
Abstract
Background and Objective
Icosapent ethyl is a high-purity prescription form of eicosapentaenoic acid ethyl ester approved as an adjunct to diet to reduce triglyceride levels in adult patients with triglyceride levels ≥500 mg/dL (≥5.65 mmol/L). The objective of this open-label, drug–drug interaction study was to examine the effects of icosapent ethyl on the steady-state pharmacokinetics of atorvastatin, a commonly prescribed medication in patients with dyslipidaemia.
Methods
Thirty healthy subjects received atorvastatin 80 mg/day on days 1–7, icosapent ethyl 4 g/day on days 8–28, and co-administration on days 29–35. Primary end-points were natural log-transformed maximum plasma concentration (C max) and area under the concentration-versus-time curve from 0 to 24 h (AUC0–24) for atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin with and without icosapent ethyl.
Results
Of the 30 subjects enrolled, 26 completed the study. The 90 % confidence intervals for C max and AUC0–24 least-squares geometric mean ratios were within the 0.80–1.25 bounds. Concomitant administration of icosapent ethyl and atorvastatin was safe and well tolerated and icosapent ethyl did not significantly change the steady state C max and AUC0–24 of atorvastatin, 2-hydroxyatorvastatin, or 4-hydroxyatorvastatin.
Conclusions
At steady-state concentrations, icosapent ethyl did not have an effect on the pharmacokinetics of atorvastatin. Co-administration of icosapent ethyl and atorvastatin was safe and well tolerated in healthy adult subjects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Patients receiving icosapent ethyl may also be receiving statin therapy including atorvastatin. |
Icosapent ethyl 4 g/day did not have an effect on the pharmacokinetics of atorvastatin. |
Co-administration of icosapent ethyl and atorvastatin was safe and well tolerated. |
1 Introduction
With the high prevalence of obesity and hypercholesterolaemia in adults in the United States, elevated serum triglycerides is common, although only a small percentage receive specific treatment [1]. In the clinical management of patients with dyslipidaemia, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) are often used and are highly effective for reducing serum cholesterol, especially in low-density lipoproteins, a major risk factor for atherosclerotic cardiovascular disease and the primary target of therapy [2, 3]. However, because statins have only modest triglyceride-lowering effects (10–30 %), hypertriglyceridaemia often persists in these individuals [4]. Additional treatment options to lower persistent triglyceride elevation include lifestyle intervention, fibrates, niacin, ezetimibe, and omega-3 fatty acids [5].
Icosapent ethyl (Vascepa® [formerly AMR101]; Amarin Pharma Inc., Bedminster, NJ, USA) is a high-purity prescription form of the omega-3 fatty acid eicosapentaenoic acid (EPA) ethyl ester approved by the United States Food and Drug Administration (FDA) as an adjunct to diet to reduce triglyceride levels in adults with severe (≥500 mg/dL [≥5.65 mmol/L]) hypertriglyceridaemia [6]. Icosapent ethyl has been studied in individuals with very high serum triglyceride levels (≥500 mg/dL [≥5.65 mmol/L] and ≤2,000 mg/dL [≤22.60 mmo/L]) [7] and, more recently, in statin-treated patients with residual high triglycerides (≥200 mg/dL [≥2.26 mmol/L] and <500 mg/dL [<5.65 mmol/L]) despite having well controlled low-density lipoprotein cholesterol ([LDL-C] ≥40 mg/dL [≥1.04 mmol/L] and <100 mg/dL [<2.59 mmol/L]) [8]. In both of these studies, icosapent ethyl at the approved dose of 4 g/day significantly reduced triglyceride levels and improved other lipid parameters without significantly increasing LDL-C levels [7, 8]. Furthermore, in the study in statin-treated patients, icosapent ethyl 4 g/day significantly decreased LDL-C by 6.2 % [8].
Following oral administration of icosapent ethyl, the active metabolite (EPA) is cleared slowly and is extensively distributed, with steady state for total (includes unesterified EPA and EPA incorporated in phospholipids, triglycerides, and cholesteryl esters) and unesterified plasma EPA reached by day 28 [9]. EPA is mostly metabolized by β-oxidation; but while EPA is also known to be metabolized by cyclooxygenases, lipoxygenases, and cytochrome P450 (CYP) enzymes, CYP-mediated metabolism is a minor pathway of elimination [6, 10]. Consequently, EPA is not expected to exhibit clinically significant drug–drug interactions due to interference with CYP-mediated metabolism of statins. However, the present study was conducted because it was expected that icosapent ethyl would often be co-administered with statins such as atorvastatin.
This study was conducted in healthy adults to examine the effect of icosapent ethyl on steady-state plasma pharmacokinetics of oral atorvastatin, a commonly prescribed statin therapy and a substrate of CYP3A4, and the potential for drug–drug interaction when administered concomitantly.
2 Study Subjects and Methods
2.1 Study Population
Healthy nonsmoking men and women aged 19–55 years were eligible to participate if they had a body mass index >18 and ≤35 kg/m2 and were in good health as determined by medical history, medical examination, and normal test results for serum biochemistry, haematology, and urinalysis at screening. Women who were pregnant, nursing, or planning a pregnancy were excluded; female subjects of childbearing potential were required to use an acceptable method of birth control. Individuals with known hypersensitivity to statins or other lipid-regulating agents were ineligible. All medications or dietary supplements with known or potential lipid-altering effects (including statins, niacin >200 mg/day, fibrates, ezetimibe, bile acid sequestrants, or medications, supplements or foods enriched with omega-3 fatty acids) were prohibited within 4 weeks prior to the first dose of study medication and until after the last pharmacokinetic sample collection. Subjects were required to discontinue any medications or substances known to be inhibitors or inducers of CYP3A4 at least 2 weeks prior to the first dose of study drug, as well as the consumption of fish or foods fortified with EPA and/or docosahexaenoic acid (DHA) at least 1 week prior to the first dose. Use of any medication or dietary supplement that may change serum lipid fractions or triglyceride levels was disallowed.
2.2 Study Design
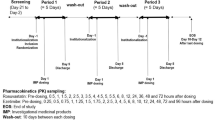
This drug–drug interaction study used an open-label crossover design to evaluate atorvastatin pharmacokinetics at steady state when given alone or with concomitant icosapent ethyl under fasting conditions. The study protocol was approved by an institutional review board (IntegReview Ethics Review Board, Austin, TX, USA) and was conducted between 9 May 2011 and 24 June 2011 at Frontage Clinical Services (a wholly-owned subsidiary of Frontage Laboratories, Inc.), Hackensack, NJ, USA. The study complied with the ethical principles of Good Clinical Practice and in accordance with the Declaration of Helsinki. All subjects provided written informed consent prior to study entry.
Eligibility assessments and clinical laboratory testing were performed within a 28-day screening period. Subjects were housed in the study unit on day 0 (night) through day 1; day 6 (night) through day 8; day 28 (night) through day 29; and day 34 (night) through day 36, during which safety evaluations and pharmacokinetics sampling were performed. Over the course of the 36-day study period, drugs were either administered by study personnel during planned site visits or self-administered by subjects while away from the study site. Compliance (calculated as actual daily dose/planned daily dose × 100) was evaluated at visits 3, 4, and 5 (days 6, 28, and 34) by reconciling the number of unused capsules or tablets against entries in the subject diary and evaluating discrepancies.
Subjects received once-daily oral doses of 80 mg atorvastatin (one tablet), given 1 h prior to the morning meal on days 1–7 and 29–35. On days 8–35, subjects received daily oral doses of 4 g icosapent ethyl (two 1-g liquid-filled gelatin capsules taken twice daily, with or following the morning and evening meals). On days scheduled for pharmacokinetic sampling, study medications were administered after a minimum 10-h overnight fast. All study drugs were taken with 240 mL water. The doses selected for study were based on established pharmacokinetic profiles and represent the maximum recommended clinical dose of atorvastatin and the FDA-approved daily dose of icosapent ethyl capsules [6, 11]. Based on a half-life of 14 h, atorvastatin was administered for 7 days at the maximum recommended dose of 80 mg [11] in order to reach maximal steady-state plasma concentrations of atorvastatin and its metabolites. Based on a half-life of 89 h and 14 days needed to reach steady-state [9], icosapent ethyl was administered for 28 days to ensure maximal steady-state levels of EPA.
2.2.1 Sampling and Bioanalytical Methods
Blood samples (6 mL) for the determination of atorvastatin and its metabolites 2-hydroxyatorvastatin and 4-hydroxyatorvastatin plasma concentrations were obtained on days 1 and 29 at time 0 (prior to dose, in fasting state) and on days 7 and 35 at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, and 24 h after the atorvastatin administration. The protocol did not specify the collection of time 0 pre-dose samples on day 7 or day 35. In order to estimate area under the plasma concentration-versus-time curve to the 24-h time point (AUC0–24), it was assumed that after 7 days of dosing, atorvastatin and its metabolites were at steady state; thus the plasma concentration value at 24 h post-dose for each day was used as the best approximation of time 0 values.
Venous blood samples for measurement of atorvastatin and its metabolites were collected into prechilled glass tubes containing dipotassium ethylenediaminetetraacetic acid and centrifuged (refrigerated; 4–8 °C) for 10 min at 2,000 g within 30 min after collection. Plasma samples were divided into two aliquots of approximately 1.5 mL each, transferred into polypropylene tubes, and stored at or below −20 °C until shipment on dry ice by overnight courier to the bioanalytical laboratory (Frontage Laboratories, Inc., Malvern, PA, USA) for analysis. Plasma concentrations of atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin were measured using a validated liquid chromatography/tandem mass spectrometry procedure. Atorvastatin, its metabolites, and the internal standards were extracted from human plasma by liquid–liquid extraction and separated by reversed-phase high-performance liquid chromatography (HPLC) with a Synergi™ Fusion-RP column (75 × 2 mm, 4 μm; Phenomenex, Torrance, CA, USA) and Shimadzu HPLC pump and autosampler (Shimadzu, Kyoto, Japan), with a flow rate of 0.8 mL/min at room temperature and elution times of 2.2, 2.0, and 1.4 min for atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin, respectively. Mobile phase A was 0.1 % formic acid in H2O and mobile phase B was 0.1 % formic acid and 2 mM ammonium formate in acetonitrile:H2O (98:2, v/v). Atorvastatin-d5, 2-hydroxyatorvastatin-d5, and 4-hydroxyatorvastatin-d5 were used as internal standards and the reference standards were atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin. Ions were monitored for atorvastatin at m/z 559.1–440.1; 2- and 4-hydroxyatorvastatin at m/z 575.1–440.1; atorvastatin-d5 at m/z 564.1–445.1, and 2- and 4-hydroxyatorvastatin-d5 at m/z 580.1–445 in positive ionization mode using the API4000™ mass spectrometer with TurboIonSpray electrospray ion source (AB Sciex, Framingham, MA, USA) at 550 °C and 3,500 V with N2. The dynamic range was 0.2–100 ng/mL for atorvastatin and its metabolites, with a lower limit of quantitation of 0.2 ng/mL. Interconversion between atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin was ≤5 %. The assay accuracy (mean determined concentration/nominal concentration) ranged from 97.0–104.0 % (intra-run) and from 97.5–103.3 % (inter-run). The assay precision (coefficient of variation of the mean determined concentration) ranged from 0.7–4.5 % (intra-run) and from 1.2–3.4 % (inter-run).
2.3 Statistical Methods and Pharmacokinetic Evaluations
Pharmacokinetic parameters were derived by noncompartmental analysis using WinNonlin version 5.0.1 (Pharsight Corporation Inc., Mountain View, CA, USA). The safety population included all subjects who received at least one dose of study drug; the pharmacokinetics analysis population included all subjects with primary pharmacokinetic end-point parameters available from days 7, 8, 35, and 36. The primary pharmacokinetic end-points for determination of drug–drug interaction were natural log (ln)-transformed AUC0–24 and maximum plasma concentration (C max) for atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin when administered at steady state either without or with icosapent ethyl (days 7 and 35, respectively). Subjects who had the protocol-defined primary pharmacokinetic parameters available from both pharmacokinetics days were included in statistical comparisons. A mixed effects analysis of variance model with treatment as a fixed effect and subject as a random effect was performed on natural ln-transformed AUC0–24 and C max parameters. Ratios of least-squares geometric means (LSGM) and corresponding 90 % confidence intervals (CIs) were calculated by exponentiation of the statistical analyses on ln-transformed pharmacokinetic parameters for the two treatments (atorvastatin with icosapent ethyl divided by atorvastatin without icosapent ethyl). A drug–drug interaction was ruled out if the 90 % CIs were within the equivalence limits of 0.80–1.25 [12, 13]. Atorvastatin alone was the reference treatment for statistical comparisons. AUC0–24 was calculated by the linear trapezoidal rule.
Secondary pharmacokinetic end-points included time of observed C max (t max) for atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin when administered at steady state either without or with icosapent ethyl (days 7 and 35, respectively). Time of observed C max was analysed without log-transformation using the nonparametric Wilcoxon signed-rank test. The corresponding 95 % CIs for the difference in medians was reported using the Walsh average and appropriate quantile of the Wilcoxon signed-rank test statistic. A significant difference for the treatment comparison was concluded if the P value was <0.05.
2.4 Safety Assessment
Safety evaluations consisted of monitoring adverse events (AEs; at each study visit after screening), clinical laboratory measurements (chemistry, haematology, and urinalysis; at screening, day 1, day 36, and end of treatment), vital signs (systolic and diastolic blood pressure, heart rate, respiratory rate, and oral body temperature; at each study visit), and physical examination findings (at screening, day 0, day 36, and end of treatment). AEs that occurred during days 1–7 prior to administration of icosapent ethyl were considered emergent to atorvastatin; all other AEs thereafter were considered emergent to administration of atorvastatin + icosapent ethyl. AEs would be considered serious if they resulted in death, were considered to be life-threatening, required hospitalization or prolongation of existing hospitalization, resulted in disability/incapacity, were considered to be a congenital anomaly/birth defect, or were considered to be an otherwise important medical event.
3 Results
3.1 Study Subjects
Thirty healthy adults were enrolled in the study (Table 1), all of whom were exposed to at least one dose of study drug and included in the safety analysis population. All subjects received at least one dose of atorvastatin and 29 subjects received at least one dose of icosapent ethyl. A total of 26 (87 %) subjects completed the study and were included in the pharmacokinetic analysis population. Three subjects withdrew consent and one had noncompliance with dosing requirements. Median compliance for icosapent ethyl (days 8–35) based on capsule counts was 98.1 % and for atorvastatin (days 1–7, 29–35) it was 100.0 %.
3.2 Pharmacokinetic Parameters
Steady-state pharmacokinetic parameters for atorvastatin and its metabolites were calculated based on individual concentration-versus-time profiles obtained when subjects had received seven consecutive daily doses of atorvastatin (days 7 and 35). Mean plasma concentration-versus-time curves for atorvastatin, 2-hydroxyatorvastatin, and 4-hydroxyatorvastatin were comparable for subjects administered atorvastatin alone or with concomitant icosapent ethyl (Fig. 1). Pharmacokinetic parameters were determined for subjects who received atorvastatin and the co-administration of atorvastatin and icosapent ethyl (Table 2). Statistical analysis indicated no significant effect on t max for atorvastatin (P = 0.127), 2-hydroxyatorvastatin (P = 0.372), and 4-hydroxyatorvastatin (P = 0.570) when atorvastatin was administered without or with icosapent ethyl.

Mean (SD) plasma concentration-versus-time curves of a atorvastatin, b 2-hydroxyatorvastatin, and c 4-hydroxyatorvastatin following oral doses of atorvastatin 80 mg/day without and with icosapent ethyl 4 g/day. Day 7 and 35 pre-dose samples were not collected (per protocol); under the assumption that atorvastatin and its metabolites were at steady state, plasma concentration data at 24 h post-dose for each day was used as the best approximation of time 0 values. SD Standard deviation
In the statistical comparisons for the primary analysis, the 90 % CI for the LSGM ratios for atorvastatin C max and AUC0–24 were within the 0.80–1.25 bounds (Table 3). Similar results were observed for 2-hydroxyatorvastatin and 4-hydroxyatorvastatin. Therefore, based upon the 90 % CI for the primary and secondary comparisons, icosapent ethyl dosed at 4 g/day at steady-state concentrations did not affect the pharmacokinetics of the CYP3A4 substrate atorvastatin.
3.3 Safety
Ten subjects (33 %) reported at least one AE during the study. Six (20.0 %) subjects reported at least one treatment-emergent AE (TEAE) during days 1–7 (atorvastatin alone), and seven (24.1 %) subjects reported at least one TEAE after day 7 (atorvastatin plus icosapent ethyl). All TEAEs were mild or moderate in intensity. AEs that occurred in two or more subjects were headache (two subjects [6.7 %] with atorvastatin alone; three subjects [10.3 %] with atorvastatin plus icosapent ethyl) and dizziness (three subjects [10.3 %] with atorvastatin plus icosapent ethyl). No subject prematurely discontinued study treatment due to an AE and no serious AEs were reported. There were no clinically significant changes in laboratory test results, vital sign assessments, or physical examination findings.
4 Discussion
The primary statistical comparisons, which utilized pharmacokinetic parameters for atorvastatin and its metabolites obtained at steady state for both study drugs, demonstrated that oral administration of 4 g/day icosapent ethyl concomitantly with the highest recommended dose of atorvastatin (80 mg/day) did not affect the pharmacokinetics of atorvastatin, a substrate of CYP3A4. C max and AUC0–24 of orally administered atorvastatin and its active metabolites were similar without or with the addition of icosapent ethyl. Both study medications were well tolerated, and no trends toward increased AEs were observed with concomitant treatment in this group of healthy men and women. The results of this study support icosapent ethyl as a potential therapeutic option for adjunct therapy in atorvastatin-treated patients.
Atorvastatin is a commonly prescribed statin therapy in patients with dyslipidaemia. Although this medication is highly effective for reducing serum LDL-C, a substantial proportion of statin-treated patients have persistent hypertriglyceridaemia and may require adjunctive therapy to lower serum triglycerides [2, 14]. Niacin and fibrates can lower serum triglyceride levels, but their use has been limited by tolerability issues, including flushing, myopathy, hepatotoxicity, and hyperglycaemia [4]. A formulation of omega-3 acid ethyl esters, which contains both EPA and DHA, is also approved in the United States for triglyceride lowering in patients with very high serum triglyceride levels [15]. A series of pharmacokinetic drug–drug interaction studies were conducted with this formulation and, consistent with results of the current study, showed a lack of pharmacokinetic drug–drug interaction with various statins, including atorvastatin, in healthy adults [16–18]. The results observed in these studies were similar to findings in the current study with icosapent ethyl.
Recent evidence suggests that formulations of EPA plus DHA may raise LDL-C, which may complicate treatment of patients with hypercholesterolaemia [19–21]. In contrast, icosapent ethyl is a high-purity form of EPA which has been shown to lower triglycerides (without elevating LDL-C) and other lipids at the approved dose of 4 g/day in patients with residual high triglycerides despite optimal statin therapy [8]. The lack of drug–drug interaction with atorvastatin demonstrated in the present study adds to the body of evidence supporting a potential adjunctive strategy in this setting. An ongoing phase III study (REDUCE-IT) [22] is evaluating the effects of concomitant therapy with icosapent ethyl and statins on cardiovascular outcomes in statin-treated patients with hypertriglyceridaemia.
The results presented here for the CYP3A4 substrate atorvastatin extend the findings to date that no clinically significant pharmacokinetic drug–drug interactions have been observed with the CYP2C19, CYP2C8 and CYP2C9 substrates omeprazole, rosiglitazone, and warfarin [6, 23, 24].
5 Conclusions
At steady-state concentrations, icosapent ethyl at the approved dose of 4 g/day did not have an effect on the single-dose pharmacokinetics of atorvastatin administered at its maximal approved dose of 80 mg/day. Co-administration of these drugs was safe and well tolerated in this study of healthy adult subjects.
References
Ford ES, Li C, Zhao G, et al. Hypertriglyceridemia and its pharmacologic treatment among US adults. Arch Intern Med. 2009;169(6):572–8.
Third Report of the National Cholesterol Education Program (NCEP). Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) final report. Circulation. 2002;106(25):3143–421.
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 report on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular disease in adults: full panel report supplement. Circulation. 2014;129(25 Suppl 2):S74–5.
Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969–89.
Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292–333.
Vascepa [package insert]. Bedminster: Amarin Pharma Inc.; 2013.
Bays HE, Ballantyne CM, Kastelein JJ, et al. Eicosapentaenoic acid ethyl ester (AMR101) therapy in patients with very high triglyceride levels (from the Multi-center, plAcebo-controlled, Randomized, double-blINd, 12-week study with an open-label Extension [MARINE] trial). Am J Cardiol. 2011;108(5):682–90.
Ballantyne CM, Bays HE, Kastelein JJ, et al. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study). Am J Cardiol. 2012;110(7):984–92.
Braeckman RA, Stirtan WG, Soni PN. Pharmacokinetics of eicosapentaenoic acid in plasma and red blood cells after multiple oral dosing with icosapent ethyl in healthy subjects. Clin Pharmacol Drug Devel. 2014;3(2):101–8.
Arnold C, Konkel A, Fischer R, et al. Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids. Pharmacol Rep. 2010;62(3):536–47.
Lipitor [package insert]. New York: Parke-Davis; 2012.
Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15(6):657–80.
Guidance for industry. Drug interaction studies—study design, data analysis, and implications for dosing and labeling. US Department of Health and Human Services; Food and Drug Administration. Available at: http://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm. Accessed 14 Oct 2014.
Wong ND, Chuang J, Wong K, et al. Residual dyslipidemia among United States adults treated with lipid modifying therapy (data from National Health and Nutrition Examination Survey 2009–2010). Am J Cardiol. 2013;112(3):373–9.
Lovaza [package insert]. Research Triangle Park: GlaxoSmithKline; 2013.
McKenney JM, Swearingen D, Di SM, et al. Study of the pharmacokinetic interaction between simvastatin and prescription omega-3-acid ethyl esters. J Clin Pharmacol. 2006;46(7):785–91.
Gosai P, Liu J, Doyle RT, et al. Effect of omega-3-acid ethyl esters on the steady-state plasma pharmacokinetics of rosuvastatin in healthy adults. Expert Opin Pharmacother. 2008;9(17):2947–53.
Di Spirito M, Morelli G, Doyle RT, et al. Effect of omega-3-acid ethyl esters on steady-state plasma pharmacokinetics of atorvastatin in healthy adults. Expert Opin Pharmacother. 2008;9(17):2939–45.
Wei MY, Jacobson TA. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: a systematic review and meta-analysis. Curr Atheroscler Rep. 2011;13(6):474–83.
Jacobson TA, Glickstein SB, Rowe JD, et al. Effects of eicosapentaenoic acid and docosahexaenoic acid on low-density lipoprotein cholesterol and other lipids: a review. J Clin Lipidol. 2012;6(1):5–18.
Maki KC, Dicklin MR, Davidson MH, et al. Baseline lipoprotein lipids and low-density lipoprotein cholesterol response to prescription omega-3 acid ethyl ester added to simvastatin therapy. Am J Cardiol. 2010;105(10):1409–12.
A study of AMR101 to evaluate its ability to reduce cardiovascular events in high risk patients with hypertriglyceridemia and on statin (REDUCE-IT). Available at: http://clinicaltrials.gov/show/NCT01492361. Accessed 14 Oct 2014.
Braeckman RA, Stirtan WG, Soni PN. Phase 1 study of the effect of icosapent ethyl on warfarin pharmacokinetic and anticoagulation parameters. Clin Drug Investig. 2014;34(7):449–56.
Braeckman RA, Stirtan WG, Soni PN. Effect of icosapent ethyl (eicosapentaenoic acid ethyl ester) on omeprazole plasma pharmacokinetics in healthy adults. Drugs R D. 2014;14(3):159–64.
Acknowledgments
This study was designed and sponsored by Amarin Pharma Inc., Bedminster, NJ, USA.
Drs. Braeckman and Soni are former employees of Amarin Pharma Inc. and were employed by Amarin Pharma Inc. during the planning, execution, and manuscript preparation of this study. Medical writing assistance was provided by Elizabeth Daro-Kaftan, PhD, of Peloton Advantage, LLC, Parsippany, NJ, USA, and funded by Amarin Pharma Inc., Bedminster, NJ, USA.
Financial disclosure
Dr. Stirtan is an employee and stock shareholder of Amarin Pharma Inc. Drs. Braeckman and Soni are former employees and current stock shareholders of Amarin Pharma Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Braeckman, R.A., Stirtan, W.G. & Soni, P.N. Effect of Concomitant Icosapent Ethyl (Eicosapentaenoic Acid Ethyl Ester) on the Pharmacokinetics of Atorvastatin. Clin Drug Investig 35, 45–51 (2015). https://doi.org/10.1007/s40261-014-0252-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-014-0252-8