuscript conception and writing (J Wang, V Kansra, X Wang)
Critical revision of the manuscript (J Wang, Zhi-Yi Zhang, Sharon Lu, D Powers, V Kansra, X Wang)
All authors were involved in the collection, analysis, and interpretation of data, in the writing of the manuscript, and in the decision to submit the manuscript for publication.
Corresponding author
Ethics declarations
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict of interest
This study was designed by the study sponsor, TESARO, Inc. Study data were collected by clinical investigators, and trial conduct was monitored by TESARO, Inc. Statistical analyses were managed by TESARO, Inc., according to a predefined statistical plan. This manuscript was developed with full author participation and assistance from a medical writer in accordance with Good Publication Practice 3 (GPP3) guidelines and International Committee of Medical Journal Editors guidelines. All authors had access to full data and analyses presented in this manuscript. All authors are employees or former employees of the study sponsor. The authors have indicated that they have no other conflicts of interest regarding the content of this article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, J., Zhang, ZY., Lu, S. et al. Effects of rolapitant administered orally on the pharmacokinetics of dextromethorphan (CYP2D6), tolbutamide (CYP2C9), omeprazole (CYP2C19), efavirenz (CYP2B6), and repaglinide (CYP2C8) in healthy subjects. Support Care Cancer 27, 819–827 (2019). https://doi.org/10.1007/s00520-018-4331-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-018-4331-x