
Abstract
Background and Objective
This study measured and compared the exposure and safety of peficitinib (ASP015K), a novel oral Janus kinase inhibitor, in subjects with normal and impaired renal function after a single oral, clinically relevant peficitinib dose.
Methods
This was an open-label, parallel-group study conducted at two centres in Japan. Subjects with normal and mildly, moderately, or severely impaired renal function received a single oral dose of peficitinib (one 150 mg tablet) under fasting conditions in a hospital setting. Blood samples were collected prior to administration and up to 72 h post-dose for pharmacokinetic assessment. Safety was assessed up to 7 days post-dose.
Results
Peficitinib plasma concentration–time profiles were similar between those with normal and impaired renal function. In subjects with impaired renal function, area under the plasma concentration–time curve and maximum concentration were 0.8- to 1.1-fold those in subjects with no impairment. Two subjects (one in the normal group and one in the mildly impaired group) each experienced a treatment-emergent adverse event (TEAE). There were no serious TEAEs, deaths or TEAEs leading to treatment withdrawal.
Conclusions
Peficitinib exposure and TEAEs were similar in subjects with and without renal impairment after a single oral 150 mg dose. Based on these findings, it is not expected that peficitinib dose adjustment will be required in clinical practice, according to the degree of renal impairment.
ClinicalTrials.gov identifier
NCT02603497.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Peficitinib exposure after a single 150 mg dose was comparable in subjects with and without impaired renal function. |
Peficitinib was well tolerated, with a similar rate of treatment-emergent adverse events in subjects with and without renal impairment. |
It is not expected that any peficitinib dose adjustment will be required in clinical practice according to the degree of renal impairment. |
1 Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that carries a significant burden for individuals and society [1,2,3]. It targets the joints, causing cartilage and bone damage; in many patients, progressive joint erosion is associated with physical disability and reduced quality of life [4, 5]. Despite the available treatments, there remains a significant unmet therapeutic need in RA, with pain, physical and mental functioning and fatigue persisting at an unacceptable level [6]. As a result, there is a drive towards the development of agents that will better address the multifactorial nature of RA and improve outcomes for patients.
The Janus kinase (JAK) family [JAK1, JAK2, JAK3, and tyrosine kinase-2 (TYK2)] of non-receptor tyrosine kinases plays a crucial role in the pro-inflammatory cytokine signalling implicated in the pathogenesis of RA, and is considered a promising alternative target for RA treatment [7, 8]. A number of JAK inhibitors have been developed in recent years, with differential specificity for one or more JAKs [8]. Peficitinib (ASP015K) is a novel, pan-JAK inhibitor that inhibits JAK1, JAK2, JAK3, and TYK2 [9]. In clinical studies, peficitinib has been shown to be efficacious as once-daily therapy for moderate-to-severe RA, with a rate of treatment-emergent adverse events (TEAEs) comparable with placebo at doses up to 150 mg [10,11,12]. This formed the basis for the recent approval of peficitinib (50 mg and 100 mg tablets) in Japan; the usual clinical dosage for adult patients with RA is 150 mg per day, which can be reduced to 100 mg per day depending on the patient’s condition [13].
In a study of peficitinib pharmacokinetics, mean urinary excretion accounted for 9–15% of a single oral dose in healthy volunteers, and 15–17% after 2 weeks of multiple dosing [14]. This may have implications for treatment in individuals with renal insufficiency. Globally, the estimated mean prevalence of chronic kidney disease [CKD; The National Kidney Disease Outcomes Quality Initiative (KDOQI) thresholds of eGFR, stages 1 to 5] is 13.4%, with prevalence typically higher in developed countries, such as North America (15.5%), Europe (18.4%) and Japan (13.7%) compared with growing economies, such as sub-Saharan Africa (8.7%) [15]. Given the high prevalence of CKD, it is assumed that a proportion of patients with RA will also have some level of renal function impairment. To determine whether peficitinib exposure is affected by the level of renal function, this study compared the pharmacokinetics and safety of a single oral dose of peficitinib in non-RA subjects with and without impaired renal function.
2 Methods
2.1 Study Design
This was an open-label, single oral dose, parallel-group comparison study conducted between November 2015 and December 2016 at two sites in Japan. Its aim was to measure and compare the pharmacokinetics and safety of peficitinib between subjects with varying degrees of renal impairment and normal renal function after administration of peficitinib at a clinically relevant dose (150 mg).
2.2 Ethical Conduct
The study was reviewed and approved by the Institutional Review Board and all subjects provided written informed consent before undergoing any study-related procedures. The study was conducted in accordance with the International Conference on Harmonization (ICH) guidelines for Good Clinical Practice (GCP), applicable regulations, and guidelines governing clinical study conduct and the ethical principles that have their origin in the Declaration of Helsinki.
2.3 Study Participants
Eligible subjects were male or female, aged ≥ 20–75 years, with a body mass index (BMI) ≥ 17.6 and < 30.0 kg/m2 at screening. Renal function was classified according to estimated glomerular filtration rate (eGFR) using a GFR predictive equation for Japanese subjects [16, 17]: for males, eGFRcreat (mL/min/1.73 m2) = 194 × [serum creatinine (mg/dL)] − 1.094 × (age) − 0.287; and for females, (male eGFR) × 0.739. Definitions for renal function were: normal, ≥ 90 mL/min/1.73 m2; mild impairment, ≥ 60 to < 90 mL/min/1.73 m2; moderate impairment, ≥ 30 to < 60 mL/min/1.73 m2; and severe impairment, ≥ 15 to < 30 mL/min/1.73 m2.
2.4 Study Drug and Administration
A single 150 mg peficitinib tablet (Astellas Pharma Inc., Tokyo, Japan) was administered orally with 150 mL of water in the morning of Day 1. Subjects had fasted overnight and drinking water was prohibited from 1 h before, until 1 h after study drug administration (except for that taken during administration).
2.5 Sample Analysis
2.5.1 Peficitinib and Metabolite H2 (M2)
The concentrations of plasma peficitinib and H2 metabolite (sulfate metabolite, equivalent to M2 identified previously [18]) were measured using validated liquid chromatography tandem mass spectrometry (LC–MS/MS). The lower limit of quantification (LLOQ) was 0.25 ng/mL when 25 μL of plasma was used. The analysis method has been published previously using rat plasma [19]. For clinical sample analysis, the method was developed using human plasma. Calibration curves for peficitinib and H2 in human plasma were linear over the concentration range 0.25–500 ng/mL, with correlation coefficients ≥ 0.9988. The intra-day accuracies were within ± 15% of the nominal concentration, and the intra-day precision did not exceed 15%.
2.5.2 Metabolites H1 (M1) and H4 (M4)
The concentration of metabolites H1 (M1; sulfate and methylated metabolite) and H4 (M4; methylated metabolite) were measured separately using two validated LC–MS/MS methods. The LLOQ was 0.25 ng/mL, when 25 μL of plasma was used. Extraction of H1 and H4 was carried out separately by solid-phase extraction. Deuterated H1 (d3-H1) was used for the internal standard (IS) of H1; a stereoisomer of peficitinib was used for the IS of H4. Plasma samples (25 µL) were mixed with 100 mM phosphate solution (pH 7; 1 mL), acetonitrile–water (1:1, v/v; 25 µL) and IS working solution (25 µL), and loaded onto the preconditioned Oasis® Hydrophilic-Lipophilic-Balanced cartridge (30 mg/1 cc; Waters Co., Milford, MA, USA). For H1 extraction, the cartridge was washed three times with 1 mL of methanol–water (5:95, v/v) and analytes were eluted twice with 0.5 mL of methanol. For H4 extraction, the cartridge was washed three times with 1 mL of methanol–water (25:75, v/v) and analytes were eluted twice with 0.5 mL of methanol–formic acid (100:0.1, v/v). Eluted solvent was evaporated at 40 °C under a stream of nitrogen gas. Residues were reconstituted with a 0.5 mL aliquot of reconstitution solution, 10 mM ammonium acetate–methanol for H1 (65:35, v/v) and H4 (60:40, v/v). A 10 or 20 µL aliquot of each sample was injected into the LC system for H1 or H4 analysis, respectively.
2.5.3 LC–MS/MS Conditions
For the H1 metabolite, LC separation was performed on an Inertsil Ph-3 column [4.6 mm I.D. × 100 mm (GL Sciences, Tokyo, Japan)] for 5 μm particle size with a mobile phase consisting of 10 mM ammonium acetate and methanol. H1 was eluted using mobile phase A (10 mM ammonium acetate solution) and mobile phase B (methanol) in the following linear gradient conditions: 0‒3.50 min, 55% B; 3.51‒4.00 min, 90% B; 4.01‒6.00 min, 90% B; 6.01‒7.00 min, 55% B; and 7.01‒7.50 min, 55% B. Flow rate was set at 0.8 mL/min except for 4.01‒7.00 min (1.5 mL/min) and column temperature was set at 40 °C. MS/MS detection was performed using an API4000 (AB SCIEX, Framingham, MA, USA). The ionspray voltage was maintained at 3000 V and temperature at 700 °C. Ion source gas 1 (air) was set at 60 psi and ion source gas 2 (air) at 60 psi. Curtain gas (nitrogen) was set at 10 psi. Selected reaction monitoring was conducted in positive ion electrospray mode. The first quadrupole (Q1) selected for the cationic moiety of each compound was as follows: H1, m/z 421; IS d3-H1, m/z 424. Product ions were generated by collision-induced dissociation within Q2 (collision gas nitrogen) and detected at the electron multiplier as follows: H1, m/z 341; IS d3-H1, m/z 344. Data were processed using Analyst Software (AB SCIEX version 1.5). Calibration curves for H1 in human plasma were linear over the concentration range 0.25‒100 ng/mL, with correlation coefficients ≥ 0.9991. The intra- and inter-day accuracies were within ± 15% of the nominal concentration, and the intra- and inter-day precision did not exceed 15%.
For analysis of the H4 metabolite, LC separation was performed on an Inertsil Ph-3 column [2.1 mm I.D. × 33 mm (GL Sciences)] for 5 μm particle size with mobile phase A [10 mM ammonium acetate–methanol (60:40, v/v)] and mobile phase B [acetonitrile–water (70:30, v/v)] in the following isocratic conditions: 0‒6.00 min, 0% B; 6.01‒9.00 min, 100% B; 9.01‒10.5 min, 0% B. Flow rate was set at 0.8 mL/min and column temperature was set at 40 °C. MS/MS detection was performed using an API4000. The ionspray voltage was maintained at 5000 V and temperature at 650 °C. Ion source gas 1 (air) was set at 60 psi and ion source gas 2 (air) at 70 psi. Curtain gas (nitrogen) was set at 10 psi. Selected reaction monitoring was conducted in positive ion electrospray mode. The Q1 selected for the cationic moiety of each compound was as follows: H4, m/z 341; IS, a stereoisomer of peficitinib, m/z 327. Product ions were generated by collision-induced dissociation within Q2 (collision gas nitrogen) and detected at the electron multiplier as follows: H4, m/z 174; IS, a stereoisomer of peficitinib, m/z 91. Data were processed using Analyst Software. Calibration curves for H4 in human plasma were linear over the concentration range 0.25‒100 ng/mL, with correlation coefficients ≥ 0.9976. The intra- and inter-day accuracies were within ± 15% of the nominal concentration, and the intra- and inter-day precision did not exceed 15%.
Chromatograms of samples at the LLOQ (0.25 ng/mL) are shown in Fig. 1. No interfering peak was detected at the retention time of peficitinib and its metabolites in blank human plasma from six subjects (data not shown).

Representative selected reaction monitoring chromatograms of blank plasma spiked with peficitinib and its metabolites (lower limit of quantification, 0.25 ng/mL). cps counts per second
2.6 Pharmacokinetic Assessments
Blood samples were collected before dosing and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, and 12 h after peficitinib administration. On Days 2 and 3, samples were collected at 24, 36, 48, and 60 h after study drug administration. On Day 4, a final blood sample was taken at 72 h after study drug administration. The anticoagulant EDTA-3K was used during collection of blood samples.
The following pharmacokinetic parameters were calculated for peficitinib and its metabolites (H1, H2 and H4) by non-compartmental analysis using WinNonlin® version 6.2 (Pharsight, Sunnyvale, CA, USA) with actual sampling time and plasma concentrations; area under the plasma concentration–time curve up to the last quantifiable time (AUClast), AUC extrapolated to infinity (AUCinf), maximum plasma concentration (Cmax), apparent total systemic clearance (CL/F), time to reach maximum plasma concentration (tmax), and terminal elimination half-life (t1/2). For peficitinib only, the apparent volume of distribution based on the terminal phase (Vz/F) was estimated and the metabolite-to-parent ratio (MPR) was estimated for H1, H2 and H4.
2.7 Safety
TEAEs, vital signs (supine blood pressure, supine pulse and axillary temperature), laboratory tests (haematology, blood biochemistry and urinalysis), and 12-lead electrocardiograms (ECG) were assessed.
A TEAE was defined as any unfavourable and unintended sign, symptom or impairment that started or worsened after administration of study drug (but did not necessarily have a causal relationship with the treatment). System organ classes and preferred terms for TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) v17.1. For each TEAE, the investigator provided an assessment of the causal relationship with peficitinib and graded the severity using the Common Terminology Criteria for Adverse Events (CTCAE) v4.0, distributed by the Japan Cooperative Oncology Group (JCOG). After subjects were discharged, a follow-up safety examination was conducted after 7 (± 2) days.
2.8 Statistical Analysis
2.8.1 Sample Size
The planned sample size was eight subjects per renal function group (normal, mild impairment, moderate impairment, and severe impairment). The sample size determination for this study was not based on statistical power.
2.8.2 Populations for Analysis
The safety analysis set included all subjects who received peficitinib. The pharmacokinetic analysis set included all those who received peficitinib and provided one or more estimable pharmacokinetic parameters.
2.8.3 Statistical Methodology
Demographic and baseline characteristics, as well as plasma concentrations and pharmacokinetic parameters, were summarised by descriptive statistics according to each renal function category (normal, mild, moderate, and severe). The natural log-transformed AUC and Cmax were used for the analysis of variance, with renal function category as a fixed effect, to estimate the geometric least-square mean ratio (GMR) and its 90% confidence interval for impaired renal function versus normal renal function.
An exploratory analysis to assess the relationship between peficitinib AUCinf and renal function was performed using the linear model with eGFR as a fixed effect.
3 Results
3.1 Participant Demographics and Baseline Characteristics
A total of 31 subjects were enrolled, received peficitinib and completed the study (Fig. 2). There were no notable differences in baseline characteristics across the renal function groups (Table 1). Most subjects were male and the mean age was 57.8 years. Mean eGFR was 77.5 mL/min/1.73 m2 in the mildly impaired renal function group, 48.5 mL/min/1.73 m2 in the moderately impaired renal function group, and 23.6 mL/min/1.73 m2 in the severely impaired renal function group.

Participant flow. PK pharmacokinetics
3.2 Pharmacokinetics
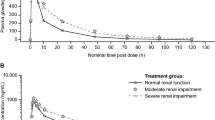
Peficitinib was rapidly absorbed, with a median tmax ranging from 1.50 h in the renal impairment groups to 1.75 h in the normal renal function group (Table 2). Plasma concentration profiles over time (0–72 h) were similar across the renal function groups (Fig. 3). There were no clear differences in pharmacokinetic parameters for parent peficitinib across the groups of subjects with renal impairment (Table 2), and the GMRs for AUCinf, AUClast and Cmax (Table 3) confirmed that no marked differences were observed between subjects in any of the renal impairment groups versus subjects with normal renal function. Linear regression analysis of peficitinib AUCinf, CL/F and eGFR across the renal function groups also confirmed that there were no statistically significant slopes and therefore no relationship between the parameters and eGFR (Fig. 4 shows the relationship between peficitinib AUCinf and eGFR).

Mean plasma concentration–time profile of peficitinib by degree of renal impairment (linear and semi-logarithmic scale). Values are mean linear (left) and semi-logarithmic (right) scales. Classification of renal impairment was based on the predictive glomerular filtration rate equation: for males, eGFRcreat (mL/min/1.73 m2) = 194 × [serum creatinine (mg/dL)] − 1.094 × (age) − 0.287; and for females, (male eGFR) × 0.739. Definitions were: normal function ≥ 90 mL/min/1.73 m2; mild impairment ≥ 60 to < 90 mL/min/1.73 m2; moderate impairment ≥ 30 to < 60 mL/min/1.73 m2; and severe impairment ≥ 15 to < 30 mL/min/1.73 m2. eGFR estimated glomerular filtration rate

The relationship between the area under the plasma concentration–time curve (AUCinf) of peficitinib and (estimated) glomerular filtration rate across the renal function groups. AUCinf area under the concentration-time curve from the time of dosing extrapolated to infinity, eGFR estimated glomerular filtration rate
Of the three conjugated peficitinib metabolites (H1, H2, H4), there was a trend for increased MPR with increasing renal impairment for the H1 and H2 metabolites, but no clear trend was observed for the H4 metabolite (Table 2).
3.3 Safety
One subject in the normal renal function group and one subject in the mildly impaired renal function group each experienced one TEAE (headache and alanine aminotransferase increased, respectively) after a single oral dose of peficitinib (Table 4). Of these TEAEs, headache in the subject in the normal renal function group was considered to be drug-related. No death, serious TEAE or TEAE leading to withdrawal of treatment was reported. Both the TEAEs were mild in intensity.
With the exception of the case of mild alanine aminotransferase increase, no clinically significant mean changes from baseline were observed for any of the clinical laboratory parameters assessed during the study.
No clinically significant mean changes from baseline were observed for any of the vital sign measurements assessed during the study.
ECG results on the day of hospital admission and the predetermined time points post-dose were considered to be normal by the investigator in all subjects. No clinically significant mean changes from baseline were observed for any of the ECG variables assessed during the study.
4 Discussion
Peficitinib is a novel oral JAK inhibitor that is efficacious for the treatment of moderate-to-severe RA. As CKD is a relatively common condition that, like RA, becomes more prevalent with age [15], it is important to understand the impact of renal impairment on peficitinib pharmacokinetics and safety, to assess whether dose adjustment may be required in clinical practice. The present study compared the pharmacokinetics and safety of a single oral dose of peficitinib in subjects with and without impaired renal function.
A single 150 mg oral dose was selected as the ‘usual’ clinical dose based on the daily dose of peficitinib in Phase III studies in Japanese patients with RA (100 mg and 150 mg) [11, 12, 20]. In this study, we evaluated the pharmacokinetic profile of a single oral dose of peficitinib 150 mg in subjects with normal renal function and impaired renal function. In previous dose-ranging studies in healthy adult volunteers, single doses of peficitinib up to 450 mg did not cause any toxicity or significant safety findings that required dose limitation. Thus, a single 150 mg dose was considered acceptable for subjects with renal impairment.
This study in 31 Japanese subjects confirmed that peficitinib plasma concentration was not significantly affected by impaired renal function. The AUCinf GMR for the severely impaired versus normal renal function group was 1.09, demonstrating that peficitinib exposure was not markedly increased, even in subjects with the most severe renal impairment. Furthermore, linear regression confirmed that there was no clear relationship between peficitinib pharmacokinetic parameters and eGFR across the renal function groups.
Previously, in studies of healthy subjects, peficitinib was shown to be rapidly absorbed, with a median tmax of 1.5 h under fasting conditions, increasing to 4 h under fed conditions [14]. Similarly, in the current study under fasting conditions, the median tmax of peficitinib was 1.5–1.75 h across the renal function groups.
Mean t1/2 in a previous study in healthy volunteers ranged from 7.7 to 12.9 h after single peficitinib doses above 100 mg [14]. In subjects with normal renal function in the present study, mean t1/2 was 6.80 h and was longer in subjects with renal impairment. However, there was no observable trend in t1/2 according to increasing severity of impairment (mild 14.96 h, moderate 14.36 h, severe 10.80 h, respectively).
While exposure to parent peficitinib remained generally consistent, irrespective of the degree of renal impairment, there were some changes in MPRs of the conjugated metabolites (H1, H2 and H4), which show very weak in vitro pharmacological action. The MPR of H1 and H2 increased with greater impairment of renal function to a greater extent than H4. This might be due to elimination delay of H1 and H2, as both of these metabolites are more susceptible to urinary elimination compared with H4, which is excreted more in feces than in urine [18].
Administration of a single oral 150 mg dose of peficitinib was well tolerated in Japanese subjects with normal renal function and impaired renal function, including those with severely impaired renal function. The only drug-related adverse event was a mild case of headache in one subject in the normal renal function group.
The findings from this pharmacokinetic study of peficitinib contrast with those from other JAK inhibitors such as tofacitinib and baricitinib, which are already on the market. A single-dose study of 10 mg tofacitinib showed that AUCinf increased 1.37-, 1.43-, and 2.23-fold in patients with mild, moderate, and severe renal impairment, respectively, relative to patients with normal renal function [21]. Dose adjustment of tofacitinib is recommended when used in patients with moderate or severe renal impairment [22, 23]. Similarly, the AUC of baricitinib was increased 1.41-, 2.22- and 4.05-fold for mild, moderate, and severe renal impairment subgroups, respectively, compared with subjects with normal renal function; Cmax was also increased 1.16-, 1.46- and 1.40-fold, respectively, for each of these subgroups [24]. Consequently, dose adjustment of baricitinib is recommended when used in patients with renal impairment, and is not recommended for use in patients with eGFR < 60 mL/min/1.73 m2 (USA) [24] or < 30 mL/min/1.73 m2 (Japan) [25].
A limitation of this study was the relatively small sample of subjects, which may affect the generalisability of the findings. Also, the subjects were mostly male, although there were no notable differences in gender across the renal function groups; a previous pharmacokinetic study in healthy male and female subjects confirmed that gender did not have a significant effect on pharmacokinetic parameters [14]. Lastly, the study did not include patients with end-stage renal disease; therefore, information about peficitinib pharmacokinetics in this group is still lacking. However, this is the first investigation into the impact of renal impairment on peficitinib pharmacokinetics and safety and, as such, it has value for informing clinical practice and ensuring patients are dosed appropriately.
5 Conclusion
The study in non-RA subjects with normal and varying degrees of renal impairment showed that after a single oral dose of 150 mg peficitinib under fasting conditions, the pharmacokinetic parameters were similar. Peficitinib was well tolerated regardless of renal function. Based on these findings, it is not expected that any peficitinib dose adjustment will be required in clinical practice according to the degree of renal impairment.
Data Availability
Access to anonymized individual participant level data will not be provided for this trial as it meets one or more of the exceptions described on http://www.clinicalstudydatarequest.com under “Sponsor Specific Details for Astellas”.
References
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388:2023–38.
Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73:1316–22.
Sokka T, Kautiainen H, Pincus T, Verstappen SMM, Aggarwal A, Alten R, et al. Work disability remains a major problem in rheumatoid arthritis in the 2000s: data from 32 countries in the QUEST-RA study. Arthritis Res Ther. 2010;12:R42.
Matcham F, Scott IC, Rayner L, Hotopf M, Kingsley GH, Norton S, et al. The impact of rheumatoid arthritis on quality-of-life assessed using the SF-36: a systematic review and meta-analysis. Semin Arthritis Rheum. 2014;44:123–30.
Ji J, Zhang L, Zhang Q, Yin R, Fu T, Li L, et al. Functional disability associated with disease and quality-of-life parameters in Chinese patients with rheumatoid arthritis. Health Qual Life Outcomes. 2017;15:89.
Taylor PC, Moore A, Vasilescu R, Alvir J, Tarallo M. A structured literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: a current perspective. Rheumatol Int. 2016;36:685–95.
O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl 2):ii111–5.
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13:234–43.
Nakayamada S, Kubo S, Iwata S, Tanaka Y. Recent progress in JAK inhibitors for the treatment of rheumatoid arthritis. Bio-Drugs. 2016;30:407–19.
Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis. 2016;75:1057–64.
Tanaka Y, Takeuchi T, Tanaka S, Kawakami A, Iwasaki M, Song YW, et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to conventional DMARDs: a randomised, double-blind, placebo-controlled phase III trial (RAJ3). Ann Rheum Dis. 2019;78:1320–32.
Takeuchi T, Tanaka Y, Tanaka S, Kawakami A, Iwasaki M, Katayama K, et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann Rheum Dis. 2019;78:1305–19.
Astellas Pharma Inc. Oral JAK inhibitor Smyraf® tablets approved in Japan for the treatment of rheumatoid arthritis (including prevention of structural joint damage) in patients who have an inadequate response to conventional therapies [Press release]; 2019; https://www.astellas.com/en/news/14651. Accessed 12 June 2019.
Cao YJ, Sawamoto T, Valluri U, Cho K, Lewand M, Swan S, et al. Pharmacokinetics, pharmacodynamics, and safety of ASP015K (peficitinib), a new Janus kinase inhibitor, in healthy subjects. Clin Pharmacol Drug Dev. 2016;5:435–49.
Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, et al. Global prevalence of chronic kidney disease—a systematic review and meta-analysis. PLoS One. 2016;11:e0158765.
Matsuo S, Imai E, Horio M, Yasuda Y, Tomita K, Nitta K, et al. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009;53:982–92.
Japanese Society of Nephrology. Essential points from Evidence-based Clinical Practice Guidelines for Chronic Kidney Disease 2018. Clin Exp Nephrol. 2019;23:1–15.
Oda K, Cao YJ, Sawamoto T, Nakada N, Fisniku O, Nagasaka Y, et al. Human mass balance, metabolite profile and identification of metabolic enzymes of [14C]ASP015K, a novel oral janus kinase inhibitor. Xenobiotica. 2015;45:887–902.
Oda K, Mera K, Nagasaka Y, Tokoro K. Simultaneous determination of a novel oral Janus kinase inhibitor ASP015K and its sulfated metabolite in rat plasma using LC–MS/MS. Biomed Chromatogr. 2015;29:967–9.
Pharmaceuticals and Medical Devices Agency (PMDA). Review Report. Peficitinib. Tokyo; 2019.
Krishnaswami S, Chow V, Boy M, Wang C, Chan G. Pharmacokinetics of tofacitinib, a janus kinase inhibitor, in patients with impaired renal function and end-stage renal disease. J Clin Pharmacol. 2014;54:46–52.
Pfizer Inc. XELJANZ® (tofacitinib) tablet: prescribing information. 2018; http://labeling.pfizer.com/showlabeling.aspx?id=959. Accessed 8 Oct 2019.
Pharmaceuticals and Medical Devices Agency (PMDA). XELJANZ® 5 mg tablets: prescribing information; 2019; http://www.info.pmda.go.jp/go/pack/3999034F1020_1_13/?view=frame&style=XML&lang=ja. Accessed 9 Oct 2019.
Eli Lilly and Company Limited. OLUMIANT® (baricitinib) tablets: prescribing information; 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207924s000lbl.pdf. Accessed 8 Oct 2019.
Pharmaceuticals and Medical Devices Agency (PMDA). OLUMIANT® tablets: prescribing information; 2019. http://www.info.pmda.go.jp/go/pack/3999043F1020_1_06/?view=frame&style=XML&lang=ja. Accessed 9 Oct 2019.
Acknowledgements
The authors would like to thank the investigators, staff, study team and volunteers who were involved in the study.
Author information
Authors and Affiliations
Contributions
Study design: DM, TS, MS, YK, TN, MK and KO. Conduct and acquisition of data: HS, AU and KF. Analysis and interpretation of data: DM, TS, MS, YK, TN, MK and KO. Writing of the manuscript: All authors.
Corresponding author
Ethics declarations
Funding and editorial support
This study was sponsored by Astellas Pharma Inc. Medical writing support was provided by Lisa O’Rourke, PhD, for Cello Health MedErgy (Europe) and funded by Astellas Pharma Inc.
Conflict of interest
DM, TS, MS, YK, TN and MK are employees of Astellas Pharma Inc. KO is an employee of Astellas Research Institute of America LLC. HS, KF and AU have no conflicts of interest to declare.
Ethics approval
The study was reviewed and approved by the Institutional Review Board. The study was conducted in accordance with the International Conference on Harmonization (ICH) guidelines for Good Clinical Practice (GCP), applicable regulations, and guidelines governing clinical study conduct and the ethical principles that have their origin in the Declaration of Helsinki.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Miyatake, D., Shibata, T., Shibata, M. et al. Pharmacokinetics and Safety of a Single Oral Dose of Peficitinib (ASP015K) in Japanese Subjects with Normal and Impaired Renal Function. Clin Drug Investig 40, 149–159 (2020). https://doi.org/10.1007/s40261-019-00873-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-019-00873-7