
Abstract
High-quality, safe, and effective biosimilars have the potential to increase access to biological therapies worldwide and to reduce cancer care costs. The European Medicines Agency (EMA) was the first regulatory authority to establish legislative procedures for the approval of biosimilars when they published their guidelines on similar biological medicinal products in 2005. Biosimilar epoetins were first approved in 2007, and a wealth of data has been collected over the last decade. Two biosimilar epoetins (under five commercial names) have been approved by the EMA so far. The availability of epoetin biosimilars generated discussion among the oncology community regarding prescribing these products, their efficacy, and their safety. These agents are approved only if they are shown in extensive analytical and clinical testing to have comparable quality, safety, and efficacy to the reference medicine, and real-world studies provide further data that biosimilar epoetins are an effective and well-tolerated option for the treatment of chemotherapy-induced anemia in patients with cancer. Other countries have adopted similar regulatory pathways to those in Europe and have approved epoetin biosimilars. The now extensive European experience with biosimilar epoetins should reassure regulators from other territories.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Biosimilar epoetin alfas have now been available in Europe for a decade. The availability of biosimilars provides an opportunity to contain spending on expensive medications while improving treatment access for patients. |
Based on the now extensive experience with biosimilar epoetins in Europe, healthcare professionals and their patients should be reassured about the therapeutic equivalence of biosimilar epoetins. |
1 Introduction
The high price of biological agents contributes to the huge healthcare burden associated with cancer treatment [1, 2]. However, expiration of patents for biological agents, including first-generation epoetins, provides an opportunity to develop and produce similar biological medicines, termed biosimilars [1]. High-quality, safe, and effective biosimilars have the potential to increase access to biological therapies worldwide and to reduce cancer care costs [3].
The European Medicines Agency (EMA) was the first regulatory authority to establish legislative procedures for the approval of biosimilars when they published their guidelines on similar biological medicinal products in 2005 [4]. The EMA regulatory pathway for the approval of biosimilars is based on demonstrating comparable quality, safety, and efficacy with no clinically meaningful differences to the originator (reference) medicine [4]. In addition to general guidelines on biosimilars, the EMA have issued guidelines for specific product classes, including biosimilar epoetins [5].
Chemotherapy-induced anemia (CIA) is a significant complication in cancer patients; however, erythropoiesis-stimulating agents (ESAs) such as epoetin alfa have been demonstrated to increase hemoglobin levels, reduce the need for transfusions, and improve quality of life in anemic patients with solid or hematologic malignancies receiving platinum or non-platinum chemotherapy [6].
Biosimilar epoetins were first approved in Europe in 2007, and a wealth of data has been collected over the last decade. Two biosimilar epoetins (under five brand names) have been approved by the EMA. HX575 is a biosimilar version of Eprex®/Erypo® (Janssen-Cilag, High Wycombe, UK), and in 2007, it became the first biosimilar epoetin to be approved in Europe. HX575 has the same international non-proprietary name (INN) as epoetin alfa and is marketed as Binocrit® (Sandoz GmbH, Kundl, Austria), Epoetin alfa HEXAL® (Hexal, Holzkirchen, Germany), and Abseamed® (Medice Arzneimittel, Iserlohn, Germany). SB309 is a biosimilar epoetin that also has Eprex®/Erypo® as its reference medicine, but the manufacturer applied for the INN epoetin zeta. It is marketed as Retacrit® (Hospira UK Limited, Maidenhead, UK) and Silapo® (STADA, Bad Vilbel, Germany). The availability of epoetin biosimilars generated discussion among the oncology community regarding possible concerns about prescribing these products. This review will discuss initial concerns raised about epoetin biosimilars, describe data gained on their use in Europe over the past 10 years, and discuss what can be learned as epoetin biosimilars are evaluated for use in other markets.
2 Initial Concerns About the Introduction of Biosimilar Epoetins
2.1 Low-Quality Medicines
One of the initial concerns raised about biosimilar medicines was that they may be of low quality, compared with the licensed reference medicine [7]. However, the same Good Manufacturing Practices (GMPs) apply equally to biosimilars and their reference medicines. In addition, extensive characterization of the biosimilar is a key requirement of the regulatory process. Characterization generally consists of analyses of primary (i.e., amino acid sequence), secondary, tertiary, and quaternary structures, including aggregation, post-translational modification (e.g., glycosylation, phosphorylation, and deamidation), chemical modification, and biological activities [8].
Biosimilar manufacturers are able to take advantage of technological improvements and use state-of-the-art systems to produce and purify biosimilar proteins. This contrasts with manufacturers of reference medicines, who may be locked in to older technologies due to the financial and regulatory impact of making changes to their methods [9]. Indeed, studies have shown the quality of biosimilar epoetins and the reference medicine to be equally as good or, in some cases, they have shown the biosimilars to have lower levels of certain impurities [10, 11]. It should be noted, however, that assured quality may not be the case for some epoetins available on the market worldwide, in particular, “intended copy” biological epoetins manufactured in countries where rigorous regulations and standardized manufacturing processes may not be in place or adhered to [12].
2.2 Lack of Similarity with the Reference Medicine
Biologics are derived from living cells or organisms and consist of relatively large and often highly complex molecular entities. There were initial concerns in some quarters that biosimilars would not be sufficiently similar to the reference medicine. A small degree of controlled variability is common with all biologics, and batches of the same medicine (whether a reference or biosimilar medicine) are never identical to each other [7]. Due to unavoidable differences in manufacturing processes, a biosimilar and the reference medicine will also not be completely identical (for example, in the levels of minor impurities or variation in the glycan profile); however, extensive characterization and comparison, as detailed above, should ensure that any microheterogeneity does not impact on the clinical performance of the biosimilar. In addition, it is a clear requirement that the protein backbone (i.e., amino acid sequence) of a biosimilar must be identical to the reference medicine.
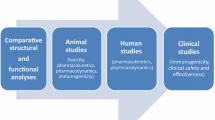
Some small differences in non-critical parameters between biosimilars and the reference epoetin have been demonstrated. For example, high mannose-6-phopshate structures have been detected in HX575 at higher levels than in reference epoetin alfa [13]. For SB309, the protein contains more glycoforms without an O-glycan chain than the reference epoetin alfa; however, there appear to be no clinical consequences [14]. Neither of these quality attributes is critical to clinical efficacy, safety, or immunogenicity, as confirmed in the clinical development programs and follow-up studies for each biosimilar that confirmed similarity with the reference medicine (Fig. 1).

Clinical development programs of European biosimilar epoetins. CIA chemotherapy-induced anemia, CKD chronic kidney disease, HD hemodialysis, IV intravenous, PD pharmacodynamic, PK pharmacokinetic, SC subcutaneous
2.3 Extrapolation of Indications
Concerns have been expressed about using biosimilars in indications or in patient populations that are approved for the reference medicine but have not been formally investigated during the clinical development of the biosimilar [7, 15]; this is known as extrapolation. Extrapolation is an important element of the biosimilarity concept. Several professional medical societies have discouraged use of biosimilars in extrapolated indications [16,17,18,19,20,21]. However, from a scientific and regulatory perspective, the active substance of the biosimilar is considered to be just another version of the active substance of the reference medicine [15].
The scientific principles behind extrapolation of data are not new for biosimilars; they also apply to the comparison of approved products before/after a change in the manufacturing process [15]. When biosimilar comparability has been demonstrated, extrapolation to other indications of the reference medicine could be acceptable, but needs to be scientifically justified and considered in light of all available (analytical, nonclinical, and clinical) data [15]. Regulators might require additional data to be provided in some instances: the active substance of the reference medicine interacts with several receptors that may have a different impact in the tested and non-tested therapeutic indications; the active substance itself has more than one active site and the sites may have a different impact in different therapeutic indications; or the studied therapeutic indication is not sensitive enough to detect differences in all relevant aspects of efficacy and safety. Since clinical studies are usually less sensitive for detecting potential differences between the biosimilar and the reference medicine, the additional data would preferably include pharmacodynamic parameters and/or specific functional assays that reflect the pharmacologic action(s) of the molecule [15]. For epoetin, the mechanism of action is the same for all currently approved indications, and there is only one known epoetin receptor; therefore, EMA states that ‘demonstration of efficacy and safety in renal anemia will allow extrapolation to other indications of the reference medicine with the same route of administration’ [5].
2.4 Concerns About Clinical Safety, Including Immunogenicity
Immunogenicity is a safety concern for all biological medicines. Antibodies generated against exogenously administered erythropoietin may, in rare cases, have a neutralizing effect and lead to pure red cell aplasia (PRCA) [7]. This is usually more of a concern in patients with chronic kidney disease (CKD) than in patients with CIA [2]. Analytical or animal data cannot predict immune responses in humans; human safety data are therefore required for EMA approval of all biosimilar products, including biosimilar epoetins [7]. This involves at least 12 months of comparative immunogenicity results, using a validated and highly sensitive assay for anti-epoetin antibody detection [5].
None of the studies in the clinical program with HX575 or SB309 administered intravenously (IV) reported the presence of neutralizing anti-erythropoietin antibodies or any signs or symptoms consistent with immune-mediated PRCA [13, 22]. The subcutaneous (SC) route of administration is usually more immunogenic than the IV route [5]. Indeed, two patients with CKD-related anemia developed neutralizing anti-erythropoietin antibodies following SC administration of HX575 in an investigational randomized clinical trial [23]. A root-cause analysis suggested that tungsten contamination of prefilled syringes may have increased the immunogenicity of a small number of study drug batches [24]. A change in the manufacturing process was implemented (introduction of low-tungsten syringes) and, following the completion of a clinical study, HX575 was approved by the EMA for SC administration in CKD patients in March 2016 [25]. Considering post-approval data for EU-authorized biosimilar epoetins, a patient with CKD-related anemia receiving SC epoetin zeta in Italy was reported to have developed immune-mediated PRCA [26]. In contrast to EMA-approved and regulated biosimilar epoetins, confirmed cases of PRCA have been reported in 23 Thai patients receiving regionally manufactured copy epoetins not approved in Europe [27]. This higher-than-expected rate corresponds to an estimated frequency of one PRCA case for every 2608 patients using epoetin.
Thrombotic complications may also be a concern with epoetins in patients with cancer, particularly if there is an exaggerated pharmacodynamic response [5]. The safety trial with SB309 was conducted in patients with CIA to provide information on the incidence of clinically significant thrombotic events, which was shown to be similar to or lower than the published rate for epoetin-treated cancer patients [22].
To further ensure safety, post-marketing, and risk management, pharmacovigilance plans must be submitted for biosimilar epoetins in Europe [28]. In a post-marketing study of IV HX575, involving more than 1700 patients with anemia due to CKD, no subjects developed anti-erythropoietin antibodies [29]. Data from the ORHEO (place of biOsimilaRs in the therapeutic management of anemia secondary to chemotherapy in HaEmatology and Oncology) observational study (n = 2333) in France, conducted primarily in patients using SB309, indicate a post-marketing safety profile consistent with the reference epoetin alfa [30, 31]. More recently, a population-based observational cohort study of more than 13,000 patients reported no difference in safety outcomes between treatment with biosimilar epoetins, reference epoetin alfa, and other ESAs [32]. Thus, for those initially concerned about the safety of epoetin biosimilars, reassurance is provided by data now accumulated, which demonstrate that there have been no specific safety signals after 10 years of clinical usage in Europe.
The current estimated exposure to HX575/Binocrit® is > 200 million patient-days; around 4000 patients have been included in clinical studies with Binocrit®, and the product is launched in > 40 countries. For SB309/Retacrit®, the total estimated population exposure (in the oncology and nephrology indication) between December 2007 and November 2013 was 54,554,947 patient-days, based on post-marketing sales, with over 35,000,000 patient-days’ experience in the oncology indication [31].
There have also been some discussions on potential safety risks associated with switching to and from biosimilar products [33]. A retrospective drug utilization study conducted in Italy quantified the occurrence of switching between different epoetins [34]. When switched from the reference epoetin alfa, 62% of patients received another patented epoetin alfa (darbepoetin alfa, epoetin beta, or methoxypolyethyleneglycol-epoetin beta) and 32% received a biosimilar epoetin alfa. Patients who initially used a biosimilar epoetin alfa were mostly switched to the reference epoetin alfa (57.5%), with 27.5% switched to another patented epoetin and 15% to another biosimilar epoetin alfa [34]. The probability of switching was associated with the duration of treatment: about 15% of users switched within 12 months and almost 25% within 2 years of observation. Switching was not restricted to the replacement of reference epoetins with biosimilar epoetins, but also extended to products that have not been directly compared in clinical studies. The authors concluded that the level of switching may provide reassurance to physicians when taken together with other sources of comparative evidence [34]. The decision to switch epoetins is usually due to a hospital or hemodialysis unit changing all of its patients from one erythropoietin to another [33]. Switching between epoetins is probably much less likely in the setting of CIA than other indications (such as CKD-related anemia), as the duration of epoetin therapy is typically much shorter in the oncology setting. A review of data from clinical trials, pharmacovigilance databases, and an overview of the literature found no evidence to suggest that switching to and from different biopharmaceuticals (including biosimilars) leads to safety concerns [33]. A retrospective analysis of 149 adult hemodialysis patients receiving stable ESA doses has reported a dose penalty when switching from the reference to a biosimilar epoetin alfa [35]. However, this report of a dose penalty is contrary to other published data. These include a large post-approval study of IV HX575 in patients (n = 1695) with CKD and a population-based analysis of real-world data from ambulatory patients (n = 6117) with CKD undergoing maintenance hemodialysis [29, 36].
3 Looking Beyond the European Experience
In the European Union (EU), the first biosimilar product was approved in 2006 and the first biosimilar epoetin in 2007 [37]. Since then, many countries and regions have developed regulatory and approval processes based on the EMA’s approach. For example, biosimilar epoetins were first approved in Australia in 2010 and in New Zealand in 2013 [28]. However, it is only relatively recently that the first biosimilar was approved in the United States (US). The US enacted the Biologics Price Competition and Innovation Act in 2010 as part of the Patient Protection and Affordable Care Act, to clarify and expedite the approval process for biosimilar agents [37]. US Food and Drug Administration (FDA) guidance on biosimilars was slow to emerge due to debates by stakeholders over how stringent the standards for new biosimilar approvals should be and whether approved biosimilars should be used interchangeably with reference medicines [38]; final guidelines on scientific and quality considerations in demonstrating biosimilarity were issued by the FDA in 2015 [39]. Unlike in Europe, the FDA pathway includes a regulatory designation on interchangeability, which, in the US, refers to the ability to automatically substitute a medicine at the pharmacy level. This requires additional data over and above what is needed for biosimilarity alone; the sponsor must demonstrate that, if administered more than once to an individual, the risk in terms of safety or diminished efficacy of switching between the biosimilar and the reference medicine is not greater than the risk of using the reference medicine alone [39]. The EMA and Australian authorities do not provide an interchangeable recommendation when approving a biosimilar. However, in a recent article authored by employees of several national regulatory agencies in Europe, the authors state their opinion that EU-approved biosimilars are considered medically interchangeable; this refers, in the EU, to the practice of changing one medicine for another that is expected to achieve the same clinical effect in a given clinical setting and in any patient, with the agreement of the prescriber [40]. The authors argue that attempts to provide data on a lack of switch-related changes in safety and efficacy with specific interchangeability studies would be demanding, and unlikely to provide definitive answers [40]; instead, state-of-the-art demonstration of biosimilarity plus heightened post-marketing surveillance should be a sufficient and realistic approach to ensuring interchangeability with supervision from prescribers. The authors conclude that EU-licensed biosimilars are interchangeable ‘if the patient is clinically monitored, will receive the necessary information, and, if needed, training on the administration of the new product’ [40]. In Europe, decisions on automatic substitution between a biological reference medicine and its biosimilar are made at the level of individual countries [5, 28].
Other additional areas of debate in the US relate to naming, payment, and pharmacovigilance [38]. Under the FDA regulatory pathway, the first biosimilar product, a granulocyte colony-stimulating factor, Zarzio® (Zarxio®, filgrastim-sndz; Hexal AG, Germany), was approved in the US in March 2015 for all the same indications as the reference medicine, Neupogen® [41]. Unlike in Europe, the FDA approved filgrastim-sndz without a requirement for post-marketing studies, and a lack of post-approval monitoring may be a concern to some healthcare professionals in the US [38]. Therefore, the provision of adequate professional and patient education regarding the equivalence of biosimilars is likely to be an important factor in acceptance [38]. In addition, the European experience should provide reassurance and the likely economic benefits may provide incentives for adoption. The expiry of patents in 2014 has potentially opened the US market to biosimilar epoetins; indeed, the FDA’s Oncology Drugs Advisory Committee has recently recommended approval of a proposed biosimilar epoetin alfa [42]. In another recent development in the US, the FDA determined that the ESA Risk Evaluation and Mitigation Strategy (which was limited to the use of ESAs to treat patients with anemia due to associated myelosuppressive chemotherapy) was no longer necessary to ensure that the benefits outweigh the risks of shortened overall survival and/or increased risk of tumor progression or recurrence in patients with cancer [43].
4 Summary
Biosimilars, including biosimilar epoetin alfas, have now been available in Europe for a decade. The availability of biosimilars offers affordable, high-quality, effective alternative treatments, and may help contain healthcare budgets, while improving treatment access for patients. These agents are approved only if they are shown in extensive analytical and clinical testing to have comparable quality, safety, and efficacy to the reference medicine, and real-world studies provide further data that biosimilar epoetins are an effective and well-tolerated option for the treatment of anemia. Other countries have adopted similar regulatory pathways to those in Europe and have approved epoetin biosimilars; however, expansion into the US market is still awaited. The now extensive European experience will reassure healthcare professionals about the therapeutic equivalence of biosimilar epoetins, and, together with comprehensive education, may help drive acceptance among payers and healthcare professionals if these agents gain approval.
Change history
28 February 2018
Figure 1, HX575 column, 5th box down, which previously read “SC HX575 vs. Eprex®/Erypo® 417 patients with CKD-related anemia” as shown here,
References
Aapro M. Biosimilars in oncology: current and future perspectives. GaBI J. 2013;2:91–3.
Bennett CL, Chen B, Hermanson T, Wyatt MD, Schulz RM, Georgantopoulos P, Kessler S, Raisch DW, Qureshi ZP, et al. Regulatory and clinical considerations for biosimilar oncology drugs. Lancet Oncol. 2014;15:e594–605.
Tabernero J, Vyas M, Giuliani R, Arnold D, Cardoso F, Casali PG, Cervantes A, Eggermont AM, Eniu A, et al. Biosimilars: a position paper of the European Society for Medical Oncology, with particular reference to oncology prescribers. ESMO Open. 2016;1:e000142.
European Medicines Agency (EMA). Guideline on similar biological medicinal products. European Medicines Agency. 2005. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf. Accessed 15 Jun 2017.
European Medicines Agency (EMA). Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant erythropoietins (Revision). European Medicines Agency. 2010. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089474.pdf. Accessed 15 Jun 2017.
Henry DH. The evolving role of epoetin alfa in cancer therapy. Oncologist. 2004;9:97–107.
Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, Gravanis I, Heim HK, Heinonen E, et al. Biosimilars: what clinicians should know. Blood. 2012;120:5111–7.
Curigliano G, O’Connor DP, Rosenberg JA, Jacobs I. Biosimilars: extrapolation for oncology. Crit Rev Oncol Hematol. 2016;104:131–7.
Schellekens H, Moors E. Clinical comparability and European biosimilar regulations. Nat Biotechnol. 2010;28:28–31.
Brinks V, Hawe A, Basmeleh AH, Joachin-Rodriguez L, Haselberg R, Somsen GW, Jiskoot W, Schellekens H. Quality of original and biosimilar epoetin products. Pharm Res. 2011;28:386–93.
Halim LA, Brinks V, Jiskoot W, Romeijn S, Haselberg R, Burns C, Wadhwa M, Schellekens H. Quality and batch-to-batch consistency of original and biosimilar epoetin products. J Pharm Sci. 2016;105:542–50.
Covic A, Abraham I. State-of-the-art biosimilar erythropoietins in the management of renal anemia: lessons learned from Europe and implications for US nephrologists. Int Urol Nephrol. 2015;47:1529–39.
European Medicines Agency (EMA). Binocrit, INN-epoetin alfa: scientific discussion. European Medicines Agency. 2007. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000725/WC500053615.pdf. Accessed 15 Jun 2017.
Mikhail A, Farouk M. Epoetin biosimilars in Europe: five years on. Adv Ther. 2013;30:28–40.
Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124:3191–6.
Shaw BE, Confer DL, Hwang WY, Pamphilon DH, Pulsipher MA. Concerns about the use of biosimilar granulocyte colony-stimulating factors for the mobilization of stem cells in normal donors: position of the World Marrow Donor Association. Haematologica. 2011;96:942–7.
Barosi G, Bosi A, Abbracchio MP, Danesi R, Genazzani A, Corradini P, Pane F, Tura S. Key concepts and critical issues on epoetin and filgrastim biosimilars. A position paper from the Italian Society of Hematology, Italian Society of Experimental Hematology, and Italian Group for Bone Marrow Transplantation. Haematologica. 2011;96:937–42.
Danese S, Gomollon F, Governing Board and Operational Board of ECCO. ECCO position statement: the use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD). J Crohns Colitis. 2013;7:586–9.
Fonseca JE, Gonçalves J, Araújo F, Cordeiro I, Teixeira F, Canhão H, da Silva JA, Garcês S, Miranda LC, et al. The Portuguese Society of Rheumatology position paper on the use of biosimilars. Acta Reumatol Port. 2014;39:60–71.
Fiorino G, Girolomoni G, Lapadula G, Orlando A, Danese S, Olivieri I, SIR, SIDeMaST, IG-IBD. The use of biosimilars in immune-mediated disease: a joint Italian Society of Rheumatology (SIR), Italian Society of Dermatology (SIDeMaST), and Italian Group of Inflammatory Bowel Disease (IG-IBD) position paper. Autoimmun Rev. 2014;13:751–5.
Mularczyk A, Gonciarz M, Bartnik W, Durlik M, Eder P, Gąsiorowska A, Linke K, Łodyga M, Łykowska-Szuber L, et al. Biosimilar medicines—their use in the treatment of inflammatory bowel diseases. Position statement of the Working Group of the Polish National Consultant in Gastroenterology. Prz Gastroenterol. 2014;9:1–3.
European Medicines Agency (EMA). Retacrit, INN-epoetin zeta: scientific discussion. European Medicines Agency. 2007. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000872/WC500054374.pdf. Accessed 15 Jun 2017.
Haag-Weber M, Eckardt KU, Hörl WH, Roger SD, Vetter A, Roth K. Safety, immunogenicity and efficacy of subcutaneous biosimilar epoetin-α (HX575) in non-dialysis patients with renal anemia: a multi-center, randomized, double-blind study. Clin Nephrol. 2012;77:8–17.
Seidl A, Hainzl O, Richter M, Fischer R, Böhm S, Deutel B, Hartinger M, Windisch J, Casadevall N, London GM, Macdougall I. Tungsten-induced denaturation and aggregation of epoetin alfa during primary packaging as a cause of immunogenicity. Pharm Res. 2012;29:1454–67.
Casadevall N, Dobronravov V, Eckardt KU, Ertürk S, Martynyuk L, Schmitt S, Schaffar G, Rudy A, Krendyukov A, Ode M. Evaluation of the safety and immunogenicity of subcutaneous HX575 epoetin alfa in the treatment of anaemia associated with chronic kidney disease in pre-dialysis and dialysis patients. Clin Nephrol. 2017;88:190–7.
Panichi V, Ricchiuti G, Scatena A, Del Vecchio L, Locatelli F. Pure red cell aplasia by epoetin zeta. Clin Kidney J. 2016;9:599–602.
Praditpornsilpa K, Tiranathanagul K, Kupatawintu P, Jootar S, Intragumtornchai T, Tungsanga K, Teerapornlertratt T, Lumlertkul D, Townamchai N, et al. Biosimilar recombinant human erythropoietin induces the production of neutralizing antibodies. Kidney Int. 2011;80:88–92.
Leung LK, Mok K, Liu C, Chan SL. What do oncologists need to know about biosimilar products? Chin J Cancer. 2016;35:91.
Hörl WH, Locatelli F, Haag-Weber M, Ode M, Roth K, Epo-PASS study group. Prospective multicenter study of HX575 (biosimilar epoetin-α) in patients with chronic kidney disease applying a target hemoglobin of 10–12 g/dl. Clin Nephrol. 2012;78:24–32.
Michallet M, Luporsi E, Soubeyran P, Amar NA, Boulanger V, Carreiro M, Dourthe LM, Labourey JL, Lepille D, et al. BiOsimilaRs in the management of anaemia secondary to chemotherapy in HaEmatology and Oncology: results of the ORHEO observational study. BMC Cancer. 2014;14:503.
Michallet M, Losem C. Biosimilar epoetin zeta in oncology and haematology: development and experience following 6 years of use. Acta Haematol. 2016;135:44–52.
Trotta F, Belleudi V, Fusco D, Amato L, Mecozzi A, Mayer F, Sansone M, Davoli M, Addis A. Comparative effectiveness and safety of erythropoiesis-stimulating agents (biosimilars vs originators) in clinical practice: a population-based cohort study in Italy. BMJ Open. 2017;7:e011637.
Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12:1473–85.
D’Amore C, Da Cas R, Rossi M, Traversa G. Switching between epoetins: a practice in support of biosimilar use. BioDrugs. 2016;30:27–32.
Minutolo R, Borzumati M, Sposini S, Abaterusso C, Carraro G, Santoboni A, Mura C, Filiberti O, Santoro D, et al. Dosing penalty of erythropoiesis-stimulating agents after switching from originator to biosimilar preparations in stable hemodialysis patients. 2016;68:170–2.
Hörbrand F, Bramlage P, Fischaleck J, Hasford J, Brunkhorst R. A population-based study comparing biosimilar versus originator erythropoiesis-stimulating agent consumption in 6,117 patients with renal anaemia. Eur J Clin Pharmacol. 2013;69:929–36.
Generics and Biosimilars Initiative (GaBI). Biosimilars approved in Europe. Generics and Biosimilars Initiative. 2016. http://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe. Accessed 15 Jun 2017.
Wish JB, Charytan C, Chertow GM, Kalantar-Zadeh K, Kliger AS, Rubin RJ, Yee J, Fishbane S. Introduction of biosimilar therapeutics into nephrology practice in the United States: report of a scientific workshop sponsored by the National Kidney Foundation. Am J Kidney Dis. 2016;68:843–52.
US Food and Drug Administration (FDA). Biosimilars: biosimilarity guidelines. US Food and Drug Administration. Nov 2015. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm290967.htm. Accessed 15 Jun 2017.
Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. BioDrugs. 2017;31:83–91.
US Food and Drug Administration (FDA). Press announcements: FDA approves first biosimilar product Zarxio. US Food and Drug Administration. 6 Mar 2015. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm436648.htm. Accessed 15 Jun 2017.
US Food and Drug Administration (FDA). FDA briefing document: BLA 125545 “Epoetin Hospira”, a proposed biosimilar to Epogen/Procrit (epoetin alfa). 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559967.pdf. Accessed 15 Jun 2017.
US Food and Drug Administration (FDA). Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). US Food and Drug Administration. 2017. https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Accessed 15 Jun 2017.
Acknowledgements
Medical writing support was provided by Tony Reardon, Spirit Medical Communications Ltd., supported by Hexal AG.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MA and PG have acted as advisors to Sandoz. MS and AK are employees of Sandoz/Hexal AG.
Funding
Medical writing support was funded by Sandoz/Hexal AG.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Aapro, M., Krendyukov, A., Schiestl, M. et al. Epoetin Biosimilars in the Treatment of Chemotherapy-Induced Anemia: 10 Years’ Experience Gained. BioDrugs 32, 129–135 (2018). https://doi.org/10.1007/s40259-018-0262-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-018-0262-9