
Abstract
Antibody, immuno- and gene therapies developed for neurological indications face a delivery challenge posed by various anatomical and physiological barriers within the central nervous system (CNS); most notably, the blood–brain barrier (BBB). Emerging delivery technologies for biotherapeutics have focused on trans-cellular pathways across the BBB utilizing receptor-mediated transcytosis (RMT). ‘Traditionally’ targeted RMT receptors, transferrin receptor (TfR) and insulin receptor (IR), are ubiquitously expressed and pose numerous translational challenges during development, including species differences and safety risks. Recent advances in antibody engineering technologies and discoveries of RMT targets and BBB-crossing antibodies that are more BBB-selective have combined to create a new preclinical pipeline of BBB-crossing biotherapeutics with improved efficacy and safety. Novel BBB-selective RMT targets and carrier antibodies have exposed additional opportunities for re-targeting gene delivery vectors or nanocarriers for more efficient brain delivery. Emergence and refinement of core technologies of genetic engineering and editing as well as biomanufacturing of viral vectors and cell-derived products have de-risked the path to the development of systemic gene therapy approaches for the CNS. In particular, brain-tropic viral vectors and extracellular vesicles have recently expanded the repertoire of brain delivery strategies for biotherapeutics. Whereas protein biotherapeutics and bispecific antibodies enabled for BBB transcytosis are rapidly heading towards clinical trials, systemic gene therapy approaches for CNS will likely remain in research phase for the foreseeable future. The promise and limitations of these emerging cross-BBB delivery technologies are further discussed in this article.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
While receptor-mediated transcytosis (RMT) has been successfully exploited to deliver biotherapeutics across the blood–brain barrier (BBB), new RMT targets and advances in antibody engineering technologies are jointly contributing to an emerging pipeline of more specific and safer brain-penetrating biotherapeutics. |
Certain viral delivery platforms capable of crossing the BBB are being refined and engineered to mitigate safety risks and enable systemic, targeted gene therapy for CNS diseases. |
Cell-derived vesicles, such as exosomes, are emerging as ‘next generation’ biotherapeutic products capable of carrying versatile therapeutic payloads to target sites. Experimental demonstration of their ability to interact with and cross the BBB raises hope that the technology can be harnessed to devise novel strategies to deliver biotherapeutics across the BBB. |
1 The Blood–Brain Barrier: A Challenge Solved?
Diseases of the CNS are among the most difficult to treat. Confounding factors include the complexity of brain physiology and pathology, as well as inadequate translational preclinical models to evaluate experimental therapeutics [1]. Despite significant progress in brain imaging surrogates for assessing therapeutic efficacy, more accessible brain-specific molecular biomarkers for early diagnosis and patient stratification for clinical trials remain sparse [1]. In addition, the delivery of therapeutics across the blood–brain barrier (BBB) remains one of the prime challenges in CNS drug development. The BBB is formed by specialized endothelial cells of brain microvessels and capillaries joined together by tight junctions that restrict paracellular transport of hydrophilic therapeutics > 500 Da [2]. Polarized efflux transporters further prevent brain access to many lipophilic synthetic molecules [2].
Biologics, pharmaceutical drug products manufactured in, extracted from, or semi-synthesized from biological sources, include vaccines, recombinant therapeutic proteins, gene therapy, and living cells or their products. Biologics are typically large and complex therapeutics, and their systemic delivery across the BBB was not considered feasible until recently. Alternative routes of delivery, including intraparenchymal pumps and intrathecal infusion, proved to be of limited value since the diffusion of these molecules within or into brain tissue, respectively, remained limited. Similarly, some clinically implemented medical procedures such as mannitol-induced osmotic BBB disruption [3] and focused ultrasound [4] for treatment of the brain have yet to show clinical benefits with biotherapeutics. Therefore, the development of novel transvascular brain delivery technologies remains critical for expanding the application of biologics, a highly successful therapeutic modality for cancer and inflammatory diseases, into diseases of the CNS.
Molecular Trojan horses are ligands or antibodies against the BBB receptors that transport essential growth factors and nutrients into the brain [5, 6]. The initial internalization and potential transport across the brain endothelium could occur via a non-specific, charge-mediated adsorptive endocytosis or via an energy-dependent and receptor-specific receptor-mediated endocytosis/transcytosis (RMT) [6]. Since the RMT process occurs via a specific receptor, it has potential for higher selectivity compared with the charge-mediated adsorptive process. The RMT process involves (i) engagement of the receptor on the luminal surface of brain endothelial cells (BEC) by the natural ligand or targeting moiety; (ii) endocytosis of the receptor-ligand complex into BEC; (iii) transcellular (transvascular) routing through complex endosomal sorting pathways, which may be receptor/cargo-specific; (iv) release of the cargo on the abluminal surface of BEC, and (v) subsequent receptor recycling to the luminal membrane of BEC [6] (Fig. 1a). The two most studied receptors undergoing RMT are transferrin receptor (TfR) and insulin receptor (IR) [6]. Antibodies against both of these receptors have been studied as molecular carriers to deliver attached therapeutic cargoes, including biologics, across the BBB. The enhanced and pharmacologically relevant brain delivery of biologics, including antibodies, growth factors, decoy receptors, and peptides has been demonstrated in pre-clinical models using various bioengineered antibodies against TfR [7,8,9,10]. Recent studies have focused on optimizing affinity of TfR antibodies [7, 10, 11] and used molecular modeling to establish the structure–function relationship of the receptor–antibody interactions (e.g., mono-valent vs bi-valent) that results in more efficient transcytosis [9]. These antibody engineering approaches resulted in significant improvements in TfR antibody pharmacokinetics [7, 10, 11], reduced lysosomal degradation within brain endothelial cells [11, 12], and enhanced efficiency of transcytosis and brain exposure [7, 9,10,11]. The molecular mechanisms that direct endosomal trafficking of receptor/antibody/cargo complexes towards the transcytosis pathway have been described in more detail recently [11,12,13,14]. Whereas TfR antibodies as molecular Trojan horses have still to enter clinical trials, a humanized mouse monoclonal antibody against IR fused to the enzyme iduronidase to enable its brain penetration in pediatric patients with severe Mucopolysaccharidosis Type I has been evaluated as safe and effective at improving biomarkers in an open-label, phase I–II clinical trial [15, 16].

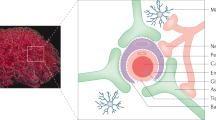
A schematic depiction of the design and mechanisms of blood–brain barrier (BBB) transmigration of three emerging brain delivery technologies: BBB-crossing antibodies (a), brain-tropic adenoviral vectors (b) and engineered extracellular vesicles (c, d). a BBB-crossing antibodies are raised against a select number of BBB receptors that undergo receptor-mediated transcytosis (RMT) (Table 1). Therapeutic antibodies (TH-Ab) or other therapeutic cargoes, bio-engineered to incorporate BBB-crossing function, internalize via clathrin-coated vesicles and are trafficked through the early endosome pathway, including multivesicular bodies (MVB). They are released on the abluminal side of the BBB, as free antibodies or, in some cases, via exosomes, where therapeutic cargoes engage central targets (such as misfolded proteins or receptors on parenchymal cells) or replace missing molecules (for example growth factors or enzymes). The carrier receptor is recycled back to the luminal membranes via recycling endosomes to accept new cargoes from the circulatory compartment. b Brain-tropic and non-brain-tropic AAVs, AAV9 and AAV2 respectively, recognize discrete receptors on brain endothelial cells that initiate internalization. Thereafter, distinct intracellular trafficking routes are utilized by AAV9 and AAV2, with the former undergoing active transcytosis across the BBB and the later peri-nuclear/nuclear localization and transduction of the endothelial cells. Following transcytosis across the BBB, AAV9 is taken up by and subsequently transduces parenchymal cells. AAV9 is also capable of spreading within the brain by anterograde, retrograde and trans-synaptic neuronal transport. c Exosomes can be used as brain delivery vehicles in either their natural or in engineered forms. They can be loaded with various gene-based (siRNA, miRNA, DNA) and protein-based (antibodies, peptides) biotherapeutics. Specific targeting ligands are genetically engineered into the exosomal membranes in producing cells to achieve BBB crossing and/or drug delivery to target cells. d Natural exosomes may internalize into brain endothelial cells by fusion with the plasma membrane and release their cargoes into endothelial cells. Exosomes expressing RMT ligands likely undergo clathrin-dependent endocytosis, trafficking and abluminal release. Transcytosing exosomes could target parenchymal cells or release their cargo into the brain extracellular space. AAV adeno-associated virus, MVB multivesicular body; TH Ab therapeutic antibody
However, the enthusiasm for TfR- and IR-targeting antibodies as BBB carriers has been dampened by translational gaps and safety concerns. Both receptors show species differences in abundance in brain endothelial cells [17], and antibodies raised against these receptors are typically species-selective, necessitating ‘surrogate’ molecules or humanization of the receptor in preclinical testing species. Additionally, translational gaps may be encountered due to neuroanatomical differences between pre-clinical animal models and humans, such as glia-neuron ratios [18]. Furthermore, safety concerns emerged during pre-clinical evaluation of TfR antibodies [19], stemming from the TfR enrichment in reticulocytes, the lungs and neurons [19, 20]. Thus, the quest continues for RMT receptors selectively expressed or highly enriched in BBB endothelial cells, which are expected to yield Trojan antibodies with better pharmacokinetic and safety profiles.
The recently demonstrated clinical benefit and subsequent FDA approval of intrathecal anti-sense oligonucleotide therapy (nusinersen, Spiranza™) to correct a genetic mutation causing spinal muscular atrophy in pediatric patients [21], as well as promising results of the Ionis-Roche anti-sense oligonucleotide (RG6043) in phase I/IIa clinical trials for Huntington’s disease, leading to PRIME (PRIority MEdicines) designation by the European Medicines Agency, sparked a renewed promise that a broader application of gene therapy to correct devastating monogenetic neurological diseases, including lysosomal storage disease, ALS and Huntington’s disease, could be realized if appropriate brain delivery technologies were introduced in gene therapy designs. A major focus of recent gene therapy research has been in developing brain-tropic adeno-associated virus (AAV) vectors and understanding the mechanisms of their passage across the BBB [22]. While AAV-mediated gene therapy has great potential, significant obstacles remain to be surmounted in their development as therapeutics, including preclinical differences in pre-existing anti-AAV antibodies [23] and differential neuron/glial selectivity [24].
Finally, the field has witnessed an explosion of various nanoparticle delivery systems using diverse materials and designs to enable more efficient gene and large protein delivery across the BBB [25]. The inherent issues associated with this approach include complexity of composition and production, the absence of toxicology studies, and size limitations for nano-formulation transport across the BBB and within the brain extracellular space (35–60 nm) [26]. One promising novel approach relies on nanovesicles derived from cells, also known as exosomes, which could be enticed to package genes or other cargoes, as well as organ-targeting address molecules, by cell engineering and ex vivo bioprocessing.
The intensified research in BBB biology and pathology and advances in drug delivery and bio-technologies have jointly created critical mass in a field that has impeded CNS drug discovery for decades. Although the problem remains formidable, solutions have emerged that promise accelerated future development of BBB-crossing biotherapeutics for diseases of the CNS. Whereas advances in engineering and development of antibodies against IR and TfR have been the subject of numerous reviews in the past and recently, in this short overview we will focus primarily on emerging targets, technologies, and innovations in cross-BBB delivery arising from wide-scale genomics and screening studies.
2 Emerging Antibody Technologies for Brain Delivery Using Receptor-Mediated Transcytosis
The discovery of novel RMT receptor–antibody pairs has broadly followed two main workflows: (a) selection of potential RMT targets based on molecular (typically ‘omics’) analyses of the BBB and subsequent generation and evaluation of antibodies against these targets, and (b) ‘function-first’ screening of antibody libraries for antibody species that can transmigrate the BBB, followed by identification/deconvolution of the target(s) they engage. Both strategies have yielded initial proof of concept of enhanced brain delivery achieved with novel RMT targets and antibody modalities. The currently used RMT targets and antibody technologies and their characteristics are summarized in Table 1.
Using transcriptomic and proteomic analyses of mouse brain endothelial cells, Zuchero et al. [27] identified multiple highly abundant BBB-expressed proteins, including basigin, Glut1, and CD98hc. Brain penetration of the antibodies generated against these targets did not correlate with target expression abundance; however, antibodies against CD98hc showed a robust accumulation in the brain after systemic administration. When CD98hc antibodies were bioengineered as heterodimerized bi-specific antibodies with anti-BACE1 antibody, a strong pharmacodynamic response—a reduction in brain Aβ levels—was observed in transgenic animals after systemic administration [27] (Table 1). However, CD98hc antibodies accelerated systemic target-mediated clearance of the cargo antibody [27], due to the ubiquitous expression of CD98hc in peripheral tissues (Table 1). Antibodies against Glut1 and basigin showed a marginal increase in brain uptake, while antibodies against IR and Lrp1, also produced in this study, did not show improved brain penetration over that observed with a control antibody [27].
Similarly, using next-generation sequencing and proteomics analyses, we have selected insulin growth factor 1 receptor (IGF1R) as a potential target for RMT based on its abundance in isolated brain vessels compared with peripheral tissues. The panel of camelid single domain antibodies (VHHs) were raised against different epitopes of the extracellular domain of human IGF1R away from the IGF1 binding site, humanized, and evaluated for BBB crossing in models in vitro and in vivo [28] (Table 1). Selected IGF1R VHHs with species cross-reactivity demonstrated a saturable, energy-dependent transport across the human BBB model in vitro [29] and highly enhanced brain and CSF exposure in rats [29]. When used in mono-valent fusion with antibody therapeutics, these VHHs had a minor impact on systemic pharmacokinetics, despite their high affinity. The transport of circulatory IGF1 across the BBB in vivo is driven by local neuronal activity [30]. Therefore, engagement of IGF1R by antibodies that do not interfere with IGF1 binding is a promising novel strategy to deliver biotherapeutic payloads across the BBB.
In separate studies using a ‘function-first’ screening approach to select BBB-crossing antibodies from high-diversity camelid VHH libraries, we identified the BBB-crossing antibody FC5 [31]. FC5 and its humanized variants have been genetically fused with various centrally acting payloads, including neuropeptides [32] and full monoclonal antibodies [33] (Table 1). FC5 had a minimal effect on circulatory pharmacokinetics of monoclonal antibodies, while increasing both their brain and CSF exposure in rats [33]. Target engagement and pharmacological effects of an FC5-delivered cargo antibody, acting as an antagonist of the G-protein-coupled metabotropic glutamate receptor 1 (mGluR1), have been observed in a rat model of thermal hyperalgesia; the bi-specific FC5-mGluR1 antibody co-localized with thalamic neurons involved in nociceptive responses through mGluR1 [33]. Deconvolution of the BBB receptor engaged by FC5 indicated a role for TMEM30A (cdc50A), a β subunit of phospholipid flippases that trigger endocytic vesicle formation by catalyzing transport of aminophospholipids from the outer to the inner leaflet of various membranes [34].
The above studies have provided a new generation of RMT targets and antibodies suitable for development of BBB delivery strategies for a wide range of biotherapeutics. Advances in the antibody engineering field have expanded formats of BBB-transporting antibodies from a full IgG, used to target IR, to antibody fragments (Fabs and ScFvs) and small monomeric single-domain antibodies of only 15 kDa, and, more recently, to antigen-binding sites incorporated into the Fc region of antibodies (Fcab™).
The engineering strategies to modulate the function of Fc fragments have also come into sharp focus. For example, an effector function of Fc may be required for therapeutic efficacy of the antibody in the brain—such is the case for amyloid β-targeting antibodies, where mobilization of the innate immune system and microglia via effector-competent Fc accelerates amyloid degradation and clearance; yet intact effector function may lead to systemic side effects when BBB carrier antibody binds peripheral targets. The example of the later is acute reticulocyte lysis observed with effector-competent TfR-BACE-1 bi-specific antibodies [19]. However, mono-valent anti-TfR Fab linked to a C-terminus of the Aβ-antibody could not trigger effector function systemically due to steric hindrance, while the bispecific antibody retained full effector function activity when engaging its brain target [33]. In addition, treatments of chronic diseases that require a constant presence of the therapeutic in the brain may benefit from an extended circulation half-life that can be achieved by Fc engineering.
Using these innovations and highly modular technologies in antibody engineering, various types of bi-specific or multi-specific antibodies that incorporate a BBB-crossing function and CNS-targeting function(s) have been engineered and expressed, as reviewed in [35], and are steadily progressing through pre-clinical development towards clinical trials.
3 Gene Therapy: Brain Tropic Adenoviral Vectors
The BBB presents a uniquely difficult challenge for the delivery of gene therapy to the CNS. Additionally, major challenges for viral gene therapy include peripheral and CNS off-target effects due to broad viral tropism, toxicity related to high gene transfer efficiency, insertional genotoxicity and anti-vector immune responses. Strategies are being developed to decrease off-target effects, including the incorporation of tissue-specific promoters for payloads [36], miRNA that suppress ectopic expression [37], as well as investigation of the role of adsorptive endocytosis in mediating brain endothelial cell transcytosis [38, 39].
Several approaches can be taken to manipulate the genome, including (i) silencing mutant alleles, (ii) replacement of dysfunctional mutated genes, (iii) introduction of a disease-modifying gene, and (iv) the correction of genetic mutations using gene editing methods. Compared with non-viral vectors, including the use of liposomes, exosomes, and polymeric nanoparticles [40, 41], viral vectors have proven more efficient in delivering gene therapy in vivo, where the goal is to transduce the maximal number of target cells [42].
Among various viral delivery systems summarized in Table 2, only select classes are suitable for CNS applications. For example, retroviruses are not suitable for use in neurological disorders because of their inability to transduce non-dividing cells. In comparison, adenoviruses have the advantages of the absence of genomic integration [43] and a very large carrying capacity, important for those diseases that require the delivery of large payloads, such as Huntington’s disease or Duchenne muscular dystrophy. However, adenoviruses are known to elicit a strong innate immune response, and severe cytotoxicity and inflammation were observed with high doses in a clinical trial [44].
AAV is a naturally replication-defective, non-pathogenic, non-enveloped single-stranded DNA parvovirus that has gained traction as a vector for CNS-targeted gene therapy [45]. Cellular transduction by AAV involves a series of events, including binding of a cell-surface receptor, internalization through endocytic uptake, endosomal/lysosomal escape, nuclear entry, capsid uncoating, and genome release [46, 47]. Events occurring during endosomal processing are indispensable to AAV transduction. Following release of the genome in the nucleus, second-strand synthesis and transcription ensue [48]. AAV exists primarily as an episomal vector that does not disrupt the host genome and is not burdened with the risk of activating oncogenes [49]. While persistent AAV expression can be achieved in non-dividing cells in the CNS, obtaining persistent expression in dividing cells requires additional modifications to the viral genome to enable chromosomal integration or episomal replication [50]. Although AAV has a small packaging capacity (4.8 kilobase pairs of nucleic acid), its ability to transduce mitotic and post-mitotic cells, tropism for neurons and glia, absence of pathogenicity or cytotoxicity, and low immunogenicity [51] are distinct benefits for CNS gene therapy [42]. A common method for optimizing gene expression is the use of self-complimentary AAV (scAAV) genomes, which can expedite expression and increase transduction efficiency but comes at a cost of a > 50% reduction in packaging capacity [52]. Additionally, the methods used for viral expression and purification have been shown to alter transduction patterns and efficiency [53], indicating a need for optimization and standardization in order to improve the reliability and predictability of AVV therapeutics.
3.1 Brain Tropism of Adeno-Associated Virus (AAV) Serotypes
AAV is a highly diverse group of viruses that include nine major serotypes (AAV1–9) and a variety of novel hybrid serotypes that are designed to alter viral tropism [54]. Typically, AAV utilizes cell-surface proteoglycans as a primary receptor along with a distinct co-receptor, and a given serotype can interact with multiple primary receptors. Thus, tissue tropism is dictated by the expression of the primary and secondary AAV receptors. A comprehensive study characterizing the in vivo biodistribution of AAV serotypes delivered via tail vein injection [55] demonstrated that the distribution and kinetics of expression varied widely among serotypes, with AAV9 found to produce the most robust tissue expression and, unlike other serotypes, was capable of producing expression in the brain.
Studies utilizing intraparenchymal injection of AAV have revealed that all serotypes are capable of transducing expression in brain cells of rodents [56, 57], non-human primates [58], and dogs [59]. However, AAV4 appeared to primarily target ependymal cells [60], AAV8 had the greatest tropism for astrocytes, microglia, and oligodendrocytes, and AAV9 had the greatest tropism for neurons [56]. Additionally, AAV9 produced the greatest distribution in brain tissue, likely related to its ability to undergo both anterograde and retrograde axonal transport as well as trans-synaptic transport (Fig. 1b) [56, 61]. AAV serotypes also display distinct transduction characteristics when administered in specific brain regions [56, 62]. While extensive variability between brains injected with AAV is reported [62], general trends are observed. Aschauer et al. [56] reported rankings of serotypes in three brain regions, finding that AAV9, AAV8, and AAV-5 produced high levels of expression, whereas AAVs consistently produced the lowest levels of expression in the striatum, hippocampus, and auditory cortex of adult mice.
Studies in rodents and non-human primates have conclusively shown that AAV9 is capable of crossing the BBB and transducing parenchymal cells, primarily neurons and glia [22, 24, 56, 63, 64], following systemic administration. Additionally, AAVrh8 and AAVrh10 have been demonstrated to produce robust expression in the CNS following intravascular administration without disruption of the BBB [63, 65, 66]. It should be noted that peripheral administration of AAV9 is also known to produce high levels of transduction in the liver and spleen [24].
AAV9 tropism is affected by both the route of administration and the age of the recipient animal. The brain regions to which AAV9 can spread following intraparenchymal injection are contingent upon the location of the injection, where spreading is dictated by the efferent and afferent projections in the injection region [67]. It has been consistently reported that intravascular administration of AAV9 transduces neurons and astrocytes in the brain and motor neurons in the spinal cord in animals (rodents and nonhuman primates); however, despite somewhat contradictory reports, the efficiency of neuronal transductions appears to decline with age [48].
Recently, progress has been made in understanding the mechanism by which AAV9 crosses the BBB. The observation that mannitol does not enhance brain delivery of AAV9, in contrast to AAV2, indicated that the mechanism of AAV9 BBB transport is via a transcellular pathway [24]. AAV particles can undergo cellular uptake through a variety of processes, including macropinocytosis, phagocytosis, and clathrin- or caveolae-mediated endocytosis, with various serotypes exhibiting distinct transduction and entry profiles [47, 68] (Fig. 1b). For example, two distinct endocytic mechanisms mediate AAV2 uptake, namely CLIC/GEEC and dynamin-dependent endocytosis, with only the former resulting in transduction [47]. In a recent study by Merkel et al. [38], the transit of AAV9 particles across the human brain microvascular endothelial cell barrier required an active transport process whereas the low level transit of AAV2 involved an energy-independent paracellular diffusion process. While both AAV2 and AAV9 were taken up by endothelial cells, AAV2 was present in endosomal structures and accumulated in the perinuclear/nuclear region, and AAV9 was almost exclusively distributed to the basolateral surface of the endothelial cells, or in funnel-like structures within the endothelial cells. The differences in the intracellular trafficking of AAV2 and AAV9 are thought to be a consequence of the differences in their receptors/co-receptors, which presumably direct the particles to different endocytic mechanisms [47].
It is conceptually possible to engineer AAV capsids that interact with receptor systems that are restricted to brain endothelial cells and are capable of triggering receptor mediated transcytosis. This principle was demonstrated using a directed evolution strategy, where AAV capsid shuffling was employed to generate vectors that selectively cross seizure-compromised BBB and exhibit a favorable biodistribution in rats [69]. Others [70,71,72] have utilized in vivo phage-display bio-panning to identify peptides that exhibit BBB specificity. Insertion of BBB-specific peptide motifs into the VP3 region of the AAV capsid enhanced the BBB tropism of AAV. Furthermore, Chen et al. [71] were able to create peptide-modified AAV with normal and disease-specific BBB tropism. Further refining this strategy to exploit known BBB transcytosis receptors should similarly enhance the specificity of BBB targeting. Consideration must also be given to the potential for adverse effects, such as increased immunogenicity or decreased transduction efficiency and BBB integrity, resulting from the introduction of non-natural sequence modifications. However, it has been demonstrated that directed evolution can be used to overcome immunogenicity [73], further indicating that the AAV capsid is amenable to engineering of beneficial functionality. Enhanced transduction of permissive and non-permissive cells following incorporation of cell-permeable peptides into the AAV capsid has been reported [74, 75]. However, this approach is not suitable for systemic administration as it currently lacks tissue specificity and would likely result in increased off-target transduction of peripheral tissues at the expense of RMT-mediated brain delivery.
3.2 Cargoes Compatible with AAV
While in vivo gene therapy based on viral vectors is thought to be best suited to gene addition/replacement [45, 76, 77], AAV can also be used to deliver gene-modifying oligonucleotides, such as short hairpin RNA (shRNA), microRNA and antisense oligonucleotides (ASOs). The approval of nusinersen in 2017 demonstrates the validity of using ASOs to regulate exon splicing in diseases such as spinal muscular atrophy, Leber congenital amaurosis, muscular dystrophy, β-thalassemia, and spinocerebellar ataxia type 3. Similarly, correcting diseases by reducing aberrant overexpression can be achieved using ASOs, shRNA, and miRNA. AAV-mediated delivery of therapeutic ASOs may represent a means to produce sustained drug delivery at a more reasonable cost. AAV delivery of shRNA and miRNA has been successfully used to decrease in vivo expression of DNM2 (myotubular myopathy) [78] and mutant huntingtin [79] in mice. SOD1 was successfully targeted for reduction with an AAV-delivered exon skipping ASO in a murine model of amyotrophic lateral sclerosis [80]. These studies indicate that AAV-mediated delivery of gene-regulating oligonucleotides represents a viable strategy for delivering cost-effective DNA therapies for a wide spectrum of CNS diseases.
4 Cell-Produced Delivery Vehicles: Promise of Extracellular Vesicles
Exosomes have recently emerged as novel lipid nanocarriers for the delivery of small molecule drugs, protein- [81, 82], and gene-based biotherapeutics [83] (Fig. 1c–d). Exosomes are a sub-population of specialized membranous extracellular vesicles (EVs) derived from endocytic compartments, 30–100 nm in size, that are actively secreted by almost all cell types [84]. They are formed in multi-vesicular bodies and released into the extracellular space by fusion with the plasma membrane [85]. Exosomes express a ubiquitous set of pan-exosome proteins such as CD9, CD81, CD63, Alix, TSG101, HSP70, and flotillin-1; they also express cell-type-specific proteins and carry cell-specific miRNA cargoes that affect their function. Exosomes produced by HEK293T cells have been characterized and a comprehensive database of protein, mRNA, and miRNA profiles has been generated [86]. Exosomes derived from human brain endothelial cells have been shown to contain distinct, possibly tissue-specific biomarkers, as well as known RMT receptors including TfR, IR, LRP, and TMEM30A [87].
Exosomes participate in inter-cellular communication and transfer of cellular contents to adjacent cells or distant organs [88], which could subsequently modify target-cell function [89, 90]. For example, glioblastoma EVs were shown to internalize into human brain microvascular endothelial cells (HBMECs), transfer ‘donor’ mRNA and miRNA, and induce angiogenic transformation [90]. These properties of exosomes have been exploited for development of delivery strategies for exogenous gene-based cargoes.
Brain- and BBB-tropism of exosomes derived from various cellular sources has been studied in model organisms, such as zebrafish [91, 92]. Interestingly, among exosomes isolated from glioblastoma (U-87MG, A-172), neuroectodermal (PFSK-1), and mouse brain endothelial (bEnd.3) cells, only brain endothelial cell-derived exosomes crossed the BBB and delivered encapsulated payloads, suggesting potential homo-tropism for the tissue of origin [92].
The mechanisms by which homo- or heterotypic exosomes interact with brain endothelial cells are not well understood. The trafficking of exosomes derived from HEK293T cells engineered to express luciferase (exo-luc) across the healthy and inflamed BBB [93] was studied in an in vitro transwell model using HBMEC. Exosomes were shown to only cross TNFα-treated HBMECs but not the healthy BBB. The uptake of exosomes by HBMEC was shown to involve an energy-dependent endocytosis process; confocal microscopy examination demonstrated that internalized exosomes colocalized with transferrin-Texas Red, an early endosome marker, and also with cholera toxin B, a late endosomal marker. Although luciferase activity was detected in the abluminal chamber, it remained uncertain if it was associated with intact exosomes. Other studies have shown that exosomes can be internalized into cells by different mechanisms, including plasma membrane fusion, clathrin-dependent endocytosis and clathrin-independent pathways such as phagocytosis, macropinocytosis, caveolin-mediated uptake, and lipid raft-mediated internalization [94,95,96,97,98].
An attractive strategy to achieve BBB crossing and cell-type-specific delivery is to genetically engineer targeting ligands known to be involved in RMT-mediated transcytosis (some shown in Table 1) into exosomal membrane proteins [83] (Fig. 1c). The preclinical proof-of-principle of this approach was demonstrated with exosomes derived from dendritic cells engineered to express lysosomal-associated membrane protein 2b (Lamp2b) fused to the neuron-specific rabies virus glycoprotein (RVG), and loaded with siRNA against BACE1 by electroporation [99]. Systemically injected RVG-siRNA exosomes silenced the mRNA and protein levels of BACE1 in neuronal cells, resulting in reduced brain amyloid levels in transgenic mice. The same strategy was also applied to knockdown the expression of α-synuclein in the mouse brain following intravenous administration of RVG-exosomes loaded with siRNA [100]. Although these preclinical studies are encouraging, many hurdles remain in the clinical translation of these findings [101].
4.1 Exosome-Associated Adeno-Associated Viruses (Exo-AAV)
Encapsulation within EVs has shown promise for increasing AAV delivery to difficult targets. During the standard AAV vector production, a portion of AAV has been shown to associate with HEK293T producer cell exosomes (exo-AAV) [102], with AAV capsids present both on the surface and encapsulated inside exosomes [103]. Exosome encapsulation shields AAV from neutralizing antibodies, while enhancing AAV9 transduction in vitro and in vivo [104, 105].
In a recent study [105], both exo-AAV8 and exo-AAV9 encoding green fluorescent protein (GFP) targeted neurons and astrocytes with higher transduction efficiency compared with conventional AAV8 or AAV9 after intravenous administration in Balb/c mice. Exo-AAV9 and exo-AAV8 had 9.3-fold and 4.3-fold increased transport, respectively, across an in vitro BBB model using bEnd.3 cells, compared with their free viral counterparts. Similarly, exo-AAV2-GFP [106] and exo-AAV1 or exo-AAV9 vectors [107] efficiently targeted and delivered transgenes across retinal and cochlear brain barriers, respectively.
Exosomes can be isolated in large quantities from cells ex vivo and engineered to package specific cargoes into secreted macrovesicles through bioprocess scale-up, similar to that of virus-like particles (VLPs). The HEK293 cell platform has been widely used for the production of AAVs and therapeutic proteins that have been approved by the FDA [108]. However, there are no cell sources or manufacturing processes that have been FDA-approved for clinical-grade exosome production [109]. The International Society for Extracellular Vesicles (ISEV) has recently summarized the criteria that need to be met by exosome-based therapeutics to turn promising preclinical compounds into drug candidates [110]. Exosomes as ‘therapeutics’ are being tested in a few clinical trials currently; notably, benefits of plant exosomes or exosomes derived from allogeneic mesenchymal stromal cells are being evaluated in acute ischemic stroke, diabetes mellitus, and several cancer indications.
5 A Glimpse into the Future of CNS Biotherapeutics
With antibody technologies coming of age, supported by unprecedented advancements and precision in antibody engineering and biomanufacturing, and progress being made in deciphering targets and mechanisms of transcytosis across the BBB, it is easy to imagine a future in which the development of all CNS biotherapeutics will be closely tied with the concurrent development and deployment of the tailored BBB-delivery strategy. The bi-specific antibodies and BBB carrier-fusion proteins targeting a variety of CNS diseases, most notably those characterized by accumulation of misfolded proteins, are advancing through pre-clinical stages and some will be entering clinical trials as early as 2019. With this burgeoning pipeline, we can expect a proof of concept of the receptor-mediated transcytosis approach in humans in the near future.
Following closely behind is the wave of gene therapies for CNS diseases. Combining the use of gene editing tools and neuro-tropic viral vectors has placed gene therapy in a position to make breakthrough advances in the treatment of currently incurable monogenetic brain diseases. Given initial successes with intrathecal administration of ASO therapies, it is likely that this route of administration will dominate subsequent clinical trials of gene therapy. However, the progress made in engineering viral vectors with brain tropism and brain delivery technologies for RNA therapeutics will eventually shift the preference towards systemic administration, the only route that can successfully address the access of these therapies to deep brain structures in humans.
As the field of biotechnology embraces the complexity of future therapeutics, engineered cell-derived targeted nanovesicles may complement antibody and gene therapy strategies to enhance the versatility of delivery options. Emerging biotechnology companies in this field, such as Evox Therapeutics (https://www.evoxtherapeutics.com/), are engineering the natural delivery capabilities of extracellular vesicles to develop an entirely novel class of biotherapeutics, notably targeting CNS indications. Lastly, RMT-targeted exo-AAVs are evolving as particularly promising novel gene therapy delivery technology, since they possess beneficial features of both exosomes, such as low immunogenicity, and viral vectors, such as robust gene delivery.
References
Butlen-Ducuing F, Pétavy F, Guizzaro L, Zienowicz M, Haas M, Alteri E, et al. Regulatory watch: Challenges in drug development for central nervous system disorders: a European Medicines Agency perspective. Nat Rev Drug Discov. 2016;15:813–4.
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25.
Bellavance M-A, Blanchette M, Fortin D. Recent advances in blood–brain barrier disruption as a CNS delivery strategy. AAPS J. 2008;10:166–77.
Poon C, McMahon D, Hynynen K. Noninvasive and targeted delivery of therapeutics to the brain using focused ultrasound. Neuropharmacology. 2017;120:20–37.
Pardridge WM. Drug targeting to the brain. Pharm Res. 2007;24:1733–44.
Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24:1759–71.
Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3:84ra44.
Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med. 2014;6:261ra154.
Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron. 2014;81:49–60.
Thom G, Burrell M, Haqqani AS, Yogi A, Lessard E, Brunette E, et al. Enhanced delivery of galanin conjugates to the brain through bioengineering of the anti-transferrin receptor antibody OX26. Mol Pharm. 2018;15:1420–31.
Haqqani AS, Thom G, Burrell M, Delaney CE, Brunette E, Baumann E, et al. Intracellular sorting and transcytosis of the rat transferrin receptor antibody OX26 across the blood–brain barrier in vitro is dependent on its binding affinity. J Neurochem. 2018. https://doi.org/10.1111/jnc.14482.
Bien-Ly N, Yu YJ, Bumbaca D, Elstrott J, Boswell CA, Zhang Y, et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med. 2014;211:233–44.
Villaseñor R, Ozmen L, Messaddeq N, Grüninger F, Loetscher H, Keller A, et al. Trafficking of endogenous immunoglobulins by endothelial cells at the blood–brain barrier. Sci Rep. 2016;6:25658.
Haqqani AS, Delaney CE, Brunette E, Baumann E, Farrington GK, Sisk W, et al. Endosomal trafficking regulates receptor-mediated transcytosis of antibodies across the blood brain barrier. J Cereb Blood Flow Metab. 2018;38:727–40.
Pardridge WM. Delivery of biologics across the blood–brain barrier with molecular Trojan horse technology. BioDrugs. 2017;31:503–19.
Giugliani R, Giugliani L, de Oliveira Poswar F, Donis KC, Corte AD, Schmidt M, et al. Neurocognitive and somatic stabilization in pediatric patients with severe mucopolysaccharidosis type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): an open label phase 1–2 trial. Orphanet J Rare Dis. 2018;13:110.
Aday S, Cecchelli R, Hallier-Vanuxeem D, Dehouck MP, Ferreira L. Stem cell-based human blood–brain barrier models for drug discovery and delivery. Trends Biotechnol. 2016;34:382–93.
Bahney J, von Bartheld CS. The cellular composition and glia-neuron ratio in the spinal cord of a human and a nonhuman primate: comparison with other species and brain regions. Anat Rec (Hoboken). 2018;301:697–710.
Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med. 2013;5:183ra57 (1–12).
Webster CI, Stanimirovic DB. A gateway to the brain: shuttles for brain delivery of macromolecules. Ther Deliv. 2015;6:1321–4.
Pane M, Palermo C, Messina S, Sansone VA, Bruno C, Catteruccia M, et al. Nusinersen in type 1 SMA infants, children and young adults: PRELIMINARY results on motor function. Neuromuscul Disord. 2018;28:582–5.
Bourdenx M, Dutheil N, Bezard E, Dehay B. Systemic gene delivery to the central nervous system using adeno-associated virus. Front Mol Neurosci. 2014;7:50.
Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene Ther Methods. 2013;24:59–67.
Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19:1058–69.
Zhou Y, Peng Z, Seven ES, Leblanc RM. Crossing the blood–brain barrier with nanoparticles. J Control Release. 2018;270:290–303.
Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci USA. 2006;103:5567–72.
Zuchero YJY, Chen X, Bien-Ly N, Bumbaca D, Tong RK, Gao X, et al. Discovery of novel blood–brain barrier targets to enhance brain uptake of therapeutic antibodies. Neuron. 2016;89:70–82.
Stanimirovic DB, Kemmerich K, Haqqani AS, Sulea T, Arbabi-Ghahroudi M, Massie B, et al. Insulin-like growth factor 1 receptor-specific antibodies and uses thereof. Patent applications: US 15/123,735, US 15/123,781, US 15/123,798; 2017.
Ribecco-Lutkiewicz M, Sodja C, Haukenfrers J, Haqqani AS, Ly D, Zachar P, et al. A novel human induced pluripotent stem cell blood–brain barrier model: Applicability to study antibody-triggered receptor-mediated transcytosis. Sci Rep. 2018;8:1873.
Nishijima T, Piriz J, Duflot S, Fernandez AM, Gaitan G, Gomez-Pinedo U, et al. Neuronal activity drives localized blood–brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron. 2010;67:834–46.
Muruganandam A, Tanha J, Narang S, Stanimirovic D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood–brain barrier endothelium. FASEB J. 2002;16:240–2.
Farrington GK, Caram-Salas N, Haqqani AS, Brunette E, Eldredge J, Pepinsky B, et al. A novel platform for engineering blood–brain barrier-crossing bispecific biologics. FASEB J. 2014;28:4764–78.
Webster CI, Caram-Salas N, Haqqani AS, Thom G, Brown L, Rennie K, et al. Brain penetration, target engagement, and disposition of the blood–brain barrier-crossing bispecific antibody antagonist of metabotropic glutamate receptor type 1. FASEB J. 2016;30:1927–40.
Coleman JA, Molday RS. Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J Biol Chem. 2011;286:17205–16.
Stanimirovic D, Kemmerich K, Haqqani AS, Farrington GK. Engineering and pharmacology of blood–brain barrier-permeable bispecific antibodies. Adv Pharmacol. 2014;71:301–35.
Dashkoff J, Lerner EP, Truong N, Klickstein JA, Fan Z, Mu D, et al. Tailored transgene expression to specific cell types in the central nervous system after peripheral injection with AAV9. Mol Ther Methods Clin Dev. 2016;3:16081.
Qiao C, Yuan Z, Li J, He B, Zheng H, Mayer C, et al. Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther. 2011;18:403–10.
Merkel SF, Andrews AM, Lutton EM, Mu D, Hudry E, Hyman BT, et al. Trafficking of adeno-associated virus vectors across a model of the blood–brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. J Neurochem. 2017;140:216–30.
Raub TJ, Audus KL. Adsorptive endocytosis and membrane recycling by cultured primary bovine brain microvessel endothelial cell monolayers. J Cell Sci. 1990;97:127–38.
Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–5.
Conceição M, Mendonça L, Nóbrega C, Gomes C, Costa P, Hirai H, et al. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials. 2016;82:124–37.
Choudhury SR, Hudry E, Maguire CA, Sena-Esteves M, Breakefield XO, Grandi P. Viral vectors for therapy of neurologic diseases. Neuropharmacology. 2017;120:63–80.
Barcia C, Jimenez-Dalmaroni M, Kroeger KM, Puntel M, Rapaport AJ, Larocque D, et al. One-year expression from high-capacity adenoviral vectors in the brains of animals with pre-existing anti-adenoviral immunity: clinical implications. Mol Ther. 2007;15:2154–63.
Wilson JM. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab. 2009;96:151–7.
Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018;2018:359.
Ding W, Zhang L, Yan Z, Engelhardt JF. Intracellular trafficking of adeno-associated viral vectors. Gene Ther. 2005;12:873–80.
Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther. 2012;19:649–58.
Saraiva J, Nobre RJ, Pereira de Almeida L. Gene therapy for the CNS using AAVs: the impact of systemic delivery by AAV9. J Control Release. 2016;241:94–109.
McCarty DM, Young SM, Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet. 2004;38:819–45.
Hagedorn C, Schnödt-Fuchs M, Boehme P, Abdelrazik H, Lipps HJ, Büning H. S/MAR element facilitates episomal long-term persistence of adeno-associated virus vector genomes in proliferating cells. Hum Gene Ther. 2017;28:1169–79.
Bessis N, GarciaCozar FJ, Boissier M-C. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004;11(Suppl 1):S10–7.
Gray SJ. Gene therapy and neurodevelopmental disorders. Neuropharmacology. 2013;68:136–42.
Wright JF. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther. 2008;15:840–8.
Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, et al. Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–8.
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–80.
Aschauer DF, Kreuz S, Rumpel S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One. 2013;8:e76310.
Xue Y-Q, Ma B-F, Zhao L-R, Tatom JB, Li B, Jiang L-X, et al. AAV9-mediated erythropoietin gene delivery into the brain protects nigral dopaminergic neurons in a rat model of Parkinson’s disease. Gene Ther. 2010;17:83–94.
Green F, Samaranch L, Zhang HS, Manning-Bog A, Meyer K, Forsayeth J, et al. Axonal transport of AAV9 in nonhuman primate brain. Gene Ther. 2016;23:520–6.
Swain GP, Prociuk M, Bagel JH, O’Donnell P, Berger K, Drobatz K, et al. Adeno-associated virus serotypes 9 and rh10 mediate strong neuronal transduction of the dog brain. Gene Ther. 2014;21:28–36.
Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, et al. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA. 2000;97:3428–32.
Castle MJ, Perlson E, Holzbaur EL, Wolfe JH. Long-distance axonal transport of AAV9 is driven by dynein and kinesin-2 and is trafficked in a highly motile Rab7-positive compartment. Mol Ther. 2014;22:554–66.
Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther. 2006;13:528–37.
Zhang H, Yang B, Mu X, Ahmed SS, Su Q, He R, et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther. 2011;19:1440–8.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65.
Yang B, Li S, Wang H, Guo Y, Gessler DJ, Cao C, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol Ther. 2014;22:1299–309.
Albright BH, Storey CM, Murlidharan G, Castellanos Rivera RM, Berry GE, Madigan VJ, et al. Mapping the structural determinants required for AAVrh.10 transport across the blood–brain barrier. Mol Ther. 2018;26:510–23.
Cearley CN, Wolfe JH. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J Neurosci. 2007;27:9928–40.
Keiser NW, Yan Z, Zhang Y, Lei-Butters DCM, Engelhardt JF. Unique characteristics of AAV1, 2, and 5 viral entry, intracellular trafficking, and nuclear import define transduction efficiency in HeLa cells. Hum Gene Ther. 2011;22:1433–44.
Gray SJ, Blake BL, Criswell HE, Nicolson SC, Samulski RJ, McCown TJ, et al. Directed evolution of a novel adeno-associated virus (AAV) vector that crosses the seizure-compromised blood–brain barrier (BBB). Mol Ther. 2010;18:570–8.
Chen YH, Claflin K, Geoghegan JC, Davidson BL. Sialic acid deposition impairs the utility of AAV9, but not peptide-modified AAVs for brain gene therapy in a mouse model of lysosomal storage disease. Mol Ther. 2012;20:1393–9.
Chen YH, Chang M, Davidson BL. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat Med. 2009;15:1215–8.
Work LM, Büning H, Hunt E, Nicklin SA, Denby L, Britton N, et al. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol Ther. 2006;13:683–93.
Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204.
Tabata K, Sugano E, Murakami F, Yamashita T, Ozaki T, Tomita H. Improved transduction efficiencies of adeno-associated virus vectors by synthetic cell-permeable peptides. Biochem Biophys Res Commun. 2016;478:1732–8.
Liu Y, Kim YJ, Ji M, Fang J, Siriwon N, Zhang LI, et al. Enhancing gene delivery of adeno-associated viruses by cell-permeable peptides. Mol Ther Methods Clin Dev. 2014;1:12.
Russell S, Bennett J, Wellman JA, Chung DC, Yu Z-F, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–60.
Morabito G, Giannelli SG, Ordazzo G, Bido S, Castoldi V, Indrigo M, et al. AAV-PHP.B-mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol Ther. 2017;25:2727–42.
Tasfaout H, Lionello VM, Kretz C, Koebel P, Messaddeq N, Bitz D, et al. Single intramuscular injection of AAV-shRNA reduces DNM2 and prevents myotubular myopathy in mice. Mol Ther. 2018;26:1082–92.
Pfister EL, Chase KO, Sun H, Kennington LA, Conroy F, Johnson E, et al. Safe and efficient silencing with a Pol II, but not a Pol lII, promoter expressing an artificial miRNA targeting human Huntingtin. Mol Ther Nucleic Acids. 2017;7:324–34.
Biferi MG, Cohen-Tannoudji M, Cappelletto A, Giroux B, Roda M, Astord S, et al. A new AAV10-U7-mediated gene therapy prolongs survival and restores function in an ALS mouse model. Mol Ther. 2017;25:2038–52.
van Dommelen SM, Vader P, Lakhal S, Kooijmans SAA, van Solinge WW, Wood MJA, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release. 2012;161:635–44.
Sun D, Zhuang X, Zhang S, Deng Z-B, Grizzle W, Miller D, et al. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv Drug Deliv Rev. 2013;65:342–7.
El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7:2112–26.
Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93.
Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–79.
Li J, Chen X, Yi J, Liu Y, Li D, Wang J, et al. Identification and characterization of 293T cell-derived exosomes by profiling the protein, mRNA and microRNA components. PLoS One. 2016;11:e0163043.
Haqqani AS, Delaney CE, Tremblay T-L, Sodja C, Sandhu JK, Stanimirovic DB. Method for isolation and molecular characterization of extracellular microvesicles released from brain endothelial cells. Fluids Barriers CNS. 2013;10:4.
Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. 2017;27:172–88.
Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9.
Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–6.
Yang T, Martin P, Fogarty B, Brown A, Schurman K, Phipps R, et al. Exosome delivered anticancer drugs across the blood–brain barrier for brain cancer therapy in Danio rerio. Pharm Res. 2015;32:2003–14.
Yang T, Fogarty B, LaForge B, Aziz S, Pham T, Lai L, et al. Delivery of small interfering RNA to inhibit vascular endothelial growth factor in zebrafish using natural brain endothelia cell-secreted exosome nanovesicles for the treatment of brain cancer. AAPS J. 2017;19:475–86.
Chen CC, Liu L, Ma F, Wong CW, Guo XE, Chacko JV, et al. Elucidation of exosome migration across the blood–brain barrier model in vitro. Cell Mol Bioeng. 2016;9:509–29.
Record M, Subra C, Silvente-Poirot S, Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem Pharmacol. 2011;81:1171–82.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83.
Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;2014:3.
McKelvey KJ, Powell KL, Ashton AW, Morris JM, McCracken SA. Exosomes: mechanisms of uptake. J Circ Biomarkers. 2015;4:7.
van Dongen HM, Masoumi N, Witwer KW, Pegtel DM. Extracellular vesicles exploit viral entry routes for cargo delivery. Microbiol Mol Biol Rev. 2016;80:369–86.
Kooijmans SAA, Stremersch S, Braeckmans K, de Smedt SC, Hendrix A, Wood MJA, et al. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release. 2013;172:229–38.
Cooper JM, Wiklander PBO, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M, et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord. 2014;29:1476–85.
Lakhal S, Wood MJA. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays. 2011;33:737–41.
Maguire CA, Balaj L, Sivaraman S, Crommentuijn MHW, Ericsson M, Mincheva-Nilsson L, et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol Ther. 2012;20:960–71.
György B, Maguire CA. Extracellular vesicles: nature’s nanoparticles for improving gene transfer with adeno-associated virus vectors. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2018;10:e1488.
György B, Fitzpatrick Z, Crommentuijn MHW, Mu D, Maguire CA. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials. 2014;35:7598–609.
Hudry E, Martin C, Gandhi S, György B, Scheffer DI, Mu D, et al. Exosome-associated AAV vector as a robust and convenient neuroscience tool. Gene Ther. 2016;23:380–92.
Wassmer SJ, Carvalho LS, György B, Vandenberghe LH, Maguire CA. Exosome-associated AAV2 vector mediates robust gene delivery into the murine retina upon intravitreal injection. Sci Rep. 2017;7:45329.
György B, Sage C, Indzhykulian AA, Scheffer DI, Brisson AR, Tan S, et al. Rescue of hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol Ther. 2017;25:379–91.
Dumont J, Euwart D, Mei B, Estes S, Kshirsagar R. Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit Rev Biotechnol. 2016;36:1110–22.
Colao IL, Corteling R, Bracewell D, Wall I. Manufacturing exosomes: a promising therapeutic platform. Trends Mol Med. 2018;24:242–56.
Lener T, Gimona M, Aigner L, Börger V, Buzas E, Camussi G, et al. Applying extracellular vesicles based therapeutics in clinical trials—an ISEV position paper. J Extracell Vesicles. 2015;4:30087.
Karaoglu Hanzatian D, Schwartz A, Gizatullin F, Erickson J, Deng K, Villanueva R, et al. Brain uptake of multivalent and multi-specific DVD-Ig proteins after systemic administration. MAbs. 2018;10:765–77.
Boado RJ, Pardridge WM. Brain and organ uptake in the rhesus monkey in vivo of recombinant iduronidase compared to an insulin receptor antibody-iduronidase fusion protein. Mol Pharm. 2017;14:1271–7.
Boado RJ, Lu JZ, Hui EK-W, Pardridge WM. Reduction in brain heparan sulfate with systemic administration of an IgG Trojan horse-sulfamidase fusion protein in the mucopolysaccharidosis type IIIA mouse. Mol Pharm. 2018;15:602–8.
Pardridge WM, Boado RJ, Giugliani R, Schmidt M. Plasma pharmacokinetics of valanafusp alpha, a human insulin receptor antibody-iduronidase fusion protein, in patients with mucopolysaccharidosis type I. BioDrugs. 2018;32:169–76.
Abulrob A, Sprong H, Van Bergen en Henegouwen P, Stanimirovic D. The blood–brain barrier transmigrating single domain antibody: mechanisms of transport and antigenic epitopes in human brain endothelial cells. J Neurochem. 2005;95:1201–14.
Acknowledgements
The authors would like to thank Caroline Sodja for preparing the artwork. Conflict of Interest: DBS, JKS and WJC are employees of the Government of Canada. All rights to inventions, including royalties and copyrights to their contributions are owned by the Crown. DBS is co-inventor on patents protecting FC5 and IGF1R single-domain antibodies. DBS, JKS, and WJC declare that they have no further conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors are employees of the Government of Canada. None have any existing conflict of interest with regard to this manuscript.
Funding
No funding was received to conduct the study described in the manuscript, or used to assist with the preparation of the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stanimirovic, D.B., Sandhu, J.K. & Costain, W.J. Emerging Technologies for Delivery of Biotherapeutics and Gene Therapy Across the Blood–Brain Barrier. BioDrugs 32, 547–559 (2018). https://doi.org/10.1007/s40259-018-0309-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-018-0309-y