
Abstract
The current problems with increasing bacterial resistance to antibacterial therapies, resulting in a growing frequency of incurable bacterial infections, necessitates the acceleration of studies on antibacterials of a new generation that could offer an alternative to antibiotics or support their action. Bacteriophages (phages) can kill antibiotic-sensitive as well as antibiotic-resistant bacteria, and thus are a major subject of such studies. Their efficacy in curing bacterial infections has been demonstrated in in vivo experiments and in the clinic. Unlike antibiotics, phages have a narrow range of specificity, which makes them safe for commensal microbiota. However, targeting even only the most clinically relevant strains of pathogenic bacteria requires large collections of well characterized phages, whose specificity would cover all such strains. The environment is a rich source of diverse phages, but due to their complex relationships with bacteria and safety concerns, only some naturally occurring phages can be considered for therapeutic applications. Still, their number and diversity make a detailed characterization of all potentially promising phages virtually impossible. Moreover, no single phage combines all the features required of an ideal therapeutic agent. Additionally, the rapid acquisition of phage resistance by bacteria may make phages already approved for therapy ineffective and turn the search for environmental phages of better efficacy and new specificity into an endless race. An alternative strategy for acquiring phages with desired properties in a short time with minimal cost regarding their acquisition, characterization, and approval for therapy could be based on targeted genome modifications of phage isolates with known properties. The first example demonstrating the potential of this strategy in curing bacterial diseases resistant to traditional therapy is the recent successful treatment of a progressing disseminated Mycobacterium abscessus infection in a teenage patient with the use of an engineered phage. In this review, we briefly present current methods of phage genetic engineering, highlighting their advantages and disadvantages, and provide examples of genetically engineered phages with a modified host range, improved safety or antibacterial activity, and proven therapeutic efficacy. We also summarize novel uses of engineered phages not only for killing pathogenic bacteria, but also for in situ modification of human microbiota to attenuate symptoms of certain bacterial diseases and metabolic, immune, or mental disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Recent progress in phage genetic engineering enables deliberate modifications of natural phage isolates to make them more suitable for therapeutic applications by modifying their host specificity and/or improving other properties on demand. |
Panels of genetically engineered phages with improved therapeutic properties, increased safety and differentiated specificity, based on the genomic scaffolds of selected well characterized phage isolates, and the ability to acquire them rapidly at a relatively low cost may soon lead to therapeutic applications. |
Progress in the understanding of the phage–bacteria interactions in the human/animal microbiome and their influence on health opens up possibilities for the application of genetically engineered phages to modify the microbiome composition and thereby attenuate symptoms of certain bacterial diseases as well as selected metabolic, mental, or immune disorders. |
1 Introduction
Until recently, the human population had been decimated for ages by epidemics of bacterial diseases and wound infections, infections during childbirths, food poisoning, and diarrhea. In 1900, the average lifespan was 31 years, and even in the richest countries, it did not exceed 50 years. The discovery of antibiotics and their introduction into medical practice in the first half of the 20th century is estimated to have increased the average lifespan by 23 years and is considered the greatest medical breakthrough of the last century [1]. However, the wide use of antibiotics in medicine, veterinary practice, animal production, and agriculture has brought about the emergence and spread of antibiotic-resistant bacterial pathogens, leading to a rising frequency of incurable bacterial infections and causing an estimated 700,000 deaths each year worldwide [2,3,4]. The most promising candidates for agents that can replace antibiotics, support their action, or revert the antibiotic resistance phenotypes are bacteriophages (phages)—viruses that specifically infect bacteria and are harmless to eukaryotic cells [5, 6]. They have been regaining interest in recent years, not only due to their proven potential in controlling bacteria, but also owing to their contribution to bacterial adaptation to diverse environments [7,8,9], their abundance and unprecedented diversity [10,11,12], their major impact on nearly all environments and forms of life [13,14,15,16,17], and the recently expanded possibilities of their targeted modification [18, 19].
The bacterial killing potential of phages, which was appreciated at the time of their discovery, is also the major driving force that has rejuvenated interest in phages today. However, the viral nature of phages makes regulations developed for chemically defined antibacterial drugs inapplicable to phages without substantial changes approved by authorized institutions [20,21,22,23,24]. This requires time. Despite that, the constantly growing incidence of prolonged, chronic, or incurable infections by antibiotic-resistant bacteria, coupled with the amassing experimental and clinical data proving the antibacterial activity and safety of phages in vivo, even upon intravenous application, urges studies on phages by themselves [25,26,27,28,29,30,31,32,33,34]. But first of all, it prompts the development of new solutions that would allow humanity to benefit from the antibacterial activity of phages in medical practice. Moreover, recent genomic and metagenomic discoveries and progress in genetic engineering and nanotechnology allow one to see the possibility of therapeutic phage applications much broader than previously anticipated.
Phages are the most abundant entities on Earth, with an estimated number of nearly 1031, exceeding that of bacterial cells by about one order of magnitude [12, 35, 36]. However, only recently has a comprehensive insight into the phageomes of various environments, including human and animal skin, gastrointestinal tract, and other body niches, become possible [17]. During that time, the number of sequenced phage genomes has been rising nearly exponentially [37]. According to the most recent classification of the International Committee for Taxonomy of Viruses, bacterial viruses are divided into 19 families [38,39,40]. The natural phage isolates selected for therapeutic use belong to the order Caudovirales (tailed phages), which currently includes ten families [38, 39]. Their virions consist of a head (capsid), packed with linear double-stranded DNA (dsDNA), and a tail. The end of the tail contains proteins allowing phage adsorption to its specific host bacterium and its penetration through the bacterial cell envelope to introduce the phage DNA [41].
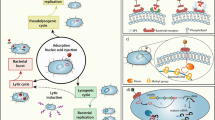
All phages can be seen as delivery vectors. Any genetic material packed within their capsid can be delivered to the phage-specific host and initiate the development of progeny phages and cell lysis or participate in recombination, transcription, or other processes leading to a phenotype change or death of the infected cell. This opens up various possibilities of phage genome modifications that either do not interfere with the natural processes of phage propagation and packaging or require in vitro packaging of the phage DNA. Some changes in the phage DNA may be designed to modify the phage capsid so that it gains affinity to certain bacterial or eukaryotic cells that normally are not the parental phage targets [42,43,44]. If capsids of such phages are additionally packed with cytotoxin-encoding genes or filled with other cytotoxic compounds or even a recombinant eukaryotic virus genome, they can serve as delivery vectors of their content when, e.g., engulfed by their new target cells [45,46,47,48,49,50,51]. Finally, the external structure of a phage virion, which in the simplest phages is composed of only a few types of proteins, enables the use of phages as molecular scaffolds for chemical modifications to conjugate them with already approved drugs (e.g., [52]; reviewed in [53]). An additional advantage of some phages is their ability to penetrate epithelial cell layers and to cross the intestine–blood or blood–brain body barriers, making them promising drug delivery platforms [54,55,56,57,58,59]. The methods of phage genome modification differ in complexity and depend on the developmental strategies of the phage of interest (depicted in Fig. 1).

Developmental strategies of tailed and filamentous phages. Objects in the scheme are not to scale
2 Engineered Phages as Future Therapeutic Options: Rationale and Perspectives
Traditionally, phages are considered potential antibacterial agents that are an alternative to antibiotics. Their advantages over antibiotics include a narrow specificity range preventing the undesired killing of commensal microbiota, the ability to multiply at the site of their host location as long as the host persists, the ability to evolve in response to host evolution, and the general lack of adverse effects on human health (reviewed in [29, 60, 61]). Most importantly, phages can cure infections with antibiotic-sensitive as well as antibiotic-resistant bacterial strains and can also be successfully used in combination with antibiotics [62]. Metagenomic data on the human and animal microbiota expand the possibilities of the therapeutic use of phages even further by demonstrating the previously unsuspected influence of the phageome on the quantitative and qualitative microbiome composition, which is critical for health and may lead to autoimmune, metabolic, or mental diseases when disturbed [63,64,65,66,67,68,69,70,71,72,73].
It is generally believed that environmental phage population is so numerous and diversified that there exists a phage for each and every bacterial strain. However, to be considered for therapeutic applications, natural phage isolates must meet certain criteria [74,75,76,77,78,79]. For obvious reasons the choice is limited to phages that are obligatorily lytic to kill every infected bacterium, do not transfer bacterial DNA, and do not encode any toxins, virulence factors, or antibiotic resistance determinants. Additionally, each phage for therapy should be well characterized at the genomic and proteomic level, as well as at the level of interaction with its host(s) and the host's host. It should efficiently lyse the target bacteria, have the widest possible strain range within a pathogenic species, be sufficiently stable under storage conditions and at the sites of infection by the bacteria, be able to overcome at least some bacterial phage-resistance mechanisms, and cause no undesired immune reactions. Although hundreds of phages targeting bacterial pathogens have been isolated (e.g., [80,81,82,83,84,85,86,87,88,89,90,91]), none of them fulfills all these criteria, and in fact, none has been characterized with respect to all of them. First, many if not most DNA phages are temperate (Fig. 1) [72, 92,93,94,95,96]. They can stay in the infected bacterium in the form of DNA (a prophage) without causing its lysis and typically encode bacterial adaptive functions, among them toxins, virulence determinants, and regulators of bacterial pathogenicity [97,98,99,100,101,102,103]. Also certain obligatorily lytic phages encode distant homologs of virulence determinants whose functions must be determined before considering such phages for medical use (see, e.g., [104]). Second, the temperate and some obligatorily lytic phages can pack bacterial DNA to their capsids in place of or in addition to their own DNA and inject it into newly infected bacteria, thereby spreading the pathogenicity determinants [98, 105,106,107,108,109]. Third, even phages with a wide strain range within a pathogenic species typically do not infect all the strains of this species, and the variability of phage receptors and anti-phage defense systems among the bacteria make this one of the most challenging problems in the development of phage preparations for therapy [110,111,112,113,114,115,116]. Numerous anti-phage defense systems act after phage DNA injection and inactivate the infecting phage DNA by cleavage (e.g., restriction-modification systems and clustered regularly interspaced palindromic repeats [CRISPR]-CRISPR-associated proteins [Cas] systems) or activate a suicidal host response or dormancy to prevent the infection spread (e.g., toxin–antitoxin [TA] systems, abortive infection systems). There is a multitude of such systems, with different structural and functional variants, and the mechanisms of action of several of them await elucidation (reviewed in [115, 117]). Moreover, new discoveries constantly expand their list. Additionally, a phage treatment of sensitive bacteria may select for resistance, and there are no standard methods to prevent the development of resistance [115, 118,119,120]. What is more, the emergence of pathogenic strains among commensal bacteria, e.g., Staphylococcus epidermidis [121], requires the use of phages that can selectively kill these strains while being safe for the non-pathogenic ones. A major problem in the introduction of phages to medical use is also the limited knowledge of their coding potential and biology. Only a few phages infecting bacteria from about a dozen genera, mostly used for model studies, have been analyzed in detail with respect to their molecular features, physiology, and interaction with the bacteria, and even for these phages the functions of about half of their genes remain unknown [122, 123].
A reasonable strategy to deal with the problem of narrow strain specificity could be the development of well characterized, single-phage preparations targeting various strains of a pathogenic species and mixing them on demand, depending on the phage sensitivity profile of the infecting bacteria [124]. Such cocktails could be adapted to a given pathogenic strain or enriched with new environmental phage isolates when needed. Although such a strategy fits the current trend to personalize medicine and has not only been widely discussed but also successfully used [28], it is commercially unattractive, as custom-made phage preparations cannot be currently approved for medical use [21, 125, 126]. Moreover, even mixtures of phages of different specificity usually do not exhibit a universal strain range [21, 91, 127,128,129], and searching for natural phages of the missing specificity is tedious, time consuming, and often either fails or leads to the isolation of only temperate phages [21, 90]. Even the so called adaptation of phages to infect resistant strains by selection of natural phage mutants has limited potential in the acquisition of phages of new specificities [76, 120, 130,131,132,133].
In view of the aforementioned problems, it is obvious that the repertoire of natural phages suitable for therapy, the possibility of ensuring their antibacterial efficacy and safety in vivo, and licensing their medical use are limited. A strategy to overcome at least some of these problems could instead rely on the construction of desired therapeutic phages by engineering selected, well characterized, natural phage isolates [122]. The introduction of genetically engineered (GE) phages into medical practice could have several advantages (Table 1).
Moreover, the recent progress in the understanding of phages and in the methods of their genetic modification may soon make construction of engineered phages a routine procedure rather than a tedious scientific undertaking. Additionally, the new discoveries concerning the interaction of phages with bacteria inside the human body and the effect of phages on human health via their influence on the microbiome open up new possibilities of therapeutic application of modified phages not only to treat infections with bacterial pathogens, but also to improve the conditions of patients suffering from diverse disorders related to a dysfunctional microbiome, by in situ microbiome engineering [69, 71–73, 156,157,158,159].
3 Phage Genetic Engineering: Classical and Modern Technologies
The acquisition of a genetically modified phage comprises two steps: (1) in vivo or in vitro replacement of the entire phage genome or its fragment with a modified one, and (2) recovery of the modified phage. Several methods of phage genome modification have been developed, but each of them has limitations and cannot be used for each and every phage (Table 2).
In the case of filamentous phages whose replicative forms (RFs) are circular dsDNA plasmids, this can be done by in vitro manipulation of the RFs and transfection of the resulting DNA to relevant host cells (see Sect. 7 of this review). A similar approach was used in early studies to modify an integration-deficient mutant of the temperate phage lambda depleted of certain restriction sites and of a large dispensable fragment of its 49-kbp genome [160]. Two essential restriction fragments of the mutant were ligated with a heterologous DNA fragment. The ligation product served to produce modified progeny phages upon transfection of Escherichia coli cells. Lambda-derived vectors with a single cloning site or with two sites flanking the dispensable DNA segment to be replaced by an intended insert were constructed in a similar way [161,162,163]. They were used, e.g., to study functions of bacterial as well as phage genes [164]. Hybrids between lambda and E. coli phages P1, T4, and T5 or Bacillus phage SPP1 constructed in that way were used in studies on the gene functions of these phages [165,166,167,168,169,170,171,172,173,174]. Overcoming the problem of low transfection efficiency of engineered lambda derivatives became possible with the development of an in vitro lambda packaging system (reviewed by Chauthaiwale et al. [164]). In this system, the lambda cos sequences, which separate the phage genome units in a substrate for DNA packaging and are cut to form cohesive ends, can serve as in vitro packaging initiation and termination signals for any 38- to 51-kbp intervening sequence. The implementation of lambda in vitro packaging was broadened by the construction of cosmids—plasmids containing a drug resistance marker, two cos sequences, and a cloning site designed for the insertion of DNA fragments of 40- to 50-kbp [163, 175]. The first E. coli–Mycobacterium shuttle plasmid, which can replicate in E. coli as a plasmid and propagate in Mycobacterium as a phage, was constructed by the insertion of a cosmid into a non-essential ~ 50-kbp region of mycobacteriophage DNA [176]. A similar shuttle plasmid was used to construct an engineered mycobacterial phage carrying a luciferase reporter gene to detect mycobacteria [177, 178]. While this strategy allows the introduction of modified mycobacterial phage DNA into E. coli cells by infection with in vitro-packed lambda virions and allows it to be isolated as a plasmid, it has serious limitations and thus has not been widely used. The recovery of a functional phage from the target bacterium requires the target to be efficiently transformed with large DNA molecules, and only functionally active phages could be recovered. Additionally, the possible expression of bactericidal and bacteriotoxic phage genes by such constructs excludes the use of this strategy for phages of bacteria more closely related to E. coli than Mycobacterium. Moreover, the use of lambda virions as delivery vectors for heterologous phage DNA for E. coli requires the availability of the lambda in vitro packaging system and experience in its use, which may be problematic in some laboratories.
More common methods of engineering phages of linear dsDNA genomes and a complex virion structure use homologous recombination in vivo (Fig. 2). The earliest ones were based on phage infection of a bacterium with a fragment of the phage DNA (donor DNA) carrying a desired modification and cloned in a plasmid [18, 179,180,181]. Recombinants were subsequently recovered from among progeny phages. If any host specificity-determining gene was exchanged into one from a different phage, they could even be directly selected as forming plaques on the new host [182, 183]. However, the use of such methods is possible only when the phage host can be transformed with the plasmid and when a restriction-deficient host mutant is available, as in the case of, e.g., E. coli K-12 or Pseudomonas aeruginosa PAO1 and also Mycobacterium smegmatis. While temperate phages modified with this technique can be recovered by polymerase chain reaction (PCR) screening of single plaques following induction of lysogens, in the case of obligatorily lytic phages that degrade host DNA at infection, the frequency of recombination is extremely low. The recovery of recombinants may be facilitated by incorporating a marker gene into the donor DNA, allowing phenotypic differentiation of modified phage plaques [184], or better, a specific host gene if such is required for phage development [185,186,187]. In the latter case, an appropriate bacterial mutant has to be used to obtain only recombinant phages.

In vivo homologous recombination-based methods of bacteriophage genome modification. Proteins depicted on the scheme of recombineering represent phage λ proteins that were originally used in this method [191]. Bet and its homologs/analogs are sufficient for recombineering with ssDNA substrates as donors (reviewed in [188]). Phage proteins depicted on the scheme of BRED represent Escherichia coli prophage Rac proteins RecE and RecT, whose mycobacterial phage Che9c homologs (gp60 and gp61) were originally used in this method [198]. Recombination with the use of RecE and RecT does not require Gam (reviewed in [188]). Bet and RecT phage λ or prophage Rac single-strand annealing proteins, BRED bacteriophage recombineering of electroporated DNA, Cas CRISPR-associated proteins, Cas9 nuclease Cas9, CRISPR clustered regularly interspaced palindromic repeats, crRNA CRISPR RNA, dsDNA double-stranded DNA, Exo and RecE phage λ or prophage Rac 5′-3' exonucleases, respectively, that degrade dsDNA to expose the created 3' single-stranded ends for binding of Bet or RecT (respectively), Gam phage λ protein increasing recombination frequency by inhibiting dsDNA substrate degradation by E. coli RecBCD, PCR polymerase chain reaction, RecA bacterial recombinase RecA, ssDNA single-stranded DNA
A technique that allows increasing the in vivo recombination frequency is based on the use of linear donor DNA and phage lambda recombination proteins Gam, Bet, and Exo, which protect linear dsDNA from host exonucleases (Gam) and efficiently promote homologous recombination between linear donor DNA and homologous target DNA [188]. This technique, known as recombineering and independent of host recombination functions, was initially applied to modify the E. coli chromosome, plasmids, and temperate phages [189,190,191,192,193]. It also increases the frequency of recombinant formation upon lytic phage infection of cells containing the donor DNA in a plasmid or in the form of a linear fragment, but a successful recovery of recombinants requires the development of their direct selection method [194, 195]. Recombineering allows the recombination to occur in E. coli with homologous DNA ends as short as 50 bp [196]. Its further modification, known as bacteriophage recombineering of electroporated DNA (BRED), was developed to facilitate the modification of obligatorily lytic phages and lytic mutants of temperate phages [197, 198]. In BRED, phage DNA and a linear fragment of homologous DNA with a desired change are electroporated together into a phage-sensitive host cell containing E. coli phage lambda or prophage Rac proteins (Exo and Bet, and RecE and RecT, respectively), or their homologs from other phages [199,200,201]. Recombinant phages are recovered upon cell lysis and infection of a sensitive host by PCR screening of plaques. However, the need for bacteria to be electroporated to carry a plasmid expressing the recombination-promoting proteins limits the application of this method to transformation-proficient bacterial strains for which relevant plasmids are available. BRED has been used for engineering of lytic phages infecting, e.g., E. coli, M. smegmatis, and Salmonella enterica, giving the recovery efficiency of recombinant phage of ca. 20%, which allowed for their identification by PCR plaque screening [199, 200, 202].
If the BRED method cannot be used and the duration of the phage and donor DNA co-existence in the cell is short, the problem of an extreme scarcity of recombinants can be circumvented by applying a counter selection of phages that did not acquire the intended modification. This can be done with the use of CRISPR-Cas systems [203,204,205]. These systems, identified in ~ 42% of bacteria, protect their native hosts from reinfection with foreign DNA [206, 207]. The CRISPR regions consist of 28- to 37-bp repeats of a conserved sequence interspersed with short spacers, which are a bank of immunological memory that stores DNA fragments from past invaders (plasmids or phages) [208]. The CRISPR regions are linked to cas genes encoding proteins responsible for RNA-guided recognition and cleavage of invading DNA sequences corresponding to the spacer regions. The guiding RNA fragments (CRISPR RNA [crRNA]) are generated by chopping CRISPR-region transcripts into units including a spacer and an upstream repeat sequence, which serves to distinguish between host and non-host sequences [209,210,211]. To achieve the elimination of unmodified phages, the cell to be infected has to contain (1) a plasmid carrying a phage genome donor fragment with the desired modification and (2) a single CRISPR repeat followed by a spacer sequence representing the wild-type phage fragment to be replaced by the donor DNA and followed by the cas gene(s) of a given CRISPR-Cas system. [203, 212, 213]. The latter can be provided on a plasmid or on the chromosome. Only recombinant phages containing the donor sequence in place of the wild-type DNA, formed via homologous recombination by the host-encoded machinery, will be resistant to crRNA-guided degradation. A customized CRISPR-Cas system from Streptococcus pyogenes, which requires only one protein (Cas9) for crRNA processing and target recognition and cleavage [214], has successfully been used to introduce point mutations, deletions, or insertions to the genomes of obligatorily lytic phages infecting Gram-negative as well as Gram-positive hosts [187, 205, 213,214,215,216,217,218]. A customized CRISPR-Cas10 system from S. epidermidis, which requires a protein complex to perform its editing function and can target only actively transcribed sequences, has been used to modify the genomes of S. epidermidis and Staphylococcus aureus lytic phages ranging in size from 18 to 148 kbp [210, 219]. The CRISPR-Cas systems allow a single-step introduction of traceless changes in phage DNA with high efficiency (40–100%) without additional screening or selection procedures [213, 215, 220].
The above methods are not applicable to phages infecting only bacteria with active restriction systems or bacteria for which efficient transformation procedures have not been developed. Additionally, none of them can be used to simultaneously edit multiple unlinked loci in a phage genome. The methods that were developed recently and revolutionized the strategy of phage genome modifications use wall-less bacteria or yeast cells to reboot the genomes of obligatorily lytic phages constructed from DNA fragments [122, 221, 222]. Jaschke et al. [222] assembled from PCR-amplified or chemically synthesized DNA fragments an engineered 5.4-kbp genome of phage φX174. The engineered DNA was ligated with a yeast plasmid vector and transformed to yeast cells. Transformants selected for the presence of the recombinant plasmid were used to isolate large quantities of plasmid DNA, which was then restriction digested to recover the phage genome. The obtained phage DNA was circularized by ligation and used to transfect cells of an E. coli strain that is normally resistant to φX174 infection, but can propagate the phage. The progeny phages could infect a φX174 sensitive host. Ando et al. [221] extended this strategy to phage T7 and its relatives whose genomes are in the range of 37–45 kbp. They used the natural ability of yeast cells to assemble linear dsDNA fragments with homologous termini into a single molecule by recombination [223]. Complete phage genomes inserted into a yeast mini-chromosome (YAC) were obtained upon transformation of yeast cells with several PCR-amplified phage DNA fragments of up to 11 kbp overlapping at the ends, and with PCR-amplified YAC DNA carrying regions of homology to the ends of the phage DNA to be assembled [221]. Swapping the genomic fragments encoding receptor-binding proteins (RBPs) or their fragments between T7 and related phages specific for different bacterial hosts allowed the construction of hybrids with a single genomic scaffold and small regions encoding specificity determinants and with different host specificity. A functional Salmonella phage FelixO1 of an 86.2-kbp genome could also be recovered from assembled genomic fragments cloned in a YAC in yeast cells [224].
Although the use of yeast cells for the recovery of engineered phage genomes has several advantages over other methods and allows constructing fully synthetic phage genomes, its disadvantage is the necessity to transform bacteria with the phage genomic DNA isolated from yeast cells to acquire functional phages. This limits the use of this method to bacteria that can be made highly competent for the uptake of large DNA molecules.
A strategy to overcome this obstacle has been developed by Kilcher et al. [225] and used to engineer temperate as well as obligatorily lytic phages with genomes of up to 154 kbp infecting Listeria monocytogenes or related Gram-positive hosts. The genomes with designed modifications were assembled enzymatically in vitro into circular forms from PCR-amplified and/or synthetic DNA fragments with overlapping ends with the Gibson method [226]. They were then used to transform so called L-forms of specific L. monocytogenes strain cells, which are deprived of the cell wall, can grow in osmotically stabilized media, and can be transformed with large DNA molecules [225]. Phages produced in the L-form cells were released by hypotonic lysis and used to infect their specific bacterial host to propagate. Surprisingly, the L-forms of L. monocytogenes could be used not only to reboot efficiently the synthetic genomes of Listeria phages, but also those of Bacillus and Staphylococcus phages, indicating a general applicability of L-form bacteria for phage genome engineering.
While the propagation of a complete engineered phage can be initiated by a single infection of a sensitive host, the dependence of such phage acquisition on living cells poses a problem. These obstacles have been overcome recently by the development of new-generation cell-free transcription and translation systems (TXTLs) that enable the synthesis of complete phages (virions) in the test tube using a suitable phage genome as a template and E. coli cytoplasmic extract enriched with certain components [227,228,229]. E. coli phages of diverse genomes, such as MS2 (3.6 kb, RNA), φX174 (5.4 kb, single-stranded DNA [ssDNA]), and T7 (40 kbp, dsDNA), could be synthesized in this way, with efficiency reaching about 0.5, 0.6, and 2.2 phages per genome copy, respectively [230]. This method appeared to be suitable even for the synthesis of the large lytic phage T4 with a 170-kbp genome, albeit with a trace efficiency only (0.001 phages per genome). Although a cell-free T4 reconstruction has so far been demonstrated from a wild-type T4 genome only, the method is suitable for the acquisition of engineered phages as well, provided that the engineered variants are functional and can be recovered upon infection of the phage host. A recently developed cell-free, de novo phage genome assembly technology from chemically synthesized oligonucleotides, which was shown to allow the assembling of a 4.3-kbp Acinetobacter phage genome at a cost below $0.015 per bp [231], may soon enable various variants of engineered phage genomes to be obtained on demand and at a low price. High throughput systems to transform such synthetic genomes into appropriate cells to recover functional phages (when possible) may soon speed up the functional analysis of phage genes as well as the acquisition of phages with desired properties for therapeutic use.
4 Phage Genetic Engineering to Modify Phages for Traditional Phage Therapy
The validity of the idea of using genomes of well characterized obligatorily lytic phages as scaffolds to construct their derivatives with desired properties with the help of genetic engineering has been proven in several studies. Numerous features of phages constructed based on such scaffolds appeared to be modifiable, with no or just slight interference with other phage properties. For instance, an engineered phage T7 expressing the biofilm-degrading enzyme dispersin reduced biofilm cell counts with an efficacy two orders of magnitude higher than the wild-type phage [232]. An engineered virulent Erwinia amylovora phage Y2 with a wide strain range that was enriched with a gene encoding the exopolysaccharide depolymerase of phage L1, showed increased infectivity and killing efficacy and the ability to reduce the E. amylovora contamination of flowers as compared to the wild-type phage [233]. Phage T7 engineered to display lipids on the capsid showed a significantly increased stability in simulated gastrointestinal tract conditions, indicating possible directions to obtaining therapeutic phages for oral delivery [234]. Phage T7 enriched with a gene encoding lactonase, an enzyme that degrades acyl homoserine lactone (a quorum-sensing signaling molecule required for biofilm formation by bacteria of many species), inhibited the biofilm formation when added to mixed-species biofilms containing E. coli and P. aeruginosa [235].
The toxin release caused by the lytic action of a phage may cause serious side effects during phage therapy. Therefore, a phage eliminating target bacteria without causing their lysis would alleviate these concerns. Paul et al. [236] developed a recombinant endolysin-deficient staphylococcal phage unable to cause lysis, but still lethal to methicillin-resistant S. aureus. The recombinant phage was highly efficient in rescuing mice infected with a lethal dose of S. aureus. In similar experiments, lysis-deficient phages infecting P. aeruginosa or E. coli provided better protection to mice from a lethal dose of these bacteria than did their wild-type counterparts [237, 238]. In all the above cases, the phages suitable for therapy could be efficiently propagated in hosts complementing their endolysin function to obtain high titer lysates.
One of the most challenging features of known phages suitable for antibacterial therapies is their limited host range. The mechanisms determining the susceptibility of bacteria to a given phage are highly differentiated, and the list of known ones is constantly expanding [112, 115, 239,240,241]. The initiation of phage infection requires recognition and binding to surface receptors on the host cell. Mapping of the variable domains of phage RBPs, which form tail fibers or tail spikes [242, 243], allows targeted mutagenesis of their genes or shuffling between phages of different host specificities to generate phage variants infecting a wider or changed repertoire of hosts without compromising their lytic activity [182, 221, 243,244,245,246,247]. Moreover, the determination of intradomain structures of RBPs allows an antibody-engineering-like approach to change the structure of their fragments interacting with the host receptors to fit them to, e.g., mutationally changed receptors conferring to bacteria resistance to a given phage [248]. Selected phages infecting E. coli, Klebsiella pneumoniae, Yersinia pseudotuberculosis, P. aeruginosa, or Acinetobacter baumannii, the model E. coli phages T2, T4, and T7 among them, have already been used as scaffolds to construct variants with modified host specificity [18, 241]. Engineering the RBPs may be especially useful for rapid acquisition of variants of well characterized phages capable of infecting emerging pathogens of new serotypes. For instance, a derivative of phage T2 infecting E. coli O157:H7 was constructed by replacing genes encoding the tip of the T2 long tail fiber with those of a new phage isolate specific for this E. coli serotype [246].
Temperate phages have generally been seen as unsuitable for traditional phage therapy due to their adaptive effects on the bacteria they infect (including pathogens) and their participation in horizontal gene transfer [74]. However, animal studies indicate that such phages can also have therapeutic potential [249, 250]. Moreover, phages of an obligatorily lytic phenotype derived from temperate phages by spontaneous mutations have been identified among environmental isolates or upon induction of lysogens [251,252,253]. The use of temperate phages in therapy need not be considered when lytic phages of a desired host specificity are available. If this is not the case, obligatorily lytic phages of the missing specificities can be obtained from appropriate temperate phages by the removal from their genomes of genomic modules essential for lysogeny and, if present, also those encoding virulence determinants [225, 250, 254, 255]. Such modified phages, although not the first choice, can be further engineered to enhance their lytic potential or broaden the host range. An advantage of temperate phages is the easiness of their genetic modifications in the lysogenic state, when their DNA is integrated with the bacterial chromosome. Several temperate phages modified to deprive them of the ability to lysogenize bacteria appeared to be effective as targeted antibacterials in in vitro and in vivo experiments [255]. For instance, the temperate Burkholderia siphovirus KS9 converted to a lytic variant by disrupting the phage repressor gene rescued Galleria mellonella infected experimentally with Burkholderia cenocepacia [250]. A recombinant of a temperate Enterococcus faecalis phage with an E. faecalis prophage, which was converted to a lysogeny-deficient variant insensitive to the phage repressor by the removal or modification of lysogeny-associated genome modules, markedly decreased the residual E. faecalis population and the biofilm biomass of both vancomycin-sensitive and resistant strains in extracted human dentin root segments [254, 256, 257]. A derivative of L. monocytogenes PSA phage depleted of genes essential for lysogeny and enriched with an additional endolysin gene from a different Listeria phage formed larger plaques than the original phage and also could lyse cells of a PSA-resistant serovar of Listeria [225].
The first case of a human phage therapy with GE phages was described by Dedrick et al. (2019) [152]. A cystic fibrosis patient underwent lung transplantation, which was complicated by Mycobacterium abscessus infection resistant to multiple antibiotics administered intravenously. Of the hundreds of mycobacterial phages tested, only one efficiently killed the infecting strain, and two others were temperate and inefficient in the infection. A lytic derivative of one of the latter obtained by genetic engineering gained high lytic activity against the infecting M. abscessus by the acquisition of a host range mutation, and an efficient lytic derivative of the second one was constructed with BRED to remove its phage repressor gene precisely [152, 258]. The three-phage cocktail (including two temperate phages engineered to become effective lytically) was administered every 12 h intravenously for 32 weeks. The treatment was well tolerated, with no major side effects, evidence of organ toxicity, or significant increase in the level of pro-inflammatory cytokines, allowing the patient to be discharged home while still on phage therapy. The therapy resulted in significant clinical improvement and eradication of infection. Phages could be detected in the patient’s serum 1 day after therapy initiation, reaching a titer of ≥ 109 PFU/ml, and became undetectable 6 days later. Although no formal proof of the phage therapy effectiveness has been provided (it was an uncontrolled study, during which the patient was also receiving other treatments, e.g., antibiotics, which could have contributed to the final outcome), the results of this experimental use of GE phages in human treatment indicated that (1) a prolonged intravenous administration of GE phage is possible without harm to the patient, while producing high therapeutic phage levels in the serum, suggesting phage multiplication in vivo, and (2) even intravenous phage administration in a human patient does not provoke antibody production that could interfere with its therapeutic efficacy. The experiment also confirmed earlier data from patients who received wild-type phages by oral or topical administration [259].
An important question is whether phages can kill intracellular bacteria. Phage genetic engineering may help to resolve this problem. Møller-Olsen et al. [260] used CRISPR-Cas-based selection to obtain a GE derivative of a T7-like phage, K1F, equipped with a gene for green fluorescence protein (GFP). The GFP-labeled phage showed some delay in infection as compared to wild-type K1F [261]. However, tracing K1F-GFP by fluorescence microscopy demonstrated its ability to penetrate human urinary bladder epithelial cells and human cerebral microvascular cells. Moreover, the phage was able to kill inside human cells a hybrid between E. coli strains K12 and K1, the latter being a nosocomial pathogen responsible for urinary tract infections, meningitis, and sepsis [260, 261].
5 Future Possibilities to Extend the Host Range of Phages by Genetic Engineering
Attempts to construct phages with extended host range by genetic engineering have been focused on manipulations with phage genes encoding RBPs, as summarized in Sect. 4 [244]. However, the phage host range depends not only on the phage’s ability to penetrate bacterial extracellular polymers or to interact with a bacterial cell receptor and inject the phage DNA, but also on phage-encoded mechanisms overcoming the post-infective bacterial anti-phage defense. Their multitude seems to correspond to the multitude of bacterial anti-phage defense mechanisms [115]. The anti-restriction and anti-CRISPR mechanisms are the best known. The former operate by avoiding relevant restriction sites in phage DNA, titrating out restriction enzymes, physically protecting phage DNA from host restrictase access, modifying the phage DNA, or stimulating the host DNA-modifying enzymes to make the DNA resistant to restriction [115, 262,263,264,265,266,267,268,269]. The anti-CRISPR defense systems act by inducing the degradation of Cas nucleases, disruption of DNA binding, or inhibition of target cleavage [270,271,272,273]. Numerous anti-CRISPR (Acrs) proteins identified so far comprise at least 46 families, and a machine-learning in silico approach allowed the identification of at least 2500 additional candidates [274,275,276,277]. When a gene encoding an Acr protein(s) is present in a phage or cloned in a plasmid in a cell with its target CRISPR-Cas system, it can suppress the action of this system [274, 277]. This opens wide possibilities to construct GE phages containing appropriate acr genes able to overcome specific types of acquired CRISPR-Cas-mediated immunity and may counteract the immunity to the phage acquired by bacteria during phage therapy [271]. Similarly, the incorporation into the phage genome of genes encoding specific anti-restriction proteins could extend the host range of such GE phages to the hosts whose restriction-modification systems would otherwise prevent the infection when the ratio of infecting phages to bacteria is not high enough to break the restriction-modification barrier.
Some phages encode homologs/analogs of antitoxins of certain bacterial TA systems [119, 278, 279]. They were shown or predicted to replace bacterial antitoxins whose supply upon phage infection could stop, leading to the death or dormancy of the infected cell. The enrichment of GE phages with genes encoding antitoxins of their host TA systems could increase the therapeutic efficacy of such phages by enabling their lytic development and facilitating their spread at the infection site.
Bacterial anti-phage defense genes including those that encode restriction-modification, TA, and CRISPR-Cas systems tend to cluster in so called defense islands [280, 281]. The systematic, ongoing analysis of other genes of these islands has brought about the discovery of new anti-phage defense systems of surprisingly differentiated mechanisms of action and high protective efficacy [117, 282,283,284,285,286,287]. Some of them stem from the same evolutionary origin as anti-viral systems of eukaryotic cells and can be complemented by relevant eukaryotic genes [287,288,289,290]. The design of therapeutic GE phages of wide strain range will benefit from future identification of phage strategies to evade these newly discovered anti-phage defense systems.
The least known repertoire of genes that could extend the host range of GE phages when inserted to their genomes comprises phage genes whose products help to evade prophage-encoded anti-phage defense mechanisms that protect lysogens from superinfection by other phages, especially unrelated ones. While a few examples of such gene products have a long history of study, how numerous and diversified the mechanisms of action of the remaining ones are and how many various stages of phage infection and development they can interfere with is only beginning to be discovered (for an extensive review, see [115]). They are typically encoded by accessory genes of temperate phages and show no homology to proteins of known function, and often only the conserved location of their genes in the genomes of related phages can serve as a guide for their identification [291, 292]. While some of them provide protection against multiple phages, others have been found to be specific only for one of the phages tested [291,292,293,294]. Notably, lysogeny is common in clinically relevant bacteria [102, 295]. Therefore, discovery and understanding of the prophage-encoded anti-phage defense system and identification of phage-encoded mechanisms to overcome them may appear to be critical for successful development of GE phages with wide strain specificity highly effective therapeutically.
6 New Directions in Phage Genetic Engineering to Expand the Possibilities of Phage Therapy
Phage replication and bacterial cell lysis, which accompany a productive phage infection and are considered to be advantages of traditional phage therapy, can be considered to be disadvantageous as well. Lysis leads to the release of bacterial toxins and can potentially cause undesired inflammatory reactions, while phage replication during therapy may raise concerns about further uncontrolled phage spread. Thus, there is an increasing interest in the use of phages to deliver to bacteria a lethal or regulatory load inserted in the phage genome by genetic engineering to achieve a therapeutic effect while attenuating lysis of bacteria, preventing phage replication, or both. Temperate phages are naturally suited for this purpose. However, obligatorily lytic phages that are defective in lytic development in a non-laboratory host can be used as well.
Among the most promising lethal loads that can specifically eradicate bacteria of certain species or strains within complex microbial communities in a sequence-specific manner without causing their massive lysis are programmable CRISPR-Cas-based gene cassettes. In a pioneering experiment, Bikard et al. [296] used an engineered temperate S. aureus φMN1 phage derivative to deliver to its host bacteria a CRISPR-Cas module with spacers targeting certain S. aureus virulence genes. The GE phage selectively killed only the S. aureus strains carrying the targeted genes when used to infect a mixed S. aureus population of virulent and avirulent strains in laboratory culture or on the skin of infected mice. Similar φMN1 derivatives targeting antibiotic resistance determinants selectively killed antibiotic-resistant S. aureus strains in a mixed population. Additionally, by lysogenization of the antibiotic susceptible cells, the phage made them immune to the acquisition of the resistance-conferring genes. Park et al. [218] integrated a CRISPR-Cas9 system targeting the nuc gene, unique to S. aureus, into the genome of a temperate S. aureus phage. To broaden the host range of the GE phage, the strain for its propagation was transformed with a plasmid carrying the tail fiber genes of a broad-range S. aureus phage. The GE phage produced in this strain showed high in vitro efficacy in eliminating S. aureus and was effective in vivo in reducing the bacterial burden of infected mouse skin. A similar approach gave promising results in a rat model of soft tissue infection where the reduction of S. aureus burden by a GE phage was similar to that obtained with a high-dose antibiotic treatment [297]. Likewise, a temperate Clostridium difficile phage was engineered by removing key lysogeny genes and arming it with a CRISPR module targeting a C. difficile genomic sequence [298]. The lytic phage obtained reduced the C. difficile burden in vivo and decreased disease severity.
In addition to targeted killing of bacteria without their lysis, the concept of using phages as delivery vectors of specifically designed genetic load opens up the possibility of modifying human or animal microbiota phenotypically, in a non-lethal way. With the increasing knowledge about the human microbiome, its association with health and diseases, and the molecular mechanisms of bacterial pathogenicity, microbiome modifications may turn out to be more effective in the long run than killing of the bacteria [299]. For instance, a temperate GE phage carrying a transcriptional repressor of Shiga toxin (Stx) gene was successfully used to repress Stx in an established E. coli population colonizing the gut in mice. The phage propagated in the gut and markedly reduced the fecal Stx concentration [300]. A novel approach to fight antibiotic-resistant bacteria with the use of a temperate GE phage was developed by Edgar et al. [301]. Lambda-derived phages carrying dominant alleles of respective genes causing sensitivity to streptomycin or nalidixic acid reverted the antibiotic resistance phenotypes caused by recessive mutations in those genes in the bacteria.
An innovative approach to deliver any programmed genetic load to bacteria by phages was proposed by Yosef et al. [302]. The method is based on the transducing capabilities of phages and not only prevents phage propagation in the target bacteria, but also allows the spectrum of target bacteria to be extended to those that can be infected but do not support phage propagation. Its utility was demonstrated with the use of T7-derived defective phages, whose certain essential functions, including tail fiber genes, were provided in trans from a plasmid carried by the phage-propagating host. The genetic load to be delivered to the target cells was provided in a separate plasmid, which also contained a site required to initiate DNA packaging to phage heads. Initial experiments on the delivery of an antibiotic resistance marker to evaluate the frequency of transduction demonstrated the great potential of this strategy for therapeutic applications upon its further development. Moreover, mutagenesis of the tail fiber genes present in the plasmid complementing the lacking functions of the phage could extend the phage host range. An additional extension of the host range for transduction can be achieved by epigenetic modifications of the DNA to be transduced to prevent its restriction in recipient cells [303].
7 Engineered Filamentous Phages in Therapies of Bacterial Infections
While the phages used in traditional phage therapy have a complex virion structure and encode from 20 to over 300 different functions, at least half of them unknown, genetic engineering has opened up the possibility of modifying phages of simple structure and smaller genomes for therapeutic use. Filamentous phages, which are workhorses in the phage display technology, appear to be the most promising candidates for this purpose [95, 304, 305]. They do not lyse bacteria, but are constantly secreted by infected cells, thereby decreasing the host growth rate (Fig. 1). Their virions, composed of a few proteins only, have the form of thin filaments 6–7 nm in diameter. Their length is not as tightly controlled as is the size of icosahaedral phage capsids, allowing the insertion of more additional DNA. The filamentous phage genome, typically comprising 11 genes, is an ssDNA molecule in the virion, but assumes a double-stranded (dsDNA) circular RF during phage propagation. It can be isolated from host bacteria by methods used for plasmid isolation, then engineered in vitro and introduced back to the cells by transformation. If it contains all the genes necessary for phage propagation and release, it can initiate the production of mature phages. Plasmids that contain an origin of dsDNA replication and an origin for ssDNA replication derived from a filamentous phage (so called phagemids) can be packed in the single-stranded form into filamentous phage virions if the other phage functions are provided in trans by a helper plasmid [306]. Filamentous phages adsorb to retractable bacterial pili and have a narrow host range [95, 305]. However, parts of their RBPs can be engineered to adsorb into bacteria of different species by replacement with suitable parts of RBPs from other filamentous phages infecting those species [307]. Alternatively, the specificity of filamentous phages can be redirected towards bacteria that are not their natural hosts by phage display-based selection of GE phages exposing peptides that bind those bacteria or that bind antibodies against them. Yacoby et al. [308] engineered M13-derived phages to bind S. aureus cells and conjugated them chemically with chloramphenicol. The S. aureus-killing efficacy of the phage-conjugated chloramphenicol was several orders of magnitude higher than that of free drug.
Filamentous phage capsids packed with phagemid DNA can deliver their genetic load to the infected cells to kill them or to modify their metabolism. For instance, M13-derived phagemids engineered to encode antimicrobial peptides (AMPs) and toxins disrupting diverse bacterial intracellular processes caused rapid, non-lytic death of bacteria in laboratory culture and were effective in vivo in a murine peritonitis infection model [309]. Another M13-derived GE phagemid specifically killed its host E. coli cells by delivering genes encoding bactericidal proteins [310]. A filamentous phage of the P. aeruginosa PAO1 strain engineered by replacing its export protein gene with a restriction endonuclease gene could not replicate in the absence of a complementing export gene nor could lyse cells, but caused chopping of their chromosomes, leading to bacterial death without a massive release of endotoxin or a significant inflammatory response [311]. More importantly, it could rescue mice infected with a lethal dose of PAO1 [237].
Lu and Collins [312] engineered an M13-based phagemid to overproduce the repressor protein of the E. coli SOS response system, which activates the pathways of oxidative DNA damage repair, e.g., in response to certain bactericidal antibiotics such as quinolones, exerting their action by inducing DNA damage and leading to cell death. Inhibiting the E. coli SOS system with such a phage enhanced the bactericidal action of quinolones in vitro. Moreover, the phage increased survival of mice with an E. coli bacteremia. It also enhanced the elimination of antibiotic-resistant bacteria, biofilm cells, and persister cells and reduced the emergence of antibiotic-resistant cells in an antibiotic-treated bacterial population. Similar effects were achieved with phages overproducing regulators of certain non-SOS gene networks, indicating the efficacy of engineered phages targeting certain genomic networks as adjuvants of antibiotic therapies. To treat Chlamydia trachomatis, an obligate intracellular pathogen, a filamentous phage was engineered to express an integrin-binding peptide (inducing phage endocytosis) and a conserved peptide from this pathogen interfering with its propagation. The engineered phage significantly reduced C. trachomatis infection in HeLa cells and primary human endocervical cells [313].
Peng et al. [314] have applied photothermal ablation of bacteria as a modification of phage therapy. Chimeric E. coli filamentous phages engineered to display RBPs of filamentous phages targeting bacteria of other genera were conjugated to gold nanorods (to form so called phanorods) [307, 314]. Upon excitation with near-infrared light, such phanorods release energy, and the resulting heat kills targeted bacteria. Moreover, phanorod irradiation efficiently eliminated bacterial cells within a biofilm, causing only minimal damage to epithelial cells.
It has also been recognized that arming filamentous phages with specific peptides offers a novel antigen delivery system for the development of new vaccine formulations. Such phages engineered to target mouse dendritic cells can strongly activate the innate and adaptive immune responses excluding the need for adjuvants. A recent article provides an overview of potential uses of such “nature-made nanoparticles” [58].
8 Engineering Phages to Control Phage Pharmacokinetics
The control of phage pharmacokinetics by engineering relies on modifying phage interactions with the immune system as the major factor determining the phage fate in vivo [315,316,317,318,319,320]. Typical modifications involve making virions ‘invisible’ to the immune system, mostly to its innate part, which is capable of removing phages without their specific recognition. Chemical modifications and microencapsulation, for instance, PEGylation or lyposomal preparations, have been used to help the phage escape filtration and metabolism in the human/animal organism. This approach will not be presented here in more detail as it does not involve specific engineering of the phage; an expert review of this topic has been published recently by Malik et al. [321].
Compared to chemical modifications, site-directed engineering of phage proteins seems less straightforward in terms of its effect on phage pharmacokinetics. In fact, effective control of the phage circulation in vivo by molecular modifications has not been achieved so far. This is probably due to the complexity of phage virions, which consist of a multitude of proteins, each potentially interacting with the phage-treated organism and affecting phage pharmacokinetics. Phages are very large as compared to conventional drugs, and this seems to be a major factor determining phages’ ability to circulate in vivo, rather than details of phage molecular structures [320]. Nevertheless, there are reports indicating that phage pharmacokinetics in animals and humans can in fact be modified (if not fully controlled) by molecular modifications of the phage capsid. Sokoloff et al. [322,323,324] used phage display to study interactions of an engineered T7 phage with the rat and human innate immunity system. Their study focused on the complement system, which is a major component of humoral (non-cellular) innate immunity. Due to its wide interactions with other elements of the immune system, both non-specific, like phagocytes, and specific, like IgM antibodies, modifying phage interactions with the complement system may affect its interactions with other types of immune response. The authors assumed that the amino acid composition of the phage surface can make the phage less or more reactive to the complement system. Indeed, they demonstrated that presenting peptides with the C-terminal lysine or arginine on the capsid changed the phage phenotype into long-circulating in rats [322]. That finding was consistent with earlier observations of Merril and coworkers who isolated from rabbits spontaneous long-circulating mutants of phage λ with the glutamic acid to lysine substitution in a part of the major capsid protein presumably located at the solvent-exposed surface in mature virions [325, 326]. They proposed that the long-circulating phenotype could be linked to lower susceptibility of the mutated phage to be captured by the mononuclear phagocytic system (referred to by them as the reticulo-endothelial system). An alternative explanation could be that the long-circulating phage was less susceptible to inactivation by the complement system, since both Sokoloff et al. [322] and Vitiello et al. [326] found that the exposure of lysine or arginine on the phage head surface facilitated phage escape from the innate immune response.
It seems likely that at least some phages whose life cycles are bound with animal- or human-associated bacteria have adapted to the complement system by minimizing their recognizability. In such cases, further improvement of the phage resistance to the immune system via genetic engineering may prove challenging, as was found by Hodyra-Stefaniak et al. [327]. In that work, coliphage T4 was engineered by phage display to present specific tissue-homing peptides expected to induce phage accumulation in specific targeted tissues (organs). However, the concentration of the engineered phages in the targeted organs was not increased, and in some cases, it was even lower than that of the wild-type phage. An analysis of the systemic pharmacokinetics of the phages revealed that their low concentration in the targeted organs was due to a lowered systemic concentration. Thus, it turned out that the engineered phages were removed from the system more rapidly than the non-engineered one, i.e., they exhibited a short-circulating phenotype. Analysis of the modifications introduced revealed that all the peptides conferring the short-circulating phenotype lacked arginine, which was present in all the others. That correlation seemed to confirm the role of arginine in protecting the phage from rapid elimination, although a similar role of lysine was not observed in that study. Apparently, engineering destroyed evolutionarily optimized properties of the phage, making it more susceptible to the immune system. In general, a short-circulating phenotype is unfavorable in therapeutic applications of phages, since their rapid elimination from the treated organism may prevent them from combating the bacterial infection [320].
9 Limitations of Phage Genetic Engineering, Potential Risks, and Future Challenges
The increasing interest in the multifarious modifications making phages safer and more suitable for defined therapeutic applications on demand has contributed to the rapid progress in the development of phage genetic engineering. While technically, phage genome modifications should soon become possible in any laboratory, phage genetic engineering has its limits [18]. Some of them result from the varying nature of phages. First, in the case of most phages, except for those with filamentous virions, the size of the genome is limited by capsid capacity. This limits the modifications to incorporation of only small fragments of extra DNA or replacement of original genomic fragments with fragments of similar size. Second, certain changes in the phage genome sequence or organization, or in the virion structure, may negatively influence phage infectivity or development [328,329,330,331]. For instance, chemical PEGylation of surface proteins of two obligatorily lytic myophages infecting Salmonella or Listeria species, which prolonged blood circulation time of those phages in mice, also decreased their infectivity [332].
The use of GE phages in therapy has raised concerns about potential effects of their release into the environment [333]. Whether such concerns are justified has not been verified yet. GE phages could influence bacterial community dynamics, genome evolution, and biogeochemistry. However, the small scale of potential use of GE phages as compared to antibiotics limits the potential risks of their spread. Nair and Khairnar [333] have asked an important question of whether the concerns associated with GE microorganisms are also relevant for GE phages. The GE phages depleted of lysogeny-associated or other undesired genes can potentially recombine with other phages, but they cannot spread such genes in the environment. Additionally, modifications of phage genomes have been shown to be associated with a fitness cost that can lead to a loss of function in GE phages or to their outcompetition by native phages [328,329,330,331, 334, 335]. However, some modified phages of reduced fitness could recover the fitness of their original phage through compensatory evolution even in less than a hundred generations. An additional concern is posed by possible interactions of GE phages with eukaryotic cells. Contrary to earlier assumptions, data accumulating in the last few years suggest such a possibility, albeit its significance is unclear at this moment [336]. It is thus obvious that despite the highly encouraging results of experimental therapeutic applications of GE phages, further progress in the development of antibacterials based on GE phages as well as wild-type phages will depend on progress in the understanding of phage structure, biology, and interactions with their hosts and other organisms.
References
Hutchings MI, Truman AW, Wilkinson B. Antibiotics: past, present and future. Curr Opin Microbiol. 2019;51:72–80. https://doi.org/10.1016/j.mib.2019.10.008.
Ventola CL. The antibiotic resistance crisis: part 1: causes and threats. P T. 2015;40(4):277–83.
Arias CA, Murray BE. Antibiotic-resistant bugs in the 21st century–a clinical super-challenge. N Engl J Med. 2009;360(5):439–43. https://doi.org/10.1056/NEJMp0804651.
Ruckert A, Fafard P, Hindmarch S, et al. Governing antimicrobial resistance: a narrative review of global governance mechanisms. J Public Health Policy. 2020;41(4):515–28. https://doi.org/10.1057/s41271-020-00248-9.
Ghosh C, Sarkar P, Issa R, Haldar J. Alternatives to conventional antibiotics in the era of antimicrobial resistance. Trends Microbiol. 2019;27(4):323–38. https://doi.org/10.1016/j.tim.2018.12.010.
Saha D, Mukherjee R. Ameliorating the antimicrobial resistance crisis: phage therapy. IUBMB Life. 2019;71(7):781–90. https://doi.org/10.1002/iub.2010.
Hambly E, Suttle CA. The viriosphere, diversity, and genetic exchange within phage communities. Curr Opin Microbiol. 2005;8(4):444–50. https://doi.org/10.1016/j.mib.2005.06.005.
Koskella B, Taylor TB. Multifaceted impacts of bacteriophages in the plant microbiome. Annu Rev Phytopathol. 2018;56:361–80. https://doi.org/10.1146/annurev-phyto-080417-045858.
Fernández L, Rodríguez A, García P. Phage or foe: an insight into the impact of viral predation on microbial communities. ISME J. 2018;12(5):1171–9. https://doi.org/10.1038/s41396-018-0049-5.
Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, et al. Uncovering Earth’s virome. Nature. 2016;536(7617):425–30.
Suttle CA. Environmental microbiology: Viral diversity on the global stage. Nat Microbiol. 2016;1(11):16205. https://doi.org/10.1038/nmicrobiol.2016.205.
Mushegian AR. Are there 1031 virus particles on earth, or more, or fewer? J Bacteriol. 2020;202:9. https://doi.org/10.1128/JB.00052-20.
Suttle CA. Marine viruses—major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–12. https://doi.org/10.1038/nrmicro1750.
Rohwer F, Thurber RV. Viruses manipulate the marine environment. Nature. 2009;459:207–12. https://doi.org/10.1038/nature08060.
Evans C, Brussaard CPD. Regional variation in lytic and lysogenic viral infection in the Southern Ocean and its contribution to biogeochemical cycling. Appl Environ Microbiol. 2012;78:6741–8. https://doi.org/10.1128/AEM.01388-12.
Carding SR, Davis N, Hoyles L. Review article: the human intestinal virome in health and disease. Aliment Pharmacol Ther. 2017;46(9):800–15. https://doi.org/10.1111/apt.14280.
Dion MB, Oechslin F, Moineau S. Phage diversity, genomics and phylogeny. Nat Rev Microbiol. 2020;18(3):125–38. https://doi.org/10.1038/s41579-019-0311-5.
Pires DP, Cleto S, Sillankorva S, Azeredo J, Lu TK. Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol Rev. 2016;80(3):523–43. https://doi.org/10.1128/MMBR.00069-15.
Chen Y, Batra H, Dong J, Chen C, Rao VB, Tao P. Genetic engineering of bacteriophages against infectious diseases. Front Microbiol. 2019;10:954. https://doi.org/10.3389/fmicb.2019.00954.
Abdelkader K, Gerstmans H, Saafan A, Dishisha T, Briers Y. The preclinical and clinical progress of bacteriophages and their lytic enzymes: the parts are easier than the whole. Viruses. 2019;11(2):96. https://doi.org/10.3390/v11020096.
Pirnay JP, Verbeken G, Ceyssens PJ, et al. The magistral phage. Viruses. 2018;10(2):64. https://doi.org/10.3390/v10020064.
Pelfrene E, Sebris Z, Cavaleri M. Comment on Fauconnier, A. Phage therapy regulation: from night to dawn. Viruses. 2019;11(9):771. https://doi.org/10.3390/v11090771.
Fauconnier A. Phage therapy regulation: from night to dawn. Viruses. 2019;11(4):352. https://doi.org/10.3390/v11040352.
Hesse S, Adhya S. Phage therapy in the twenty-first century: Facing the decline of the antibiotic era; is it finally time for the age of the phage? Annu Rev Microbiol. 2019;73:155–74. https://doi.org/10.1146/annurev-micro-090817-062535.
Sybesma W, Rohde C, Bardy P, Pirnay JP, Cooper I, Caplin J, Chanishvili N, Coffey A, De Vos D, Scholz AH, McCallin S, Püschner HM, Pantucek R, Aminov R, Doškař J, Kurtbӧke Dİ. Silk route to the acceptance and re-implementation of bacteriophage therapy-Part II. Antibiotics (Basel). 2018;7(2):35. https://doi.org/10.3390/antibiotics7020035.
Furfaro LL, Payne MS, Chang BJ. Bacteriophage therapy: clinical trials and regulatory hurdles. Front Cell Infect Microbiol. 2018;8:376. https://doi.org/10.3389/fcimb.2018.00376.
Jennes S, Merabishvili M, Soentjens P, Pang KW, Rose T, Keersebilck E, et al. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury—a case report. Crit Care. 2017;21:129. https://doi.org/10.1186/s13054-017-1709-y.
Schooley RT, Biswas B, Gill JJ, et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob Agents Chemother. 2017;61(10):e00954-e1017. https://doi.org/10.1128/AAC.00954-17.
Międzybrodzki R, Borysowski J, Weber-Dąbrowska B, et al. Clinical aspects of phage therapy. Adv Virus Res. 2012;83:73–121. https://doi.org/10.1016/B978-0-12-394438-2.00003-7.
Rubalskii E, Ruemke S, Salmoukas C, et al. Bacteriophage therapy for critical infections related to cardiothoracic surgery. Antibiotics (Basel). 2020;9(5):E232. https://doi.org/10.3390/antibiotics9050232.
Mendes JJ, Leandro C, Corte-Real S, et al. Wound healing potential of topical bacteriophage therapy on diabetic cutaneous wounds. Wound Repair Regen. 2013;21(4):595–603. https://doi.org/10.1111/wrr.12056.
Seth AK, Geringer MR, Nguyen KT, et al. Bacteriophage therapy for Staphylococcus aureus biofilm-infected wounds: a new approach to chronic wound care. Plast Reconstr Surg. 2013;131(2):225–34. https://doi.org/10.1097/PRS.0b013e31827e47cd.
Fish R, Kutter E, Wheat G, Blasdel B, Kutateladze M, Kuhl S. Compassionate use of bacteriophage therapy for foot ulcer treatment as an effective step for moving toward clinical trials. Methods Mol Biol. 2018;1693:159–70. https://doi.org/10.1007/978-1-4939-7395-8_14.
Pires DP, Costa AR, Pinto G, Meneses L, Azeredo J. Current challenges and future opportunities of phage therapy. FEMS Microbiol Rev. 2020;44(6):684–700. https://doi.org/10.1093/femsre/fuaa017.
Bergh O, Børsheim KY, Bratbak G, Heldal M. High abundance of viruses found in aquatic environments. Nature. 1989;340(6233):467–8. https://doi.org/10.1038/340467a0.
Wommack KE, Colwell RR. Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev. 2000;64(1):69–114. https://doi.org/10.1128/mmbr.64.1.69-114.2000.
Kropinski AM. Bacteriophage research—what we have learnt and what still needs to be addressed. Res Microbiol. 2018;169(9):481–7. https://doi.org/10.1016/j.resmic.2018.05.002.
Adriaenssens EM, Wittmann J, Kuhn JH, et al. Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch Virol. 2018;163(4):1125–9. https://doi.org/10.1007/s00705-018-3723-z.
Adriaenssens EM, Sullivan MB, Knezevic P, et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch Virol. 2020;165(5):1253–60. https://doi.org/10.1007/s00705-020-04577-8.
Mäntynen S, Sundberg LR, Oksanen HM, Poranen MM. Half a century of research on membrane-containing bacteriophages: bringing new concepts to modern virology. Viruses. 2019;11(1):76. https://doi.org/10.3390/v11010076.
Ackermann HW. Tailed bacteriophages: the order Caudovirales. Adv Virus Res. 1998;51:135–201. https://doi.org/10.1016/s0065-3527(08)60785-x.
Dąbrowska K, Kaźmierczak Z, Majewska J, et al. Bacteriophages displaying anticancer peptides in combined antibacterial and anticancer treatment. Future Microbiol. 2014;9(7):861–9. https://doi.org/10.2217/fmb.14.50.
Shoae-Hassani A, Mortazavi-Tabatabaei SA, Sharif S, Madadi S, Rezaei-Khaligh H, Verdi J. Recombinant λ bacteriophage displaying nanobody towards third domain of HER-2 epitope inhibits proliferation of breast carcinoma SKBR-3 cell line. Arch Immunol Ther Exp (Warsz). 2013;61(1):75–83. https://doi.org/10.1007/s00005-012-0206-x.
Peng H, Chen IA. Phage engineering and the evolutionary arms race. Curr Opin Biotechnol. 2020;68:23–9. https://doi.org/10.1016/j.copbio.2020.09.009.
Poul MA, Marks JD. Targeted gene delivery to mammalian cells by filamentous bacteriophage. J Mol Biol. 1999;288(2):203–11. https://doi.org/10.1006/jmbi.1999.2678.
Phumyen A, Jantasorn S, Jumnainsong A, Leelayuwat C. Doxorubicin-conjugated bacteriophages carrying anti-MHC class I chain-related A for targeted cancer therapy in vitro. Onco Targets Ther. 2014;7:2183–95. https://doi.org/10.2147/OTT.S69315.
Park J, Chariou PL, Steinmetz NF. Site-specific antibody conjugation strategy to functionalize virus-based nanoparticles. Bioconjug Chem. 2020. https://doi.org/10.1021/acs.bioconjchem.0c00118.10.1021/acs.bioconjchem.0c00118.
Kolesanova EF, Melnikova MV, Bolshakova TN, Rybalkina EY, Sivov IG. Bacteriophage MS2 as a tool for targeted delivery in solid tumor chemotherapy. Acta Naturae. 2019;11(2):98–101. https://doi.org/10.32607/20758251-2019-11-2-98-101.
Garg P. Filamentous bacteriophage: a prospective platform for targeting drugs in phage-mediated cancer therapy. J Cancer Res Ther. 2019;15(Supplement):S1–10. https://doi.org/10.4103/jcrt.JCRT_218_18.
Hwang YJ, Myung H. Engineered bacteriophage T7 as a potent anticancer agent in vivo. Front Microbiol. 2020;11:491001. https://doi.org/10.3389/fmicb.2020.491001.
Przystal JM, Waramit S, Pranjol MZI, et al. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol Med. 2019;11(4):e8492. https://doi.org/10.15252/emmm.201708492.
Vaks L, Benhar I. In vivo characteristics of targeted drug-carrying filamentous bacteriophage nanomedicines. J Nanobiotechnology. 2011;9:58. https://doi.org/10.1186/1477-3155-9-58.
Sokullu E, Soleymani Abyaneh H, Gauthier MA. Plant/bacterial virus-based drug discovery, drug delivery, and therapeutics. Pharmaceutics. 2019;11(5):211. https://doi.org/10.3390/pharmaceutics11050211.
Nguyen S, Baker K, Padman BS, et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. MBio. 2017;8(6):e01874-17. https://doi.org/10.1128/mBio.01874-17.
Międzybrodzki R, Kłak M, Jończyk-Matysiak E, et al. Means to facilitate the overcoming of gastric juice barrier by a therapeutic staphylococcal bacteriophage A5/80. Front Microbiol. 2017;8:467. https://doi.org/10.3389/fmicb.2017.00467.
Schubbert R, Lettmann C, Doerfler W. Ingested foreign (phage M13) DNA survives transiently in the gastrointestinal tract and enters the bloodstream of mice. Mol Gen Genet. 1994;242(5):495–504. https://doi.org/10.1007/BF00285273.
Frenkel D, Solomon B. Filamentous phage as vector-mediated antibody delivery to the brain. Proc Natl Acad Sci USA. 2002;99(8):5675–9. https://doi.org/10.1073/pnas.072027199.
Sartorius R, D’Apice L, Prisco A, De Berardinis P. Arming filamentous bacteriophage, a nature-made nanoparticle, for new vaccine and immunotherapeutic strategies. Pharmaceutics. 2019;11(9):437. https://doi.org/10.3390/pharmaceutics11090437.
Apawu AK, Curley SM, Dixon AR, et al. MRI compatible MS2 nanoparticles designed to cross the blood-brain-barrier: providing a path towards tinnitus treatment. Nanomedicine. 2018;14(7):1999–2008. https://doi.org/10.1016/j.nano.2018.04.003.
Loc-Carrillo C, Abedon ST. Pros and cons of phage therapy. Bacteriophage. 2011;1(2):111–4. https://doi.org/10.4161/bact.1.2.14590.
Vandenheuvel D, Lavigne R, Brüssow H. Bacteriophage therapy: advances in formulation strategies and human clinical trials. Annu Rev Virol. 2015;2(1):599–618. https://doi.org/10.1146/annurev-virology-100114-054915.
Abedon ST. Phage-antibiotic combination treatments: antagonistic impacts of antibiotics on the pharmacodynamics of phage therapy? Antibiotics (Basel). 2019;8(4):182. https://doi.org/10.3390/antibiotics8040182.
Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018;24:392–400. https://doi.org/10.1038/nm.4517.
Barko PC, McMichael MA, Swanson KS, Williams DA. The gastrointestinal microbiome: a review. J Vet Intern Med. 2018;32:9–25. https://doi.org/10.1111/jvim.14875.
Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. 2016;167:915–32. https://doi.org/10.1016/j.cell.2016.10.027.
Martens EC, Neumann M, Desai MS. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat Rev Microbiol. 2018;16:457. https://doi.org/10.1038/s41579-018-0036-x.
Manrique P, Bolduc B, Walk ST, Oost J, Vos WM, Young MJ. Healthy human gut phageome. Proc Natl Acad Sci USA. 2016;113:10400–5. https://doi.org/10.1073/pnas.1601060113.
Dalmasso M, Hill C, Ross RP. Exploiting gut bacteriophages for human health. Trends Microbiol. 2014;22:399–405. https://doi.org/10.1016/j.tim.2014.02.010.
Stern J, Miller G, Li X, Saxena D. Virome and bacteriome: two sides of the same coin. Curr Opin Virol. 2019;37:37–43. https://doi.org/10.1016/j.coviro.2019.05.007.
Ruff WE, Greiling TM, Kriegel MA. Host-microbiota interactions in immune-mediated diseases. Nat Rev Microbiol. 2020;18(9):521–38. https://doi.org/10.1038/s41579-020-0367-2.
Mirzaei MK, Maurice CF. Ménage à trois in the human gut: interactions between host, bacteria and phages. Nat Rev Microbiol. 2017;15(7):397–408. https://doi.org/10.1038/nrmicro.2017.30.
Keen EC, Dantas G. Close encounters of three kinds: bacteriophages, commensal bacteria, and host immunity. Trends Microbiol. 2018;26(11):943–54. https://doi.org/10.1016/j.tim.2018.05.009.
Çolakoğlu M, Xue J, Trajkovski M. Bacteriophage prevents alcoholic liver disease. Cell. 2020;180(2):218–20. https://doi.org/10.1016/j.cell.2019.12.034.
Łobocka M, Hejnowicz MS, Gągała U, Weber-Dąbrowska B, Wêgrzyn G, Dadlez M, et al. The first step to bacteriophage therapy—how to choose the correct phage. In: Borysowski J, Międzybrodzki R, Górski A, editors., et al., Phage therapy: current research and applications. Norfolk: Caister Academic Press; 2014. p. 23–69.
Philipson CW, Voegtly LJ, Lueder MR, et al. Characterizing phage genomes for therapeutic applications. Viruses. 2018;10(4):188. https://doi.org/10.3390/v10040188.
Merabishvili M, Pirnay JP, De Vos D. Guidelines to compose an ideal bacteriophage cocktail. Methods Mol Biol. 2018;1693:99–110. https://doi.org/10.1007/978-1-4939-7395-8_9.
Fernández L, Gutiérrez D, García P, Rodríguez A. The perfect bacteriophage for therapeutic applications—a quick guide. Antibiotics (Basel). 2019;8(3):126. https://doi.org/10.3390/antibiotics8030126.
Reuter M, Kruger DH. Approaches to optimize therapeutic bacteriophage and bacteriophage-derived products to combat bacterial infections. Virus Genes. 2020;56(2):136–49. https://doi.org/10.1007/s11262-020-01735-7.
Casey E, van Sinderen D, Mahony J. In vitro characteristics of phages to guide “real life” phage therapy suitability. Viruses. 2018;10(4):163. https://doi.org/10.3390/v10040163.
Łobocka M, Hejnowicz MS, Dąbrowski K, et al. Genomics of staphylococcal Twort-like phages–potential therapeutics of the post-antibiotic era. Adv Virus Res. 2012;83:143–216. https://doi.org/10.1016/B978-0-12-394438-2.00005-0.
Krylov VN. Bacteriophages of Pseudomonas aeruginosa: long-term prospects for use in phage therapy. Adv Virus Res. 2014;88:227–78. https://doi.org/10.1016/B978-0-12-800098-4.00005-2.
Mattila S, Ruotsalainen P, Jalasvuori M. On-demand isolation of bacteriophages against drug-resistant bacteria for personalized phage therapy. Front Microbiol. 2015;6:1271. https://doi.org/10.3389/fmicb.2015.01271.
Herridge WP, Shibu P, O’Shea J, Brook TC, Hoyles L. Bacteriophages of Klebsiella spp., their diversity and potential therapeutic uses. J Med Microbiol. 2020;69(2):176–94. https://doi.org/10.1099/jmm.0.001141.
Fong K, Tremblay DM, Delaquis P, et al. Diversity and host specificity revealed by biological characterization and whole genome sequencing of bacteriophages infecting Salmonella enterica. Viruses. 2019;11(9):854. https://doi.org/10.3390/v11090854.
Attai H, Brown PJB. Isolation and characterization T4- and T7-like phages that infect the bacterial plant pathogen Agrobacterium tumefaciens. Viruses. 2019;11(6):528. https://doi.org/10.3390/v11060528.
McShan WM, McCullor KA, Nguyen SV. The bacteriophages of Streptococcus pyogenes. Microbiol Spectr. 2019;7:3. https://doi.org/10.1128/microbiolspec.GPP3-0059-2018.
Grose JH, Casjens SR. Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology. 2014;468–470:421–43. https://doi.org/10.1016/j.virol.2014.08.024.
Bolocan AS, Upadrasta A, Bettio PHA, et al. Evaluation of phage therapy in the context of Enterococcus faecalis and its associated diseases. Viruses. 2019;11(4):366. https://doi.org/10.3390/v11040366.
Oliveira H, Sampaio M, Melo LDR, et al. Staphylococci phages display vast genomic diversity and evolutionary relationships. BMC Genomics. 2019;20(1):357. https://doi.org/10.1186/s12864-019-5647-8.
Korf IHE, Meier-Kolthoff JP, Adriaenssens EM, et al. Still something to discover: novel insights into Escherichia coli phage diversity and taxonomy. Viruses. 2019;11(5):454. https://doi.org/10.3390/v11050454.
Kaczorowska J, Casey E, Neve H, et al. A Quest of great importance-developing a broad spectrum Escherichia coli phage collection. Viruses. 2019;11(10):899. https://doi.org/10.3390/v11100899.
Ingmer H, Gerlach D, Wolz C. Temperate phages of Staphylococcus aureus. Microbiol Spectr. 2019;7:5. https://doi.org/10.1128/microbiolspec.GPP3-0058-2018.
Ghosh D, Roy K, Williamson KE, et al. Prevalence of lysogeny among soil bacteria and presence of 16S rRNA and trzN genes in viral-community DNA. Appl Environ Microbiol. 2008;74(2):495–502. https://doi.org/10.1128/AEM.01435-07.
Derbise A, Chenal-Francisque V, Pouillot F, et al. A horizontally acquired filamentous phage contributes to the pathogenicity of the plague bacillus. Mol Microbiol. 2007;63(4):1145–57. https://doi.org/10.1111/j.1365-2958.2006.05570.x.
Mai-Prochnow A, Hui JG, Kjelleberg S, Rakonjac J, McDougald D, Rice SA. Big things in small packages: the genetics of filamentous phage and effects on fitness of their host. FEMS Microbiol Rev. 2015;39(4):465–87. https://doi.org/10.1093/femsre/fuu007.
Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49(2):277–300. https://doi.org/10.1046/j.1365-2958.2003.03580.x.
Boyd EF. Bacteriophage-encoded bacterial virulence factors and phage-pathogenicity island interactions. Adv Virus Res. 2012;82:91–118. https://doi.org/10.1016/B978-0-12-394621-8.00014-5.
Davies EV, Winstanley C, Fothergill JL, James CE. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol Lett. 2016;363(5):fnw015. https://doi.org/10.1093/femsle/fnw015.
Bondy-Denomy J, Davidson AR. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol. 2014;52(3):235–42. https://doi.org/10.1007/s12275-014-4083-3.
Fortier LC, Sekulovic O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence. 2013;4(5):354–65. https://doi.org/10.4161/viru.24498.
Tinsley CR, Bille E, Nassif X. Bacteriophages and pathogenicity: more than just providing a toxin? Microbes Infect. 2006;8(5):1365–71. https://doi.org/10.1016/j.micinf.2005.12.013.
Brüssow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68(3):560–602. https://doi.org/10.1128/MMBR.68.3.560-602.2004.
Argov T, Azulay G, Pasechnek A, et al. Temperate bacteriophages as regulators of host behavior. Curr Opin Microbiol. 2017;38:81–7. https://doi.org/10.1016/j.mib.2017.05.002.
Głowacka-Rutkowska A, Ulatowska M, Empel J, Kowalczyk M, Boreczek J, Łobocka M. A Kayvirus distant homolog of staphylococcal virulence determinants and VISA biomarker is a phage lytic enzyme. Viruses. 2020;12(3):292. https://doi.org/10.3390/v12030292.
Schicklmaier P, Schmieger H. Frequency of generalized transducing phages in natural isolates of the Salmonella typhimurium complex. Appl Environ Microbiol. 1995;61(4):1637–40. https://doi.org/10.1128/AEM.61.4.1637-1640.1995.
Torres-Barceló C. The disparate effects of bacteriophages on antibiotic-resistant bacteria. Emerg Microbes Infect. 2018;7(1):168. https://doi.org/10.1038/s41426-018-0169-z.
Penadés JR, Chen J, Quiles-Puchalt N, Carpena N, Novick RP. Bacteriophage-mediated spread of bacterial virulence genes. Curr Opin Microbiol. 2015;23:171–8. https://doi.org/10.1016/j.mib.2014.11.019.
Chiang YN, Penadés JR, Chen J. Genetic transduction by phages and chromosomal islands: The new and noncanonical. PLoS Pathog. 2019;15(8):e1007878. https://doi.org/10.1371/journal.ppat.1007878.
Masters M. Transduction: host DNA transfer by bacteriophages. In: Schaechter M, editor. The desk encyclopedia of microbiology. London: Elsevier Academic Press; 2004. p. 1000–12.
Bertozzi Silva J, Storms Z, Sauvageau D. Host receptors for bacteriophage adsorption. FEMS Microbiol Lett. 2016;363(4):fnw002. https://doi.org/10.1093/femsle/fnw002.
Dunne M, Hupfeld M, Klumpp J, Loessner MJ. Molecular basis of bacterial host interactions by Gram-positive targeting bacteriophages. Viruses. 2018;10(8):397. https://doi.org/10.3390/v10080397.
Doron S, Melamed S, Ofir G, et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359(6379):eaar4120. https://doi.org/10.1126/science.aar4120.
Ofir G, Sorek R. Contemporary phage biology: from classic models to new Insights. Cell. 2018;172(6):1260–70. https://doi.org/10.1016/j.cell.2017.10.045.
Bernheim A, Sorek R. The pan-immune system of bacteria: antiviral defence as a community resource. Nat Rev Microbiol. 2020;18(2):113–9. https://doi.org/10.1038/s41579-019-0278-2.
Hampton HG, Watson BNJ, Fineran PC. The arms race between bacteria and their phage foes. Nature. 2020;577(7790):327–36. https://doi.org/10.1038/s41586-019-1894-8.
Lu TK, Koeris MS. The next generation of bacteriophage therapy. Curr Opin Microbiol. 2011;14(5):524–31. https://doi.org/10.1016/j.mib.2011.07.028.
Rostøl JT, Marraffini L. (Ph)ighting phages: how bacteria resist their parasites. Cell Host Microbe. 2019;25(2):184–94. https://doi.org/10.1016/j.chom.2019.01.009.
Głowacka-Rutkowska A, Gozdek A, Empel J, et al. The ability of lytic staphylococcal podovirus vB_SauP_phiAGO1.3 to coexist in equilibrium with its host facilitates the selection of host mutants of attenuated virulence but does not preclude the phage antistaphylococcal activity in a nematode infection model. Front Microbiol. 2019;9:3227. https://doi.org/10.3389/fmicb.2018.03227.
Kwiatek M, Parasion S, Rutyna P, et al. Isolation of bacteriophages and their application to control Pseudomonas aeruginosa in planktonic and biofilm models. Res Microbiol. 2017;168(3):194–207. https://doi.org/10.1016/j.resmic.2016.10.009.
Rohde C, Resch G, Pirnay JP, et al. Expert opinion on three phage therapy related topics: bacterial phage resistance, phage training and prophages in bacterial production strains. Viruses. 2018;10(4):178. https://doi.org/10.3390/v10040178.
Argemi X, Hansmann Y, Prola K, Prévost G. Coagulase-negative staphylococci pathogenomics. Int J Mol Sci. 2019;20(5):1215. https://doi.org/10.3390/ijms20051215.
Young R, Gill JJ. Microbiology. Phage therapy redux—what is to be done? Science. 2015;350(6265):1163–4. https://doi.org/10.1126/science.aad6791.
Kilcher S, Loessner MJ. Engineering bacteriophages as versatile biologics. Trends Microbiol. 2019;27(4):355–67. https://doi.org/10.1016/j.tim.2018.09.006.
Pirnay JP, De Vos D, Verbeken G, et al. The phage therapy paradigm: prêt-à-porter or sur-mesure? Pharm Res. 2011;28(4):934–7. https://doi.org/10.1007/s11095-010-0313-5.
Debarbieux L, Pirnay JP, Verbeken G, et al. A bacteriophage journey at the European Medicines Agency. FEMS Microbiol Lett. 2016;363(2):fnv225. https://doi.org/10.1093/femsle/fnv225.
Cui Z, Guo X, Feng T, Li L. Exploring the whole standard operating procedure for phage therapy in clinical practice. J Transl Med. 2019;17(1):373. https://doi.org/10.1186/s12967-019-2120-z.
Bourdin G, Navarro A, Sarker SA, et al. Coverage of diarrhoea-associated Escherichia coli isolates from different origins with two types of phage cocktails. Microb Biotechnol. 2014;7(2):165–76. https://doi.org/10.1111/1751-7915.12113.
Gibson SB, Green SI, Liu CG, et al. Constructing and characterizing bacteriophage libraries for phage therapy of human infections. Front Microbiol. 2019;10:2537. https://doi.org/10.3389/fmicb.2019.02537.
Styles KM, Thummeepak R, Leungtongkam U, et al. Investigating bacteriophages targeting the opportunistic pathogen Acinetobacter baumannii. Antibiotics (Basel). 2020;9(4):E200. https://doi.org/10.3390/antibiotics9040200.
Sergueev KV, Filippov AA, Farlow J, et al. Correlation of host range expansion of therapeutic bacteriophage Sb-1 with allele state at a hypervariable repeat locus. Appl Environ Microbiol. 2019;85(22):e01209-e1219. https://doi.org/10.1128/AEM.01209-19.
Botka T, Pantůček R, Mašlaňová I, et al. Lytic and genomic properties of spontaneous host-range Kayvirus mutants prove their suitability for upgrading phage therapeutics against staphylococci. Sci Rep. 2019;9(1):5475. https://doi.org/10.1038/s41598-019-41868-w.
Friman VP, Soanes-Brown D, Sierocinski P, et al. Pre-adapting parasitic phages to a pathogen leads to increased pathogen clearance and lowered resistance evolution with Pseudomonas aeruginosa cystic fibrosis bacterial isolates. J Evol Biol. 2016;29(1):188–98. https://doi.org/10.1111/jeb.12774.
Betts A, Vasse M, Kaltz O, Hochberg ME. Back to the future: evolving bacteriophages to increase their effectiveness against the pathogen Pseudomonas aeruginosa PAO1. Evol Appl. 2013;6(7):1054–63. https://doi.org/10.1111/eva.12085.
Hyman P. Phages for phage therapy: isolation, characterization, and host range breadth. Pharmaceuticals (Basel). 2019;12(1):35. https://doi.org/10.3390/ph12010035.
Mutti M, Corsini L. Robust approaches for the production of active ingredient and drug product for human phage therapy. Front Microbiol. 2019;10:2289. https://doi.org/10.3389/fmicb.2019.02289.
Schmidt C. Phage therapy’s latest makeover. Nat Biotechnol. 2019;37(6):581–6. https://doi.org/10.1038/s41587-019-0133.
Ahiwale S, Tamboli N, Thorat K, Kulkarni R, Ackermann H, Kapadnis B. In vitro management of hospital Pseudomonas aeruginosa biofilm using indigenous T7-like lytic phage. Curr Microbiol. 2011;62(2):335–40. https://doi.org/10.1007/s00284-010-9710-6.
Turner D, Ackermann HW, Kropinski AM, Lavigne R, Sutton JM, Reynolds DM. Comparative analysis of 37 Acinetobacter bacteriophages. Viruses. 2017;10(1):5. https://doi.org/10.3390/v10010005.
Jansen M, Wahida A, Latz S, et al. Enhanced antibacterial effect of the novel T4-like bacteriophage KARL-1 in combination with antibiotics against multi-drug resistant Acinetobacter baumannii. Sci Rep. 2018;8(1):14140. https://doi.org/10.1038/s41598-018-32344-y.
Huang K, Brady A, Mahurkar A, et al. MetaRef: a pan-genomic database for comparative and community microbial genomics. Nucleic Acids Res. 2014;42(Database issue):D617–24. https://doi.org/10.1093/nar/gkt1078.
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15(6):589–94. https://doi.org/10.1016/j.gde.2005.09.006.
Laing CR, Zhang Y, Thomas JE, Gannon VP. Everything at once: comparative analysis of the genomes of bacterial pathogens. Vet Microbiol. 2011;153(1–2):13–26. https://doi.org/10.1016/j.vetmic.2011.06.014.
Krupovic M, Prangishvili D, Hendrix RW, Bamford DH. Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol Mol Biol Rev. 2011;75(4):610–35. https://doi.org/10.1128/MMBR.00011-11.
Devoto AE, Santini JM, Olm MR, et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat Microbiol. 2019;4(4):693–700. https://doi.org/10.1038/s41564-018-0338-9.
Al-Shayeb B, Sachdeva R, Chen LX, et al. Clades of huge phages from across Earth’s ecosystems. Nature. 2020;578(7795):425–31. https://doi.org/10.1038/s41586-020-2007-4.
Aiewsakun P, Adriaenssens EM, Lavigne R, Kropinski AM, Simmonds P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: steps towards a unified taxonomy. J Gen Virol. 2018;99(9):1331–43. https://doi.org/10.1099/jgv.0.001110.
Payne RJ, Jansen VA. Understanding bacteriophage therapy as a density-dependent kinetic process. J Theor Biol. 2001;208(1):37–48. https://doi.org/10.1006/jtbi.2000.2198.
Sabouri S, Sepehrizadeh Z, Amirpour-Rostami S, Skurnik M. A minireview on the in vitro and in vivo experiments with anti-Escherichia coli O157:H7 phages as potential biocontrol and phage therapy agents. Int J Food Microbiol. 2017;243:52–7. https://doi.org/10.1016/j.ijfoodmicro.2016.12.004.
Łobocka MB, Głowacka A, Dąbrowski K, Hejnowicz MS, Gozdek A, Weber-Dąbrowska B et al. A method of evaluating the therapeutic efficacy of bacteriophages. 2012. Pat. UPRP PL219654 B1, Pat. EP2872156 B1; Pat. US 9678063 B2, WO2014/012872 A1.
Tsonos J, Oosterik LH, Tuntufye HN, et al. A cocktail of in vitro efficient phages is not a guarantee for in vivo therapeutic results against avian colibacillosis. Vet Microbiol. 2014;171(3–4):470–9. https://doi.org/10.1016/j.vetmic.2013.10.021.
Badawy S, Pajunen MI, Haiko J, Baka ZAM, Abou-Dobara MI, El-Sayed AKA, Skurnik M. Identification and functional analysis of temperate Siphoviridae bacteriophages of Acinetobacter baumannii. Viruses. 2020;12(6):604. https://doi.org/10.3390/v12060604.
Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25(5):730–3. https://doi.org/10.1038/s41591-019-0437-z.
Dahlman S, Avellaneda-Franco L, Barr JJ. Phages to shape the gut microbiota? Curr Opin Biotechnol. 2020;68:89–95. https://doi.org/10.1016/j.copbio.2020.09.016.
Marônek M, Link R, Monteleone G, Gardlík R, Stolfi C. Viruses in cancers of the digestive system: active contributors or idle bystanders? Int J Mol Sci. 2020;21(21):8133. https://doi.org/10.3390/ijms21218133.
Rasmussen TS, Koefoed AK, Jakobsen RR, Deng L, Castro-Mejía JL, Brunse A, et al. Bacteriophage-mediated manipulation of the gut microbiome—promises and presents limitations. FEMS Microbiol Rev. 2020;44(4):507–21. https://doi.org/10.1093/femsre/fuaa020.
Zárate S, Taboada B, Yocupicio-Monroy M, Arias CF. Human virome. Arch Med Res. 2017;48(8):701–16. https://doi.org/10.1016/j.arcmed.2018.01.005.
Sutton TDS, Hill C. Gut bacteriophage: current understanding and challenges. Front Endocrinol (Lausanne). 2019;10:784. https://doi.org/10.3389/fendo.2019.00784.
Nakatsu G, Zhou H, Wu WKK, Wong SH, Coker OO, Dai Z, et al. Alterations in enteric virome are associated with colorectal cancer and survival outcomes. Gastroenterology. 2018;155(2):529-541.e5. https://doi.org/10.1053/j.gastro.2018.04.018.
Yang J, Zheng P, Li Y, et al. Landscapes of bacterial and metabolic signatures and their interaction in major depressive disorders. Sci Adv. 2020;6(49):eaba8555. https://doi.org/10.1126/sciadv.aba8555.
Murray NE, Murray K. Manipulation of restriction targets in phage lambda to form receptor chromosomes for DNA fragments. Nature. 1974;251(5475):476–81. https://doi.org/10.1038/251476a0.
Thomas M, Cameron JR, Davis RW. Viable molecular hybrids of bacteriophage lambda and eukaryotic DNA. Proc Natl Acad Sci USA. 1974;71(11):4579–83. https://doi.org/10.1073/pnas.71.11.4579.
Murray NE. Lambda II. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA (eds) Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1983. pp. 395–431.
Murray NE. The impact of phage lambda: from restriction to recombineering. Biochem Soc Trans. 2006;34(2):203–7. https://doi.org/10.1042/BST20060203.
Chauthaiwale VM, Therwath A, Deshpande VV. Bacteriophage lambda as a cloning vector. Microbiol Rev. 1992;56(4):577–91.
Sternberg N. A characterization of bacteriophage P1 DNA fragments cloned in a lambda vector. Virology. 1979;96(1):129–42. https://doi.org/10.1016/0042-6822(79)90179-x.
Sternberg N, Austin S. Isolation and characterization of P1 minireplicons, lambda-P1:5R and lambda-P1:5L. J Bacteriol. 1983;153(2):800–12. https://doi.org/10.1128/JB.153.2.800-812.1983.
Velten J, Fukada K, Abelson J. In vitro construction of bacteriophage lambda and plasmid DNA molecules containing DNA fragments from bacteriophage T4. Gene. 1976;1(1):93–106. https://doi.org/10.1016/0378-1119(76)90009-3.
Wilson GG, Tanyashin VI, Murray NE. Molecular cloning of fragments of bacteriophage T4 DNA. Mol Gen Genet. 1977;156(2):203–14. https://doi.org/10.1007/BF00283493.
Vorozheikina D, Glinskaite I, Tikhomirova L, Bayev A. Complementation of T4 phage am mutations by hybrid phages lambda-T4. Mol Gen Genet. 1980;178(3):655–61. https://doi.org/10.1007/BF00337875.
Revel HR. Molecular cloning of the T4 genome: organization and expression of the tail fiber gene cluster 34–38. Mol Gen Genet. 1981;182(3):445–55. https://doi.org/10.1007/BF00293934.
Revel HR. Organization of the bacteriophage T4 tail fiber gene cluster 34–38. Prog Clin Biol Res. 1981;64:353–64.
Brunel F, Davison J, Merchez M. Cloning of bacteriophage T5 DNA fragments in plasmid pBR322 and bacteriophage lambda gtWES. Gene. 1979;8(1):53–68. https://doi.org/10.1016/0378-1119(79)90007-6.
Ksenzenko VN, Kamynina TP, Kazantsev SI, Shlyapnikov MG, Kryukov VM, Bayev AA. Cloning of genes for bacteriophage T5 stable RNAs. Biochim Biophys Acta. 1982;697(2):235–42. https://doi.org/10.1016/0167-4781(82)90082-3.
Amann EP, Reeve JN, Morelli G, Behrens B, Trautner TA. Cloning and expression of the Bacillus subtilis phage SPP1 in E. coli. I. Construction and characterization of lambda/SPP1 hybrids. Mol Gen Genet. 1981;182(2):293–8. https://doi.org/10.1007/BF00269673.
Collins J, Hohn B. Cosmids: a type of plasmid gene-cloning vector that is packageable in vitro in bacteriophage lambda heads. Proc Natl Acad Sci USA. 1978;75(9):4242–6. https://doi.org/10.1073/pnas.75.9.4242.
Jacobs WR Jr, Tuckman M, Bloom BR. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature. 1987;327(6122):532–5. https://doi.org/10.1038/327532a0.
Jacobs WR Jr, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, et al. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science. 1993;260(5109):819–922. https://doi.org/10.1126/science.8484123.
Pearson RE, Jurgensen S, Sarkis GJ, Hatfull GF, Jacobs WR Jr. Construction of D29 shuttle phasmids and luciferase reporter phages for detection of mycobacteria. Gene. 1996;183(1–2):129–36. https://doi.org/10.1016/s0378-1119(96)00530-6.
Ehretsmann CP, Carpousis AJ, Krisch HM. Specificity of Escherichia coli endoribonuclease RNase E: in vivo and in vitro analysis of mutants in a bacteriophage T4 mRNA processing site. Genes Dev. 1992;6(1):149–59. https://doi.org/10.1101/gad.6.1.149.
Karam JD, Drake JW, Kreuzer KN, Mosig G, Hall DH, Eiserling FA, et al. Molecular biology of bacteriophage T4. Washington, DC: American Society for Microbiology; 1994.
Sarkis GJ, Jacobs WR Jr, Hatfull GF. L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol Microbiol. 1995;15(6):1055–67. https://doi.org/10.1111/j.1365-2958.1995.tb02281.x.
Mahichi F, Synnott AJ, Yamamichi K, Osada T, Tanji Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol Lett. 2009;295(2):211–7. https://doi.org/10.1111/j.1574-6968.2009.01588.x.
Le S, He X, Tan Y, et al. Mapping the tail fiber as the receptor binding protein responsible for differential host specificity of Pseudomonas aeruginosa bacteriophages PaP1 and JG004. PLoS ONE. 2013;8(7):e68562. https://doi.org/10.1371/journal.pone.0068562.
Loessner MJ, Rees CE, Stewart GS, Scherer S. Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl Environ Microbiol. 1996;62(4):1133–40. https://doi.org/10.1128/AEM.62.4.1133-1140.1996.
Qimron U, Marintcheva B, Tabor S, Richardson CC. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc Natl Acad Sci USA. 2006;103(50):19039–44. https://doi.org/10.1073/pnas.0609428103.
Tran NQ, Rezende LF, Qimron U, Richardson CC, Tabor S. Gene 1.7 of bacteriophage T7 confers sensitivity of phage growth to dideoxythymidine. Proc Natl Acad Sci USA. 2008;105(27):9373–8. https://doi.org/10.1073/pnas.0804164105.
Grigonyte AM, Harrison C, MacDonald PR, et al. Comparison of CRISPR and marker-based methods for the engineering of phage T7. Viruses. 2020;12(2):193. https://doi.org/10.3390/v12020193.
Murphy KC. Phage recombinases and their applications. Adv Virus Res. 2012;83:367–414. https://doi.org/10.1016/B978-0-12-394438-2.00008-6.
Murphy KC. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol. 1998;180(8):2063–71. https://doi.org/10.1128/JB.180.8.2063-2071.1998.
Murphy KC, Campellone KG, Poteete AR. PCR-mediated gene replacement in Escherichia coli. Gene. 2000;246(1–2):321–30. https://doi.org/10.1016/s0378-1119(00)00071-8.
Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97(11):5978–83. https://doi.org/10.1073/pnas.100127597.
Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci USA. 2001;98(12):6742–6. https://doi.org/10.1073/pnas.121164898.
Oppenheim AB, Rattray AJ, Bubunenko M, Thomason LC, Court DL. In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology. 2004;319(2):185–9. https://doi.org/10.1016/j.virol.2003.11.007.
Pan YJ, Lin TL, Chen CC, et al. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. J Virol. 2017;91(6):e02457-e2516. https://doi.org/10.1128/JVI.02457-16.
Jensen JD, Parks AR, Adhya S, Rattray AJ, Court DL. λ Recombineering used to engineer the genome of phage T7. Antibiotics (Basel). 2020;9(11):805. https://doi.org/10.3390/antibiotics9110805.
Muyrers JP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999;27(6):1555–7. https://doi.org/10.1093/nar/27.6.1555.
Marinelli LJ, Hatfull GF, Piuri M. Recombineering: a powerful tool for modification of bacteriophage genomes. Bacteriophage. 2012;2(1):5–14. https://doi.org/10.4161/bact.18778.
Marinelli LJ, Piuri M, Swigonová Z, Balachandran A, Oldfield LM, van Kessel JC, et al. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE. 2008;3(12):e3957. https://doi.org/10.1371/journal.pone.0003957.
Fehér T, Karcagi I, Blattner FR, Pósfai G. Bacteriophage recombineering in the lytic state using the lambda red recombinases. Microb Biotechnol. 2012;5(4):466–76. https://doi.org/10.1111/j.1751-7915.2011.00292.x.
da Silva JL, Piuri M, Broussard G, et al. Application of BRED technology to construct recombinant D29 reporter phage expressing EGFP. FEMS Microbiol Lett. 2013;344(2):166–72. https://doi.org/10.1111/1574-6968.12171.
Datta S, Costantino N, Zhou X, Court DL. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc Natl Acad Sci USA. 2008;105(5):1626–31. https://doi.org/10.1073/pnas.0709089105.
Shin H, Lee JH, Yoon H, Kang DH, Ryu S. Genomic investigation of lysogen formation and host lysis systems of the Salmonella temperate bacteriophage SPN9CC. Appl Environ Microbiol. 2014;80(1):374–84. https://doi.org/10.1128/AEM.02279-13.
Martel B, Moineau S. CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014;42(14):9504–13. https://doi.org/10.1093/nar/gku628.
Hatoum-Aslan A. Phage genetic engineering using CRISPR- Cas systems. Viruses. 2018;10(6):335. https://doi.org/10.3390/v10060335.
Lemay ML, Tremblay DM, Moineau S. Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS Synth Biol. 2017;6(7):1351–8. https://doi.org/10.1021/acssynbio.6b00388.
Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18(2):67–83. https://doi.org/10.1038/s41579-019-0299-x.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12. https://doi.org/10.1126/science.1138140.
Barrangou R, Marraffini LA. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell. 2014;54(2):234–44. https://doi.org/10.1016/j.molcel.2014.03.011.
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155(Pt 3):733–40. https://doi.org/10.1099/mic.0.023960-0.
Bari SMN, Walker FC, Cater K, Aslan B, Hatoum-Aslan A. Strategies for editing virulent staphylococcal phages using CRISPR-Cas10. ACS Synth Biol. 2017;6(12):2316–25. https://doi.org/10.1021/acssynbio.7b00240.
Loureiro A, da Silva GJ. CRISPR-Cas: converting a bacterial defence mechanism into a state-of-the-art genetic manipulation tool. Antibiotics (Basel). 2019;8(1):18. https://doi.org/10.3390/antibiotics8010018.
Ramachandran G, Bikard D. Editing the microbiome the CRISPR way. Philos Trans R Soc Lond B Biol Sci. 2019;374(1772):20180103. https://doi.org/10.1098/rstb.2018.0103.
Schilling T, Dietrich S, Hoppert M, Hertel R. A CRISPR-Cas9-based toolkit for fast and precise in vivo genetic engineering of Bacillus subtilis phages. Viruses. 2018;10(5):241. https://doi.org/10.3390/v10050241.
Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. https://doi.org/10.1016/j.mib.2017.05.008.
Shen J, Zhou J, Chen GQ, Xiu ZL. Efficient genome engineering of a virulent Klebsiella bacteriophage using CRISPR-Cas9. J Virol. 2018;92(17):e00534-e618. https://doi.org/10.1128/JVI.00534-18.
Tao P, Wu X, Tang WC, Zhu J, Rao V. Engineering of bacteriophage T4 genome using CRISPR-Cas9. ACS Synth Biol. 2017;6(10):1952–61. https://doi.org/10.1021/acssynbio.7b00179.
Hoshiga F, Yoshizaki K, Takao N, Miyanaga K, Tanji Y. Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol Lett. 2019;366(4):fnz041. https://doi.org/10.1093/femsle/fnz041.
Park JY, Moon BY, Park JW, Thornton JA, Park YH, Seo KS. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci Rep. 2017;7:44929. https://doi.org/10.1038/srep44929.
Liu TY, Liu JJ, Aditham AJ, Nogales E, Doudna JA. Target preference of type III-A CRISPR-Cas complexes at the transcription bubble. Nat Commun. 2019;10(1):3001. https://doi.org/10.1038/s41467-019-10780-2.
Bari SMN, Hatoum-Aslan A. CRISPR-Cas10 assisted editing of virulent staphylococcal phages. Methods Enzymol. 2019;616:385–409. https://doi.org/10.1016/bs.mie.2018.10.023.
Ando H, Lemire S, Pires DP, Lu TK. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 2015;1(3):187–96. https://doi.org/10.1016/j.cels.2015.08.013.
Jaschke PR, Lieberman EK, Rodriguez J, Sierra A, Endy D. A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology. 2012;434(2):278–84. https://doi.org/10.1016/j.virol.2012.09.020.
DeMarini DJ, Creasy CL, Lu Q, et al. Oligonucleotide-mediated, PCR-independent cloning by homologous recombination. Biotechniques. 2001;30(3):520–3. https://doi.org/10.2144/01303st02.
Lu TKT, et al. Institute For Environmental Health Inc. Recombinant Phage and Methods; 2011. US20130122549A1.
Kilcher S, Studer P, Muessner C, Klumpp J, Loessner MJ. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc Natl Acad Sci USA. 2018;115(3):567–72. https://doi.org/10.1073/pnas.1714658115.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–5. https://doi.org/10.1038/nmeth.1318.
Shin J, Jardine P, Noireaux V. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth Biol. 2012;1(9):408–13. https://doi.org/10.1021/sb300049p.
Garamella J, Marshall R, Rustad M, Noireaux V. The All E. coli TX-TL Toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth Biol. 2016;5(4):344–55. https://doi.org/10.1021/acssynbio.5b00296.
Rustad M, Eastlund A, Marshall R, Jardine P, Noireaux V. Synthesis of infectious bacteriophages in an E. coli-based cell-free expression system. J Vis Exp. 2017;126:56144. https://doi.org/10.3791/56144.
Rustad M, Eastlund A, Jardine P, Noireaux V. Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth Biol. 2018;3(1):ysy02. https://doi.org/10.1093/synbio/ysy002.
Yeom H, Ryu T, Lee AC, et al. Cell-free bacteriophage genome synthesis using low-cost sequence-verified array-synthesized oligonucleotides. ACS Synth Biol. 2020;9(6):1376–84. https://doi.org/10.1021/acssynbio.0c00051.
Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 2007;104(27):11197–202. https://doi.org/10.1073/pnas.0704624104.
Born Y, Fieseler L, Thöny V, Leimer N, Duffy B, Loessner MJ. Engineering of bacteriophages Y2::dpoL1-C and Y2::luxAB for efficient control and rapid detection of the fire blight pathogen, Erwinia amylovora. Appl Environ Microbiol. 2017;83(12):e00341-e417. https://doi.org/10.1128/AEM.00341-17.
Nobrega FL, Costa AR, Santos JF, et al. Genetically manipulated phages with improved pH resistance for oral administration in veterinary medicine. Sci Rep. 2016;6:39235. https://doi.org/10.1038/srep39235.
Pei R, Lamas-Samanamud GR. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Appl Environ Microbiol. 2014;80(17):5340–8. https://doi.org/10.1128/AEM.01434-14.
Paul VD, Sundarrajan S, Rajagopalan SS, et al. Lysis-deficient phages as novel therapeutic agents for controlling bacterial infection. BMC Microbiol. 2011;11:195. https://doi.org/10.1186/1471-2180-11-195.
Hagens S, Habel A, von Ahsen U, von Gabain A, Bläsi U. Therapy of experimental Pseudomonas infections with a nonreplicating genetically modified phage. Antimicrob Agents Chemother. 2004;48(10):3817–22. https://doi.org/10.1128/AAC.48.10.3817-3822.2004.
Matsuda T, Freeman TA, Hilbert DW, et al. Lysis-deficient bacteriophage therapy decreases endotoxin and inflammatory mediator release and improves survival in a murine peritonitis model. Surgery. 2005;137(6):639–46. https://doi.org/10.1016/j.surg.2005.02.012.
Nobrega FL, Vlot M, de Jonge PA, et al. Targeting mechanisms of tailed bacteriophages. Nat Rev Microbiol. 2018;16(12):760–73. https://doi.org/10.1038/s41579-018-0070-8.
de Jonge PA, Nobrega FL, Brouns SJJ, Dutilh BE. Molecular and evolutionary determinants of bacteriophage host range. Trends Microbiol. 2019;27(1):51–63. https://doi.org/10.1016/j.tim.2018.08.006.
Dams D, Brøndsted L, Drulis-Kawa Z, Briers Y. Engineering of receptor-binding proteins in bacteriophages and phage tail-like bacteriocins. Biochem Soc Trans. 2019;47(1):449–60. https://doi.org/10.1042/BST20180172.
Pouillot F, Blois H, Iris F. Genetically engineered virulent phage banks in the detection and control of emergent pathogenic bacteria. Biosecur Bioterror. 2010;8(2):155–69. https://doi.org/10.1089/bsp.2009.0057.
Dunne M, Prokhorov NS, Loessner MJ, Leiman PG. Reprogramming bacteriophage host range: design principles and strategies for engineering receptor binding proteins. Curr Opin Biotechnol. 2021;68:272–81. https://doi.org/10.1016/j.copbio.2021.02.006.
Tétart F, Desplats C, Krisch HM. Genome plasticity in the distal tail fiber locus of the T-even bacteriophage: recombination between conserved motifs swaps adhesin specificity. J Mol Biol. 1998;282(3):543–56. https://doi.org/10.1006/jmbi.1998.2047.
Yehl K, Lemire S, Yang AC, et al. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell. 2019;179(2):459-469.e9. https://doi.org/10.1016/j.cell.2019.09.015.
Yoichi M, Abe M, Miyanaga K, Unno H, Tanji Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157:H7. J Biotechnol. 2005;115(1):101–7. https://doi.org/10.1016/j.jbiotec.2004.08.003.
Golomidova AK, Kulikov EE, Prokhorov NS, et al. Branched lateral tail fiber organization in T5-Like bacteriophages DT57C and DT571/2 is revealed by genetic and functional analysis. Viruses. 2016;8(1):26. https://doi.org/10.3390/v8010026.
Russell J, Bikard D. Learning from antibodies: Phage host-range engineering. Cell Host Microbe. 2019;26(4):445–6. https://doi.org/10.1016/j.chom.2019.09.010.
Matsuzaki S, Yasuda M, Nishikawa H, Kuroda M, Ujihara T, Shuin T, et al. Experimental protection of mice against lethal Staphylococcus aureus infection by novel bacteriophage phi MR11. J Infect Dis. 2003;187(4):613–24. https://doi.org/10.1086/374001.
Lynch KH, Seed KD, Stothard P, Dennis JJ. Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J Virol. 2010;84(3):1276–88. https://doi.org/10.1128/JVI.01843-09.
Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF. Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol. 1998;279(1):143–64. https://doi.org/10.1006/jmbi.1997.1610.
Brown ER, Cherry WB. Specific identification of Bacillus anthracis by means of a variant bacteriophage. J Infect Dis. 1955;96(1):34–9. https://doi.org/10.1093/infdis/96.1.34.
Schuch R, Fischetti VA. Detailed genomic analysis of the Wbeta and gamma phages infecting Bacillus anthracis: implications for evolution of environmental fitness and antibiotic resistance. J Bacteriol. 2006;188(8):3037–51. https://doi.org/10.1128/JB.188.8.3037-3051.2006.
Zhang H, Fouts DE, DePew J, Stevens RH. Genetic modifications to temperate Enterococcus faecalis phage Ef11 that abolish the establishment of lysogeny and sensitivity to repressor, and increase host range and productivity of lytic infection. Microbiology. 2013;159(Pt 6):1023–35. https://doi.org/10.1099/mic.0.067116-0.
Monteiro R, Pires DP, Costa AR, Azeredo J. Phage therapy: Going temperate? Trends Microbiol. 2019;27(4):368–78. https://doi.org/10.1016/j.tim.2018.10.008.
Tinoco JM, Buttaro B, Zhang H, Liss N, Sassone L, Stevens R. Effect of a genetically engineered bacteriophage on Enterococcus faecalis biofilms. Arch Oral Biol. 2016;71:80–6. https://doi.org/10.1016/j.archoralbio.2016.07.001.
Tinoco JM, Liss N, Zhang H, Nissan R, Gordon W, Tinoco E, et al. Antibacterial effect of genetically-engineered bacteriophage ϕEf11/ϕFL1C(Δ36)P(nisA) on dentin infected with antibiotic-resistant Enterococcus faecalis. Arch Oral Biol. 2017;82:166–70. https://doi.org/10.1016/j.archoralbio.2017.06.005.
Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell. 2013;49(2):237–48. https://doi.org/10.1016/j.molcel.2012.11.012.
Łusiak-Szelachowska M, Żaczek M, Weber-Dąbrowska B, et al. Antiphage activity of sera during phage therapy in relation to its outcome. Future Microbiol. 2017;12:109–17. https://doi.org/10.2217/fmb-2016-0156.
Møller-Olsen C, Ho SFS, Shukla RD, Feher T, Sagona AP. Engineered K1F bacteriophages kill intracellular Escherichia coli K1 in human epithelial cells. Sci Rep. 2018;8(1):17559. https://doi.org/10.1038/s41598-018-35859-6.
Møller-Olsen C, Ross T, Leppard KN, et al. Bacteriophage K1F targets Escherichia coli K1 in cerebral endothelial cells and influences the barrier function. Sci Rep. 2020;10(1):8903. https://doi.org/10.1038/s41598-020-65867-4.
Spoerel N, Herrlich P, Bickle TA. A novel bacteriophage defence mechanism: the anti-restriction protein. Nature. 1979;278(5699):30–4. https://doi.org/10.1038/278030a0.
Iida S, Streiff MB, Bickle TA, Arber W. Two DNA antirestriction systems of bacteriophage P1, darA, and darB: characterization of darA- phages. Virology. 1987;157(1):156–66. https://doi.org/10.1016/0042-6822(87)90324-2.
King G, Murray NE. Restriction alleviation and modification enhancement by the Rac prophage of Escherichia coli K-12. Mol Microbiol. 1995;16(4):769–77. https://doi.org/10.1111/j.1365-2958.1995.tb02438.x.
Walkinshaw MD, Taylor P, Sturrock SS, Atanasiu C, Berge T, Henderson RM, et al. Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol Cell. 2002;9(1):187–94. https://doi.org/10.1016/s1097-2765(02)00435-5.
Rusinov IS, Ershova AS, Karyagina AS, Spirin SA, Alexeevski AV. Avoidance of recognition sites of restriction-modification systems is a widespread but not universal anti-restriction strategy of prokaryotic viruses. BMC Genomics. 2018;19(1):885. https://doi.org/10.1186/s12864-018-5324-3.
Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8(5):317–27. https://doi.org/10.1038/nrmicro2315.
Samson JE, Magadán AH, Sabri M, Moineau S. Revenge of the phages: defeating bacterial defences. Nat Rev Microbiol. 2013;11(10):675–87. https://doi.org/10.1038/nrmicro3096.
Hutinet G, Kot W, Cui L, et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat Commun. 2019;10(1):5442. https://doi.org/10.1038/s41467-019-13384-y.
Stanley SY, Maxwell KL. Phage-encoded anti-CRISPR defenses. Annu Rev Genet. 2018;52:445–64. https://doi.org/10.1146/annurev-genet-120417-031321.
Davidson AR, Lu WT, Stanley SY, Wang J, Mejdani M, Trost CN, et al. Anti-CRISPRs: protein inhibitors of CRISPR-Cas systems. Annu Rev Biochem. 2020;89:309–32. https://doi.org/10.1146/annurev-biochem-011420-111224.
Osuna BA, Karambelkar S, Mahendra C, Christie KA, Garcia B, Davidson AR, et al. Listeria phages induce Cas9 degradation to protect lysogenic genomes. Cell Host Microbe. 2020;28(1):31-40.e9. https://doi.org/10.1016/j.chom.2020.04.001.
Cui YR, Wang SJ, Chen J, Li J, Chen W, Wang S, et al. Allosteric inhibition of CRISPR-Cas9 by bacteriophage-derived peptides. Genome Biol. 2020;21(1):51. https://doi.org/10.1186/s13059-020-01956-x.
Gussow AB, Park AE, Borges AL, Shmakov SA, Makarova KS, Wolf YI, et al. Machine-learning approach expands the repertoire of anti-CRISPR protein families. Nat Commun. 2020;11(1):3784. https://doi.org/10.1038/s41467-020-17652-0.
Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493(7432):429–32. https://doi.org/10.1038/nature11723.
Borges AL, Davidson AR, Bondy-Denomy J. The discovery, mechanisms, and evolutionary impact of anti-CRISPRs. Annu Rev Virol. 2017;4(1):37–59. https://doi.org/10.1146/annurev-virology-101416-041616.
Eitzinger S, Asif A, Watters KE, Iavarone AT, Knott GJ, Doudna JA, et al. Machine learning predicts new anti-CRISPR proteins. Nucleic Acids Res. 2020;48(9):4698–708. https://doi.org/10.1093/nar/gkaa219.
Otsuka Y, Yonesaki T. Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol Microbiol. 2012;83(4):669–81. https://doi.org/10.1111/j.1365-2958.2012.07975.x.
Blower TR, Evans TJ, Przybilski R, Fineran PC, Salmond GP. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 2012;8(10):e1003023. https://doi.org/10.1371/journal.pgen.1003023.
Makarova KS, Wolf YI, Snir S, Koonin EV. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J Bacteriol. 2011;193(21):6039–56. https://doi.org/10.1128/JB.05535-11.
Koonin EV. Hunting for treasure chests in microbial defense islands. Mol Cell. 2018;70(5):761–2. https://doi.org/10.1016/j.molcel.2018.05.025.
Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, Sorek R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359(6379):eaar4120. https://doi.org/10.1126/science.aar4120.
Goldfarb T, Sberro H, Weinstock E, Cohen O, Doron S, Charpak-Amikam Y, Afik S, Ofir G, Sorek R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015;34(2):169–83. https://doi.org/10.15252/embj.201489455.
Ofir G, Melamed S, Sberro H, Mukamel Z, Silverman S, Yaakov G, Doron S, Sorek R. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat Microbiol. 2018;3(1):90–8. https://doi.org/10.1038/s41564-017-0051-0.
Depardieu F, Didier JP, Bernheim A, Sherlock A, Molina H, Duclos B, Bikard D. A Eukaryotic-like serine/threonine kinase protects staphylococci against phages. Cell Host Microbe. 2016;20(4):471–81. https://doi.org/10.1016/j.chom.2016.08.010.
Millman A, Bernheim A, Stokar-Avihail A, Fedorenko T, Voichek M, Leavitt A, Oppenheimer-Shaanan Y, Sorek R. Bacterial retrons function in anti-phage defense. Cell. 2020;183(6):1551-1561.e12. https://doi.org/10.1016/j.cell.2020.09.065.
Bernheim A, Millman A, Ofir G, Meitav G, Avraham C, Shomar H, Rosenberg MM, Tal N, Melamed S, Amitai G, Sorek R. Prokaryotic viperins produce diverse antiviral molecules. Nature. 2021;589(7840):120–4. https://doi.org/10.1038/s41586-020-2762-2.
Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer-Shaanan Y, Kacen A, Doron S, Amitai G, Sorek R. Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature. 2019;574(7780):691–5. https://doi.org/10.1038/s41586-019-1605-5.
Morehouse BR, Govande AA, Millman A, Keszei AFA, Lowey B, Ofir G, Shao S, Sorek R, Kranzusch PJ. STING cyclic dinucleotide sensing originated in bacteria. Nature. 2020;586(7829):429–33. https://doi.org/10.1038/s41586-020-2719-5.
Ye Q, Lau RK, Mathews IT, Birkholz EA, Watrous JD, Azimi CS, Pogliano J, Jain M, Corbett KD. HORMA domain proteins and a trip13-like ATPase regulate bacterial cGAS-like enzymes to mediate bacteriophage immunity. Mol Cell. 2020;77(4):709-722.e7. https://doi.org/10.1016/j.molcel.2019.12.009.
Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, Davidson AR, Maxwell KL. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016;10(12):2854–66. https://doi.org/10.1038/ismej.2016.79.
Dedrick RM, Jacobs-Sera D, Bustamante CA, et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat Microbiol. 2017;2:16251. https://doi.org/10.1038/nmicrobiol.2016.251.
Gentile GM, Wetzel KS, Dedrick RM, Montgomery MT, Garlena RA, Jacobs-Sera D, Hatfull GF. More evidence of collusion: a new prophage-mediated viral defense system encoded by mycobacteriophage Sbash. MBio. 2019;10(2):e00196-e219. https://doi.org/10.1128/mBio.00196-19.
Montgomery MT, Guerrero Bustamante CA, Dedrick RM, Jacobs-Sera D, Hatfull GF. Yet more evidence of collusion: a new viral defense system encoded by Gordonia phage CarolAnn. MBio. 2019;10(2):e02417-e2418. https://doi.org/10.1128/mBio.02417-18.
Touchon M, Bernheim A, Rocha EP. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016;10(11):2744–54. https://doi.org/10.1038/ismej.2016.47.
Bikard D, Euler CW, Jiang W, et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol. 2014;32(11):1146–50. https://doi.org/10.1038/nbt.3043.
Cobb LH, Park J, Swanson EA, et al. CRISPR-Cas9 modified bacteriophage for treatment of Staphylococcus aureus induced osteomyelitis and soft tissue infection. PLoS ONE. 2019;14(11):e0220421. https://doi.org/10.1371/journal.pone.0220421.
Selle K, Fletcher JR, Tuson H, et al. In vivo targeting of Clostridioides difficile using phage-delivered CRISPR-Cas3 Antimicrobials. MBio. 2020;11(2):e00019-20. https://doi.org/10.1128/mBio.00019-20.
Voorhees PJ, Cruz-Teran C, Edelstein J, Lai SK. Challenges & opportunities for phage-based in situ microbiome engineering in the gut. J Control Release. 2020;326:106–19. https://doi.org/10.1016/j.jconrel.2020.06.016.
Hsu BB, Way JC, Silver PA. Stable neutralization of a virulence factor in bacteria using temperate phage in the mammalian gut. mSystems. 2020;5(1):e00013-20. https://doi.org/10.1128/mSystems.00013-20.
Edgar R, Friedman N, Molshanski-Mor S, Qimron U. Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl Environ Microbiol. 2012;78(3):744–51. https://doi.org/10.1128/AEM.05741-11.
Yosef I, Goren MG, Globus R, Molshanski-Mor S, Qimron U. Extending the host range of bacteriophage particles for DNA transduction. Mol Cell. 2017;66(5):721-728.e3. https://doi.org/10.1016/j.molcel.2017.04.025.
Auster O, Globus R, Yosef I, Qimron U. Optimizing DNA transduction by selection of mutations that evade bacterial defense systems. RNA Biol. 2019;16(4):595–9. https://doi.org/10.1080/15476286.2018.1514235.
Rakonjac J, Russel M, Khanum S, Brooke SJ, Rajič M. Filamentous phage: Structure and biology. Adv Exp Med Biol. 2017;1053:1–20. https://doi.org/10.1007/978-3-319-72077-7_1.
Ju Z, Sun W. Drug delivery vectors based on filamentous bacteriophages and phage-mimetic nanoparticles. Drug Deliv. 2017;24(1):1898–908. https://doi.org/10.1080/10717544.2017.1410259.
Specthrie L, Bullitt E, Horiuchi K, Model P, Russel M, Makowski L. Construction of a microphage variant of filamentous bacteriophage. J Mol Biol. 1992;228(3):720–4. https://doi.org/10.1016/0022-2836(92)90858-h.
Peng H, Chen IA. Rapid colorimetric detection of bacterial species through the capture of gold nanoparticles by chimeric phages. ACS Nano. 2019;13(2):1244–52. https://doi.org/10.1021/acsnano.8b06395.
Yacoby I, Shamis M, Bar H, Shabat D, Benhar I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob Agents Chemother. 2006;50(6):2087–97. https://doi.org/10.1128/AAC.00169-06.
Krom RJ, Bhargava P, Lobritz MA, Collins JJ. Engineered phagemids for nonlytic, targeted antibacterial therapies. Nano Lett. 2015;15(7):4808–13. https://doi.org/10.1021/acs.nanolett.5b01943A.
Westwater C, Kasman LM, Schofield DA, Werner PA, Doaln JW, Schmidt MG, et al. Use of genetically engineered phage to deliver antimicrobial agents to bacteria: an alternative therapy for treatment of bacterial infections. Antimicrob Agents Chemother. 2003;47(4):1301–7. https://doi.org/10.1128/aac.47.4.1301-1307.2003.
Hagens S, Bläsi U. Genetically modified filamentous phage as bactericidal agents: a pilot study. Lett Appl Microbiol. 2003;37(4):318–23. https://doi.org/10.1046/j.1472-765x.2003.01400.x.
Lu TK, Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci USA. 2009;106(12):4629–34. https://doi.org/10.1073/pnas.0800442106.
Bhattarai SR, Yoo SY, Lee SW, Dean D. Engineered phage-based therapeutic materials inhibit Chlamydia trachomatis intracellular infection. Biomaterials. 2012;33(20):5166–74. https://doi.org/10.1016/j.biomaterials.2012.03.054.
Peng H, Borg RE, Dow LP, Pruitt BL, Chen IA. Controlled phage therapy by photothermal ablation of specific bacterial species using gold nanorods targeted by chimeric phages. Proc Natl Acad Sci USA. 2020;117(4):1951–61. https://doi.org/10.1073/pnas.1913234117.
Hodyra-Stefaniak K, Miernikiewicz P, Drapała J, et al. Mammalian host-versus-phage immune response determines phage fate in vivo. Sci Rep. 2015;5:14802. https://doi.org/10.1038/srep14802.
Van Belleghem JD, Clement F, Merabishvili M, Lavigne R, Vaneechoutte M. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci Rep. 2017;7(1):8004.
Górski A, Dąbrowska K, Międzybrodzki R, Weber-Dąbrowska B, Łusiak-Szelachowska M, Jończyk-Matysiak E, et al. Phages and immunomodulation. Future Microbiol. 2017;12:905–14. https://doi.org/10.2217/fmb-2017-0049.
Górski A, Międzybrodzki R, Łobocka M, Głowacka-Rutkowska A, Bednarek A, Borysowski J, et al. Phage therapy: what have we learned? Viruses. 2018;10(6):288. https://doi.org/10.3390/v10060288.
Dąbrowska K. Phage therapy: what factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med Res Rev. 2019;39(5):2000–25. https://doi.org/10.1002/med.21572.
Dąbrowska K, Abedon ST. Pharmacologically aware phage therapy: pharmacodynamic and pharmacokinetic obstacles to phage antibacterial action in animal and human bodies. Microbiol Mol Biol Rev. 2019;83(4):e00012-19. https://doi.org/10.1128/MMBR.00012-19.
Malik DJ, Sokolov IJ, Vinner GK, Mancuso F, Cinquerrui S, Vladisavljevic GT, et al. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv Colloid Interface Sci. 2017;249:100–33. https://doi.org/10.1016/j.cis.2017.05.014.
Sokoloff AV, Bock I, Zhang G, Sebestyen MG, Wolff JA. The interactions of peptides with the innate immune system studied with use of T7 phage peptide display. Mol Ther. 2000;2:131–9. https://doi.org/10.1006/mthe.2000.0110.
Sokoloff AV, Bock I, Zhang G, Hoffman S, Dama J, Ludtke JJ, et al. Specific recognition of protein carboxy-terminal sequences by natural IgM antibodies in normal serum. Mol Ther. 2001;3:821–30. https://doi.org/10.1006/mthe.2001.0340.
Sokoloff AV, Puckett M, Ludtke JJ, Fetterly B. Sequence-specific binding of normal serum immunoglobulin M to exposed protein C-termini. Immunology. 2004;112:237–49. https://doi.org/10.1111/j.1365-2567.2004.01868.x.
Merril CR, Biswas B, Carlton R, Jensen NC, Creed GJ, Zullo S, et al. Long-circulating bacteriophage as antibacterial agents. Proc Natl Acad Sci USA. 1996;93:3188–92. https://doi.org/10.1073/pnas.93.8.3188.
Vitiello CL, Merril CR, Adhya S. An amino acid substitution in a capsid protein enhances phage survival in mouse circulatory system more than a 1000-fold. Virus Res. 2005;114:101–3. https://doi.org/10.1016/j.virusres.2005.05.014.
Hodyra-Stefaniak K, Lahutta K, Majewska J, Kaźmierczak Z, Lecion D, Harhala M, et al. Bacteriophages engineered to display foreign peptides may become short-circulating phages. Microb Biotechnol. 2019;12(4):730–41. https://doi.org/10.1111/1751-7915.13414.
Cecchini N, Schmerer M, Molineux IJ, Springman R, Bull JJ. Evolutionarily stable attenuation by genome rearrangement in a virus. G3 (Bethesda). 2013;3(8):1389–97. https://doi.org/10.1534/g3.113.006403.
Springman R, Molineux IJ, Duong C, Bull RJ, Bull JJ. Evolutionary stability of a refactored phage genome. ACS Synth Biol. 2012;1:425–30. https://doi.org/10.1021/sb300040v.
Springman R, Kapadia-Desai DS, Molineux IJ, Bull JJ. Evolutionary recovery of a recombinant viral genome. G3 (Bethesda). 2012;2(7):825–30. https://doi.org/10.1534/g3.112.002758.
Bull JJ, Molineux IJ, Wilke CO. Slow fitness recovery in a codon-modified viral genome. Mol Biol Evol. 2012;29(10):2997–3004. https://doi.org/10.1093/molbev/mss119.
Kim KP, Cha JD, Jang EH, et al. PEGylation of bacteriophages increases blood circulation time and reduces T-helper type 1 immune response. Microb Biotechnol. 2008;1(3):247–57. https://doi.org/10.1111/j.1751-7915.2008.00028.x.
Nair A, Khairnar K. Genetically engineered phages for therapeutics: proceed with caution. Nat Med. 2019;25(7):1028. https://doi.org/10.1038/s41591-019-0506-3.
Rokyta DR, Wichman HA. Genic incompatibilities in two hybrid bacteriophages. Mol Biol Evol. 2009;26(12):2831–9. https://doi.org/10.1093/molbev/msp199.
Gladstone EG, Molineux IJ, Bull JJ. Evolutionary principles and synthetic biology: avoiding a molecular tragedy of the commons with an engineered phage. J Biol Eng. 2012;6:13. https://doi.org/10.1186/1754-1611-6-13.
Bodner K, Melkonian AL, Covert MW. The enemy of my enemy: new insights regarding bacteriophage-mammalian cell interactions. Trends Microbiol. 2020. https://doi.org/10.1016/j.tim.2020.10.014.
Acknowledgements
The authors acknowledge support from the Polish Academy of Sciences and the National Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by funds from the National Science Foundation, Grant no. 2019/33/B/NZ2/02006.
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Author contributions
MŁ: conceptualization, preparation of the major part of the manuscript and artwork, reviewing, and editing; KD: writing part of the manuscript; AG: writing parts of the manuscript and support and advice during manuscript preparation.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Łobocka, M., Dąbrowska, K. & Górski, A. Engineered Bacteriophage Therapeutics: Rationale, Challenges and Future. BioDrugs 35, 255–280 (2021). https://doi.org/10.1007/s40259-021-00480-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-021-00480-z